
Synthesis, Structural Characterization and Anticancer Activity of New 5-Trifluoromethyl-2-thioxo-thiazolo[4,5-d]pyrimidine Derivatives


Abstract
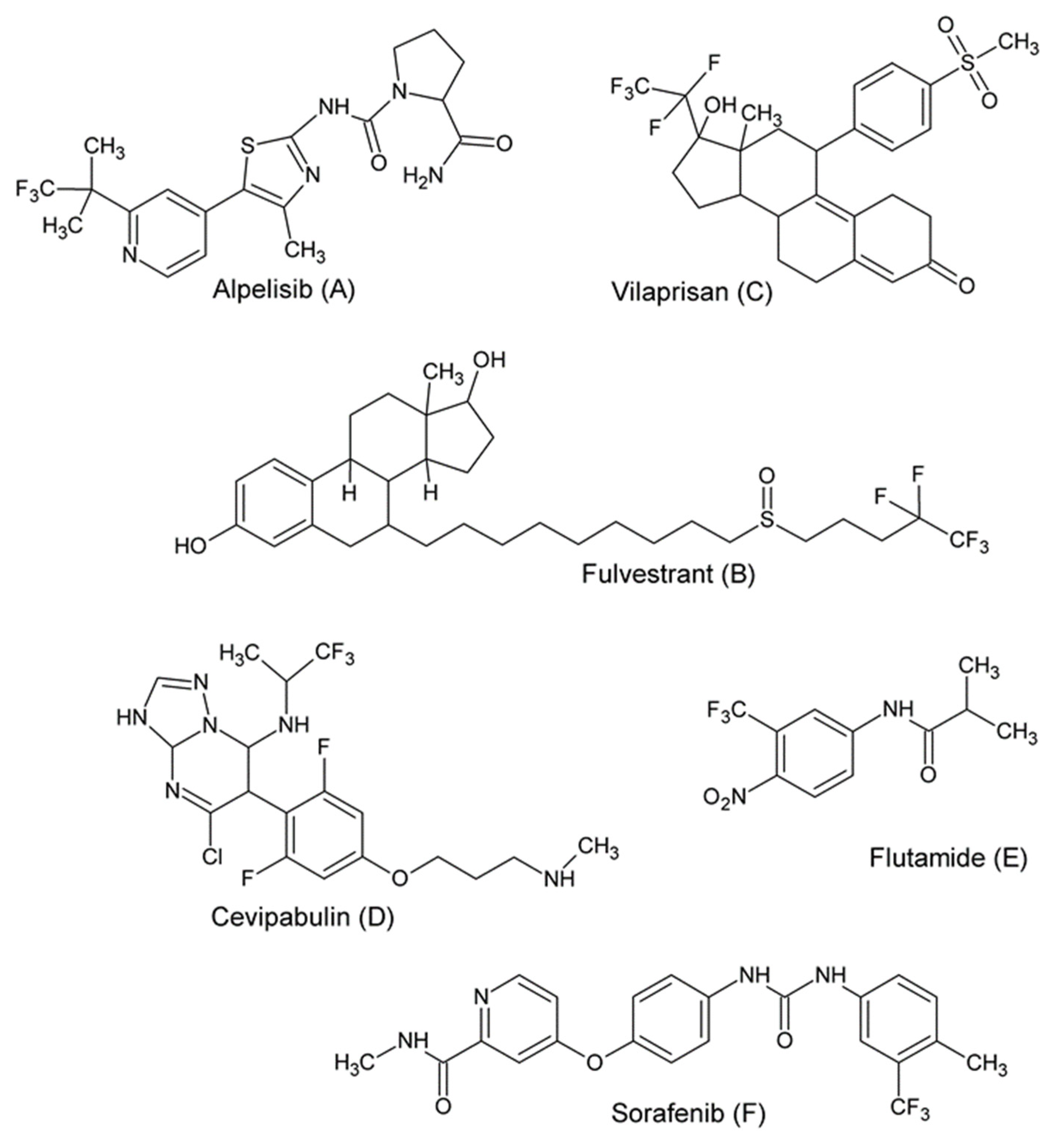
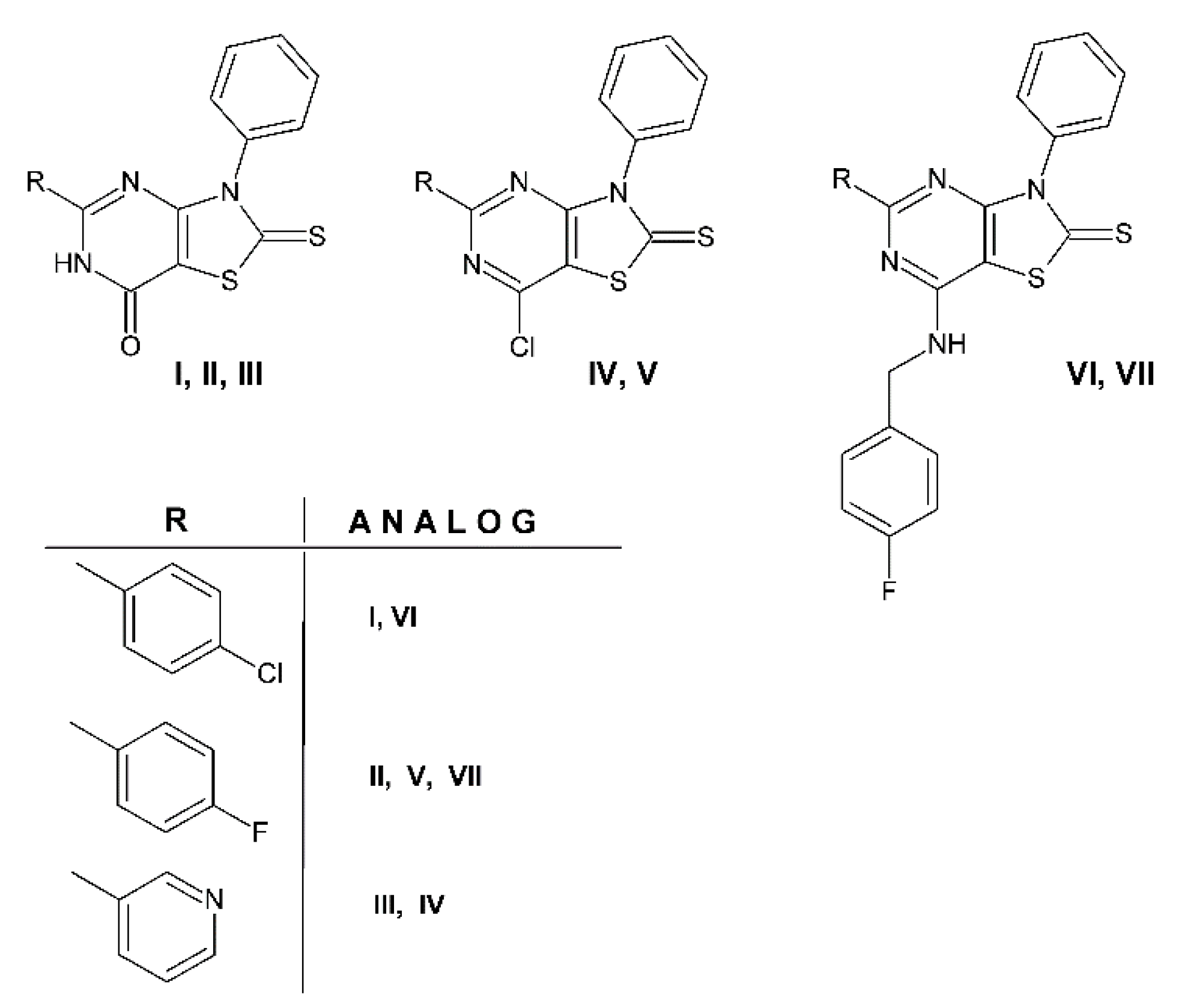
:1. Introduction
2. Results and Discussion
2.1. Chemistry
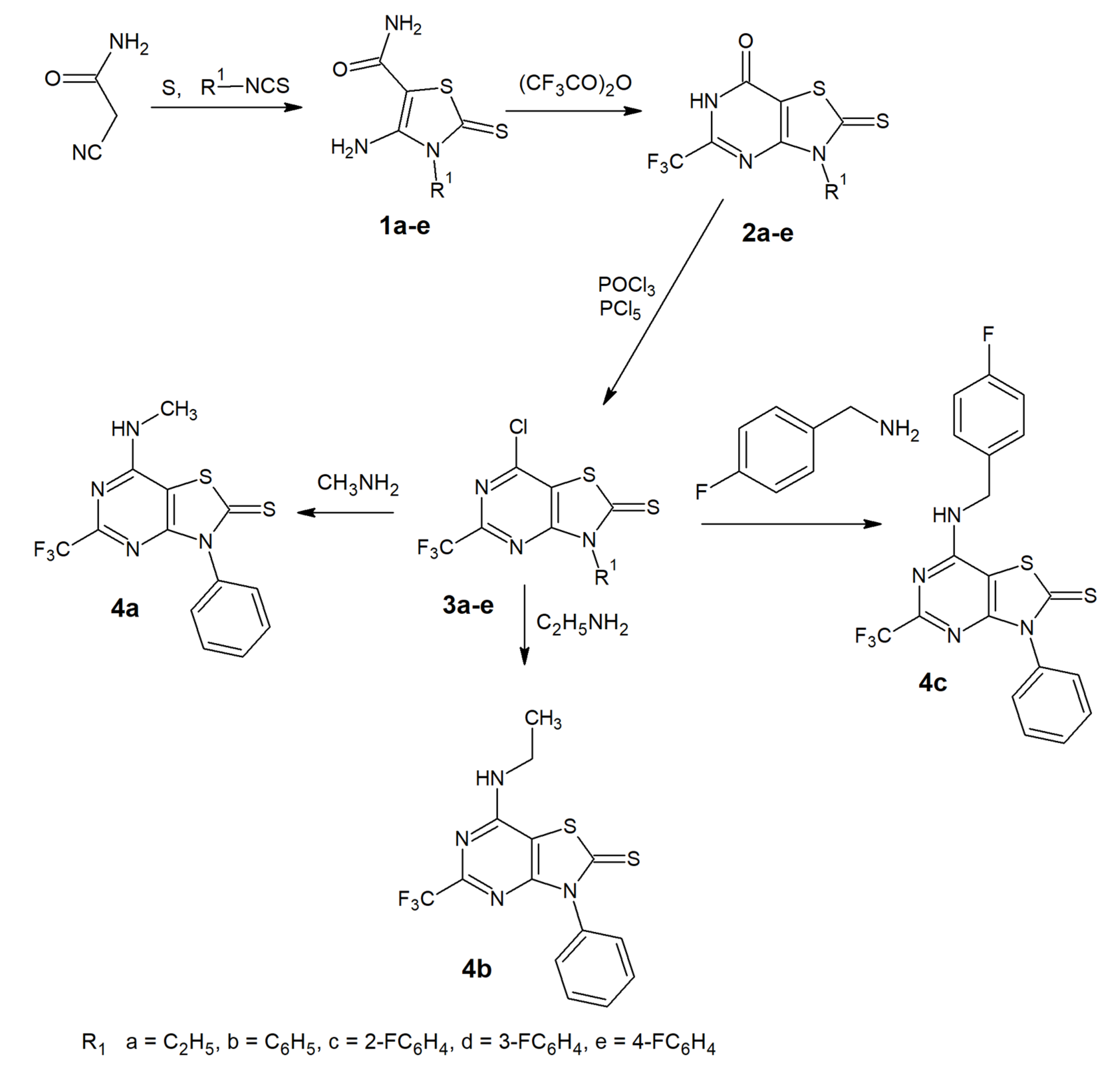
2.1.1. Synthesis and Physiochemical Properties
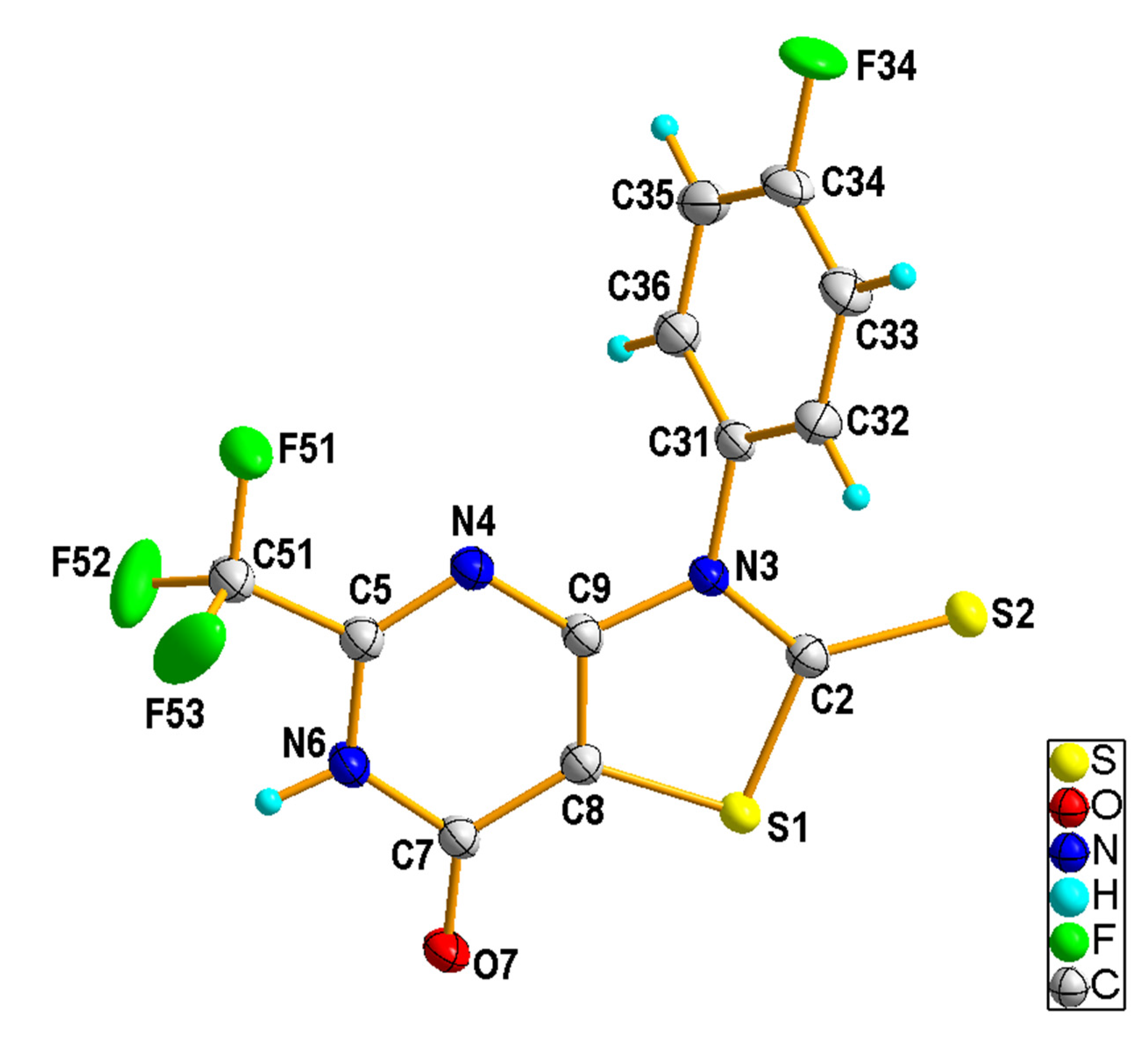
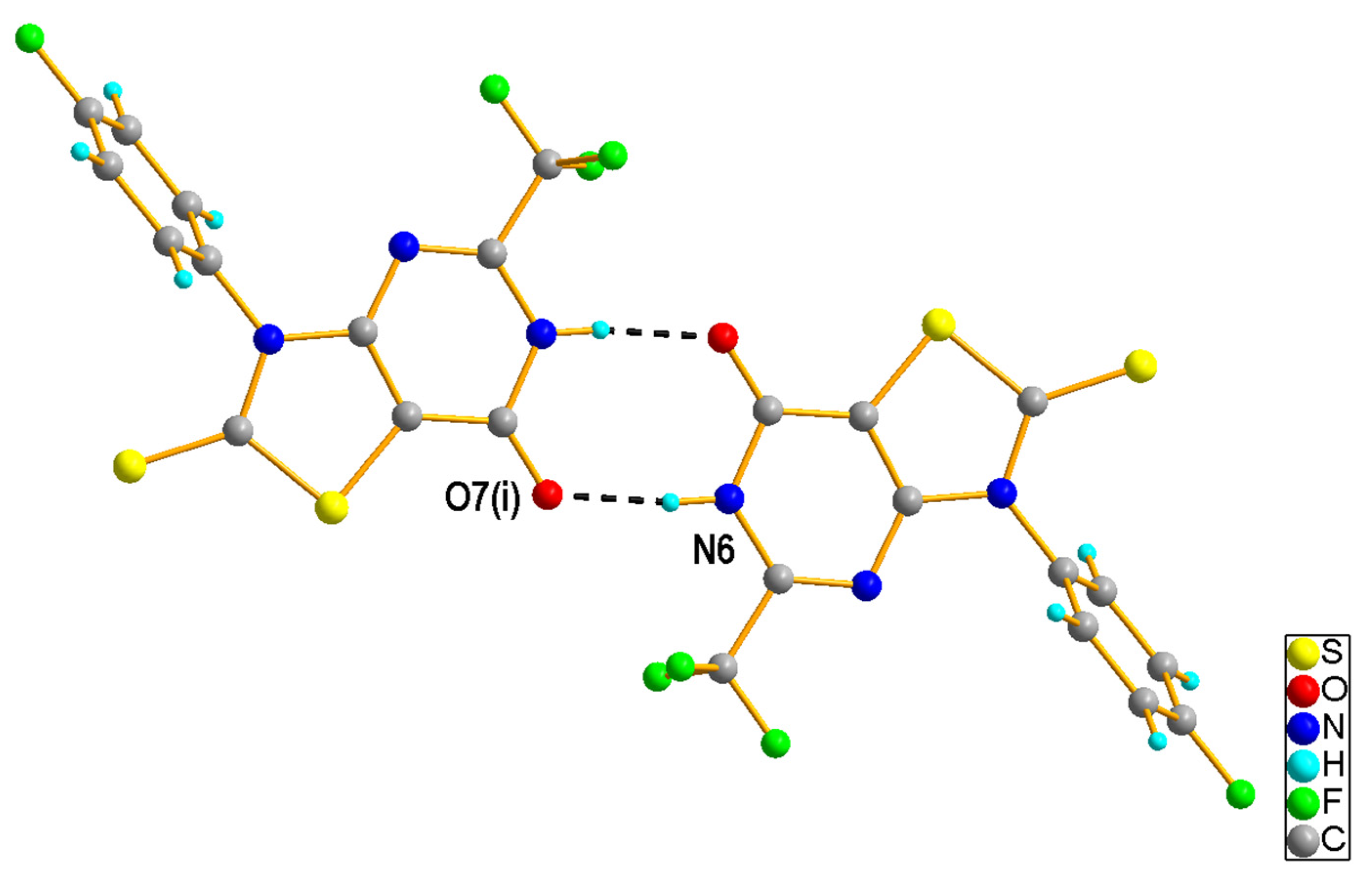
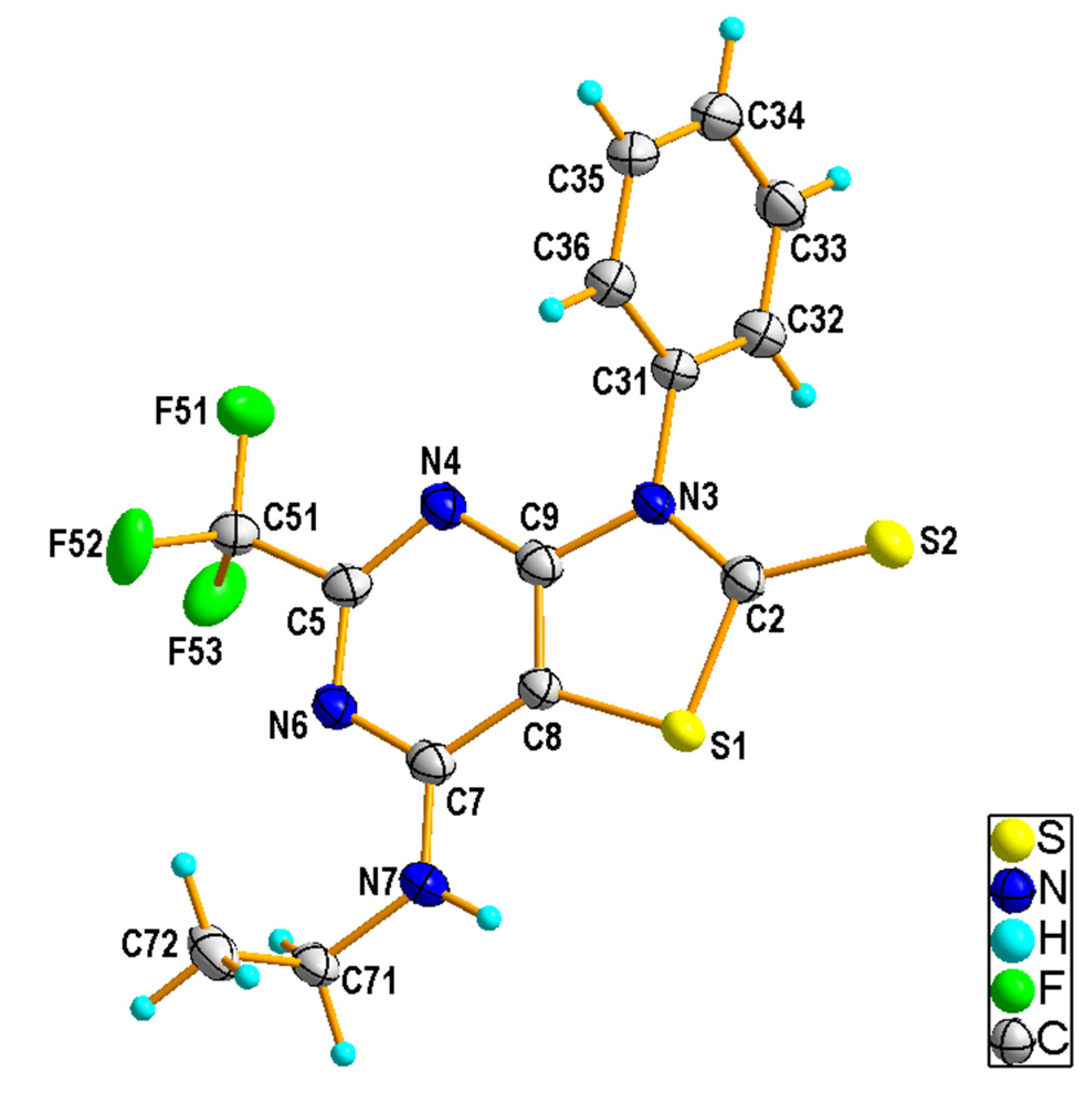
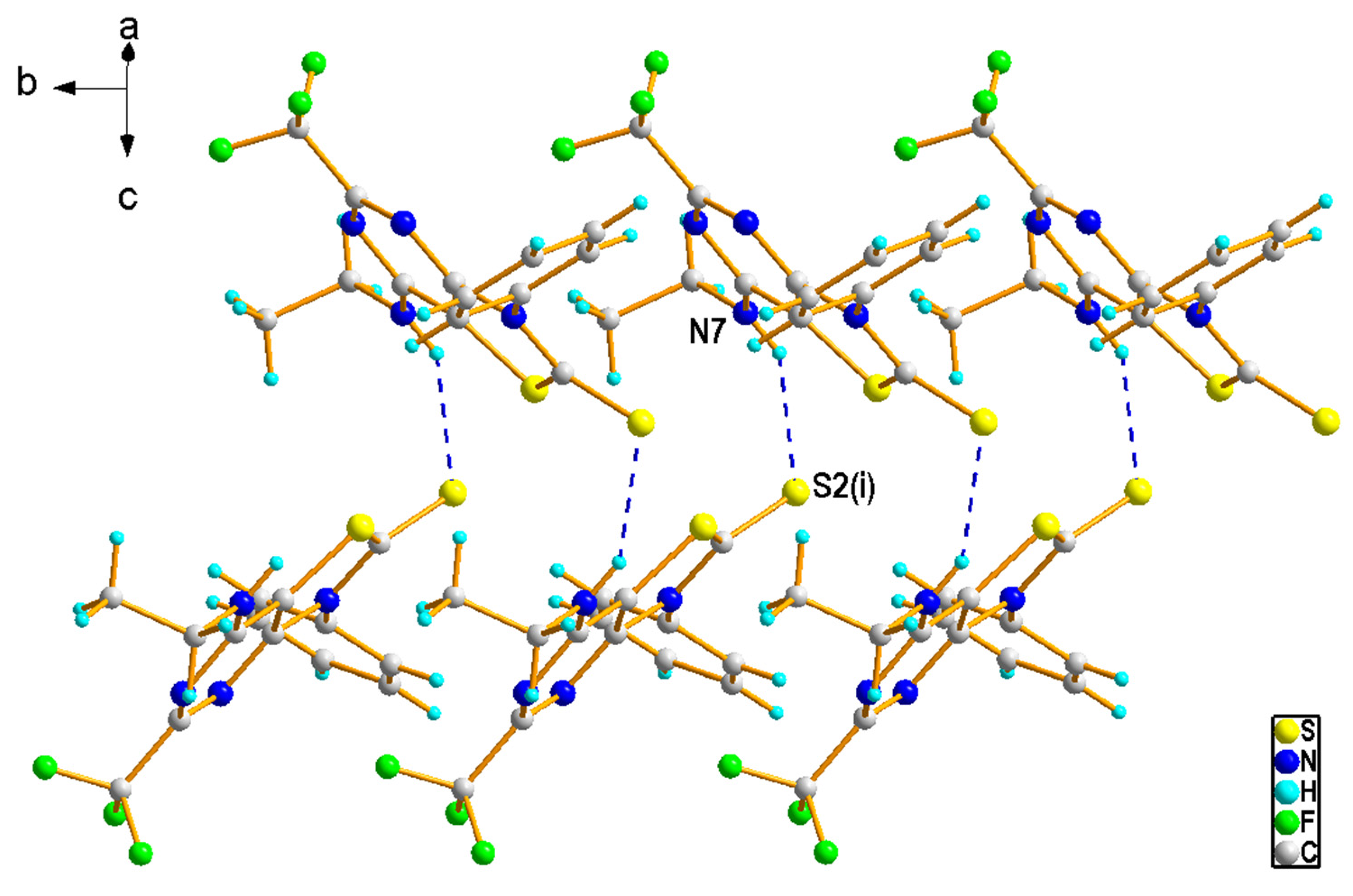
2.1.2. Crystal Structures of the Compounds 2e and 4b
2.2. Biological Activity
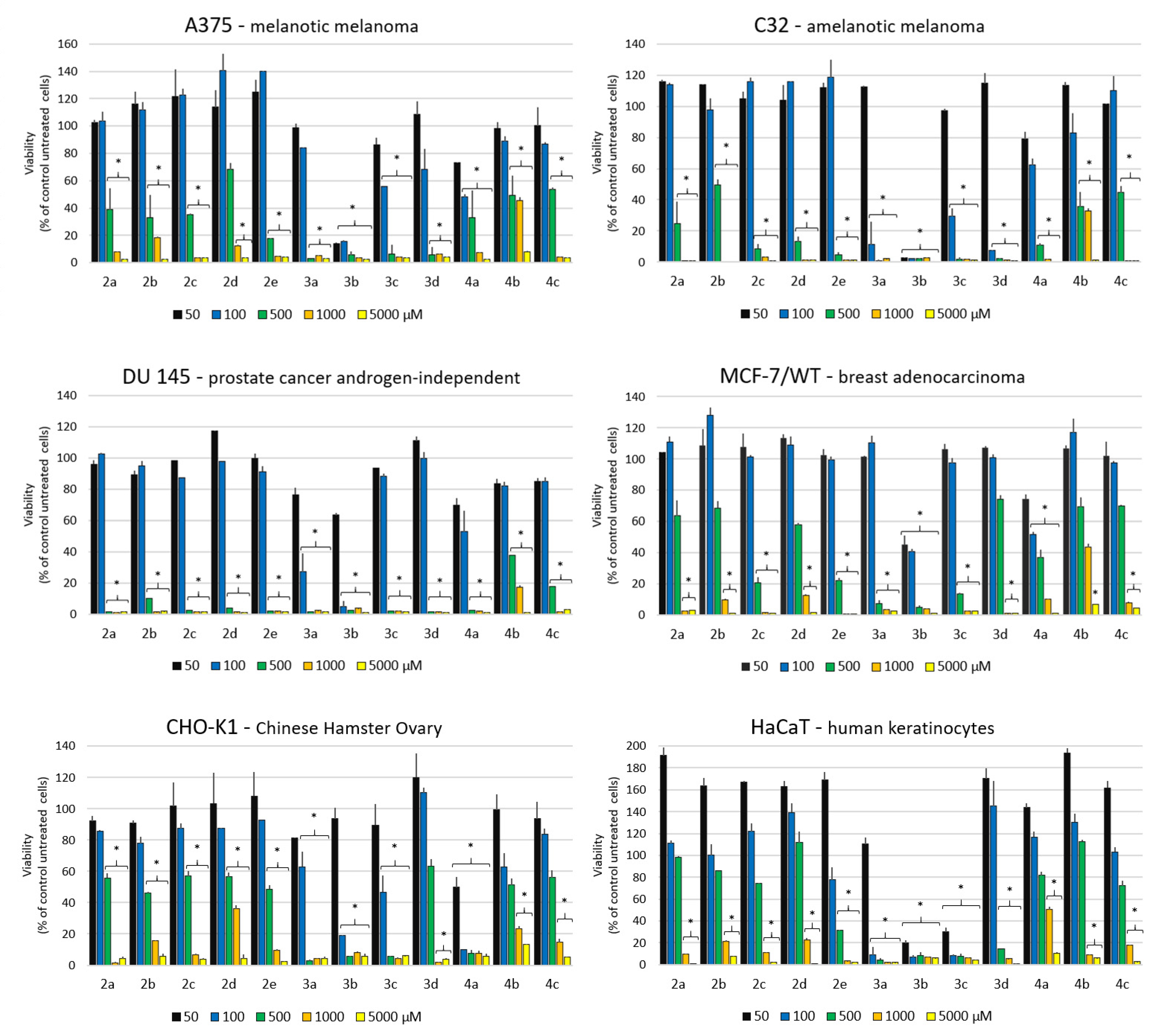
2.2.1. Antiproliferative Activity
2.2.2. Anticancer Screening Data Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. Preparation of Compounds 1a-1e
3.1.2. Preparation of Compounds 2a-2e
3.1.3. Preparation of Compounds 3a-3e
3.1.4. Preparation of Compounds 4a-4c
3.1.5. X-ray Structural Studies
3.2. Biological Activity
3.2.1. Antiproliferative Activity
Cell Lines
Cell Toxicity Test
Cell Viability Assays
3.2.2. Anticancer Screening Methodology
Primary Anticancer Assay and Determination of GI50, TGI and LC50 Values
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Swallow, S. Chapter Two—Fluorine in Medicinal Chemistry. In Progress in Medicinal Chemistry, 1st ed.; Lawton, G., Witty, D.R., Eds.; Elsevier B.V.: London, UK, 2015; Volume 54, pp. 65–133. [Google Scholar] [CrossRef]
- Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in Medicinal Chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Kirk, K.L. Fluorine in Medicinal Chemistry: Recent Therapeutic Applications of Fluorinated Small Molecules. J. Fluor. Chem. 2006, 127, 1013–1029. [Google Scholar] [CrossRef]
- Isanbor, C.; O’Hagan, D. Fluorine in Medicinal Chemistry: A Review of Anti-Cancer Agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The Role of Fluorine in Medicinal Chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Wolters, L.P.; Schyman, P.; Pavan, M.J.; Jorgensen, W.L.; Bickelhaupt, F.M.; Kozuch, S. The Many Faces of Halogen Bonding: A Review of Theoretical Models and Methods. WIREs Comput. Mol. Sci. 2014, 4, 523–540. [Google Scholar] [CrossRef]
- Park, B.K.; Kitteringham, N.R.; O’Neill, O.P. Metabolism of Fluorine-Containing Drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- McClinton, M.A.; McClinton, D.A. Trifluoromethylations and Related Reactions in Organic Chemistry. Tetrahedron 1992, 48, 6555–6666. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib Plus Fulvestrant in PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer after a CDK4/6 Inhibitor (BYLieve): One Cohort of a Phase 2, Multicentre, Open-Label, Non-Comparative Study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef]
- Möller, C.; Bone, W.; Cleve, A.; Klar, U.; Rotgeri, A.; Rottmann, A.; Schultze-Mosgau, M.H.; Wagenfeld, A.; Schwede, W. Discovery of Vilaprisan (BAY 1002670): A Highly Potent and Selective Progesterone Receptor Modulator Optimized for Gynecologic Therapies. ChemMedChem 2018, 13, 2271–2280. [Google Scholar] [CrossRef]
- Rubaiya Nasrin, S.; Ishihara, T.; Md Rashedul Kabir, A.; Konagaya, A.; Sada, K.; Kakugo, A. Comparison of Microtubules Stabilized with the Anticancer Drugs Cevipabulin and Paclitaxel. Polym. J. 2020, 52, 969–976. [Google Scholar] [CrossRef]
- Helsen, C.; Van Den Broeck, T.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen Receptor Antagonists for Prostate Cancer Therapy. Endocr. Relat. Cancer 2014, 21, T105–T118. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; Mcnabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; Mchugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Kuppast, B.; Fahmy, H. Thiazolo[4,5-d]Pyrimidines as a Privileged Scaffold in Drug Discovery. Eur. J. Med. Chem. 2016, 113, 198–213. [Google Scholar] [CrossRef]
- Becan, L.; Wojcicka, A. 5-(3-Pyridinyl)Thiazole[4,5-d]Pyrimidine Derivatives: Synthesis and in Vitro Anticancer Evaluation. Acta Pol. Pharm. Drug Res. 2018, 75, 349–357. [Google Scholar]
- Becan, L.; Wagner, E. Synthesis and Anticancer Evaluation of Novel 3,5-Diaryl-Thiazolo[4,5-d]Pyrimidin-2-One Derivatives. Med. Chem. Res. 2013, 22, 2376–2384. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, H.T.Y.; Rostom, S.A.F.; Saudi, M.N.; Zjawiony, J.K.; Robins, D.J. Synthesis and In Vitro Evaluation of the Anticancer Activity of Novel Fluorinated Thiazolo[4,5-d]Pyrimidines. Arch. Pharm. 2003, 336, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Becan, L.; Wagner, E. Synthesis and Antitumor Screening of Novel 3-Phenylthiazolo[4,5-d]Pyrimidin-2-Thione Derivatives. Arzneim. Forsch. 2008, 58, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Gewald, K. Heterocyclen aus CH-aciden Nitrilen. VI. Reaktion von methylenaktiven Nitrilen mit Senfölen und Schwefel. J. Prakt. Chem. 1966, 32, 26–30. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics. Available online: https://www.molinspiration.com/cgi-bin/properties (accessed on 1 August 2021).
- Speak, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Grever, M.; Schepartz, S.; Chabner, B. The National Cancer Institute: Cancer Drug Discovery and Development Program. Semin. Oncol. 1992, 19, 622–638. [Google Scholar] [PubMed]
- Boyd, M.R. The NCI In Vitro Anticancer Drug Discovery Screen. In Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval; Teicher, B.A., Ed.; Humana Press: Totowa, NJ, USA, 1997; pp. 23–42. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.; Boyd, M.R. Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay. Cancer Res. 1988, 48, 584–588. [Google Scholar]
- Devani, M.; Shishoo, C.; Pathak, U.; Parikh, S.; Radhakrishnan, A.; Padhya, A. Synthesis and Pharmacological Properties of Some 4-Amino-5-Substituted Thiazole-2(3H)-Thiones and Thiazolo(4,5-d)Pyrimidin-7(6H) One-2(3H)-Thiones. Arzneim. Forsch. 1977, 27, 1652–1655. [Google Scholar] [CrossRef]
- CrysAlis PRO; Rigaku Oxford Diffraction: Yarnton, UK, 2020.
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2012. [Google Scholar]
- Drąg-Zalesinska, M.; Drąg, M.; Poręba, M.; Borska, S.; Kulbacka, J.; Saczko, J. Anticancer properties of ester derivatives of betulin in human metastatic melanoma cells (Me-45). Cancer Cell Int. 2017, 17, 4. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW | LogP | H-Bond Acceptor | H-Bond Donor |
|---|---|---|---|---|
| 2a | 281.28 | 1.45 | 4 | 1 |
| 2b | 329.33 | 2.34 | 4 | 1 |
| 2c | 347.32 | 2.67 | 4 | 1 |
| 2d | 347.32 | 2.69 | 4 | 1 |
| 2e | 347.32 | 2.51 | 4 | 1 |
| 3a | 299.73 | 2.80 | 3 | 0 |
| 3b | 347.77 | 3.70 | 3 | 0 |
| 3c | 365.76 | 4.02 | 3 | 0 |
| 3d | 365.76 | 4.05 | 3 | 0 |
| 3e | 365.76 | 3.86 | 3 | 0 |
| 4a | 342.37 | 2.88 | 4 | 1 |
| 4b | 365.40 | 3.25 | 4 | 1 |
| 4c | 436.46 | 4.44 | 4 | 1 |
| Compound | A375 | C32 | DU145 | MCF-7/WT | CHO-K1 | HaCaT | Mean A ** |
|---|---|---|---|---|---|---|---|
| 2a | 429.2 ± 17.5 | 434.3 ± 17.7 | 251.4 ± 10.2 | 519.0 ± 21.1 | 520.7 ± 21.2 | 615.1 ± 25.0 | 461.6 |
| 2b | 366.8 ± 15.1 | 495.7 ± 20.4 | 356.3 ± 14.7 | 557.0 ± 22.9 | 318.8 ± 13.1 | 712.0 ± 29.3 | 467.8 |
| 2c | 450.2 ± 17.6 | 364.2 ± 14.3 | 162.8 ± 6.4 | 420.2 ± 16.5 | 525.2 ± 20.6 | 542.9 ± 21.3 | 410.9 |
| 2d | 531.1 ± 22.4 | 383.9 ± 16.2 | 120.8 ± 5.1 | 518.7 ± 21.9 | 552.7 ± 23.3 | 724.7 ± 30.6 | 472.0 |
| 2e | 379.2 ± 13.5 | 354.9 ± 12.6 | 139.2 ± 5.0 | 434.4 ± 15.5 | 482.6 ± 17.2 | 234.2 ± 8.3 | 337.4 |
| 3a | 141.8 ± 3.6 | 89.3 ± 2.3 | 72.5 ± 1.9 | 355.5 ± 9.1 | 121.6 ± 3.1 | 88.3 ± 2.3 | 144.8 |
| 3b | 25.4 ± 0.5 | 24.4 ± 0.5 | 55.1 ± 1.1 | 49.0 ± 1.0 | 75.5 ± 1.6 | 33.5 ± 0.7 | 43.8 |
| 3c | 107.6 ± 2.6 | 86.4 ± 2.1 | 168.1 ± 4.0 | 248.3 ± 6.0 | 93.8 ± 2.3 | 37.8 ± 0.9 | 123.6 |
| 3d | 103.9 ± 3.4 | 87.4 ± 2.9 | 126.4 ± 4.2 | 540.0 ± 17.9 | 512.3 ± 17.0 | 359.8 ± 11.9 | 288.3 |
| 4a | 113.7 ± 3.5 | 132.3 ± 4.1 | 96.9 ± 3.0 | 143.6 ± 4.4 | 48.5 ± 1.5 | 747.5 ± 22.9 | 213.7 |
| 4b | 523.6 ± 22.3 | 355.7 ± 15.2 | 322.9 ± 13.8 | 686.4 ± 29.3 | 236.9 ± 10.1 | 729.9 ± 31.1 | 475.9 |
| 4c | 506.6 ± 22.1 | 485.2 ± 21.2 | 236.6 ± 10.3 | 568.1 ± 24.8 | 548.6 ± 24.0 | 565.0 ± 24.7 | 485.0 |
| Mean B *** | 306.6 | 274.5 | 175.8 | 420.0 | 336.4 | 449.2 |
| Panel/Cell Line | Growth of Cells (%) 2 10−5 M | log GI50 3 | log TGI 4 | log LC50 5 | |||
|---|---|---|---|---|---|---|---|
| 2b | 3b | 4b | 4c | 3b | |||
| Leukemia | |||||||
| CCRF-CEM | 96.48 | −51.41 | - 6 | 102.97 | −6.03 | −5.50 | >−4.00 |
| HL-60(TB) | 149.39 | −41.20 | 98.91 | 93.41 | −5.81 | −5.41 | - |
| K-562 | 115.47 | 33.07 | 102.59 | 107.45 | −6.16 | −5.36 | >−4.00 |
| MOLT-4 | 117.56 | −27.71 | 81.83 | 96.58 | −5.51 | −5.07 | >−4.00 |
| RPMI-8226 | 108.14 | 44.45 | - | 104.96 | −5.60 | −5.11 | >−4.00 |
| SR | 105.93 | - | 83.46 | 94.72 | −6.36 | −5.70 | >−4.00 |
| Non-SmallCell lung Cancer | |||||||
| A549/ATCC | 103.88 | 91.16 | 74.36 | 98.32 | −5.12 | −4.66 | −4.27 |
| HOP-62 | - | 95.98 | 86.06 | 92.88 | −5.73 | −5.46 | −5.20 |
| HOP-92 | 97.54 | −21.23 | - | 106.96 | −5.90 | −5.56 | −5.23 |
| NCI-H226 | 103.60 | 89.12 | 88.51 | 112.41 | −4.86 | −4.31 | >−4.00 |
| NCI-H23 | 112.30 | 99.73 | 93.44 | - | −5.51 | −4.95 | −4.03 |
| NCI-H322M | 128.80 | 101.54 | - | - | −5.42 | −4.90 | −4.42 |
| NCI-H460 | 123.50 | 68.89 | 100.34 | 109.73 | −5.49 | −4.99 | −4.30 |
| NCI-H522 | 90.35 | −67.57 | 70.40 | 90.73 | −6.69 | −6.33 | −5.82 |
| Colon Cancer | |||||||
| COLO-205 | 114.26 | 30.63 | 105.73 | 112.07 | −5.71 | −5.44 | −5.18 |
| HCT-116 | 102.27 | −27.21 | 94.34 | 111.01 | −5.76 | −5.47 | −5.18 |
| HCT-15 | 106.95 | 2.98 | 94.67 | 103.39 | −5.73 | −5.26 | >−4.00 |
| HCC-2998 | 116.48 | −26.89 | 115.06 | 118.24 | −5.65 | −5.31 | −4.89 |
| HT29 | 100.96 | −71.75 | 88.76 | 103.27 | −5.72 | −5.40 | - |
| KM12 | 100.96 | 90.93 | 89.17 | 113.23 | −5.64 | −5.30 | −4.84 |
| SW620 | 95.09 | −63.05 | 106.24 | 114.74 | −5.67 | −5.32 | −4.67 |
| CNS Cancer | |||||||
| SF-268 | 101.77 | 84.73 | 104.72 | 109.32 | −5.75 | 5.46 | −5.18 |
| SF-295 | 116.63 | - | 94.05 | 91.10 | −4.91 | −4.59 | −4.28 |
| SF-539 | 114.10 | 75.80 | 100.98 | 102.28 | −5.78 | −5.50 | −5.23 |
| SNB-19 | 102.20 | 98.46 | 106.58 | 106.48 | −5.75 | −5.49 | −5.23 |
| SNB-75 | 91.21 | - | 99.24 | 103.11 | −5.70 | −5.38 | −5.05 |
| U251 | 101.63 | - | 93.27 | - | −5.76 | −5.50 | −5.24 |
| Melanoma | |||||||
| LOX IMVI | 84.58 | 3.11 | 88.57 | 109.59 | −5.75 | −5.48 | −5.21 |
| MALME-3M | 121.70 | 40.72 | 144.23 | 106.31 | −5.64 | −5.37 | −5.09 |
| M14 | 112.91 | 26.96 | 107.61 | 112.06 | −5.70 | −5.39 | −5.08 |
| MDA-MB-435 | 114.64 | 6.26 | 112.84 | 102.86 | −5.73 | −5.43 | −5.13 |
| SK-MEL-2 | 97.36 | 55.66 | 88.08 | 95.77 | −5.70 | −5.42 | −5.14 |
| SK-MEL-28 | 102.86 | −62.53 | 112.46 | 110.02 | −5.71 | −5.45 | −5.19 |
| SK-MEL-5 | - | 81.95 | 87.63 | 105.71 | −5.66 | −5.31 | −4.91 |
| UACC-257 | 107.08 | 19.38 | 98.66 | 96.72 | −5.76 | −5.43 | −5.10 |
| UACC-62 | 109.52 | 79.14 | 93.86 | 105.93 | - | - | - |
| Ovarian Cancer | |||||||
| IGROV1 | −5.14 | 63.57 | 101.57 | 109.98 | −5.71 | −5.41 | −5.12 |
| OVCAR-3 | 110.35 | −41.27 | 106.99 | 111.62 | −5.72 | −5.47 | −5.22 |
| OVCAR-4 | 117.19 | 23.73 | 98.24 | 125.04 | −5.75 | −5.44 | −5.14 |
| OVCAR-5 | 101.99 | 86.56 | 112.27 | 102.72 | −5.06 | −4.60 | −4.19 |
| OVCAR-8 | 103.45 | 4.40 | 96.11 | 98.68 | −5.71 | −5.27 | >−4.00 |
| NCI/ADR-RES | 25.77 | 109.92 | 92.88 | 114.00 | −5.48 | −4.96 | >−4.00 |
| SK-OV-3 | 108.53 | 101.94 | 101.70 | 99.96 | −5.31 | −4.97 | >−4.00 |
| Renal Cancer | |||||||
| 786-0 | 104.97 | 90.12 | 101.39 | 99.38 | −5.71 | −5.42 | −5.13 |
| A498 | 90.58 | 91.46 | - | 121.43 | −4.87 | −4.57 | −4.28 |
| ACHN | 110.42 | 27.40 | 93.13 | 99.14 | −5.76 | −5.48 | −5.20 |
| CAKI-1 | 124.11 | 81.74 | 93.63 | 94.38 | −5.78 | −5.51 | −5.24 |
| SN12C | 106.95 | 85.61 | 100.30 | 101.41 | −5.45 | −4.88 | −4.39 |
| TK-10 | 130.06 | 51.24 | 88.79 | 130.70 | −5.70 | −5.39 | −5.08 |
| UO-31 | 91.22 | −82.97 | 103.75 | 82.87 | −5.77 | −5.49 | −5.22 |
| RXF 393 | 95.34 | −16.27 | 100.09 | 104.44 | −5.74 | −5.47 | −5.21 |
| Prostate C. | |||||||
| PC-3 | 100.88 | 53.66 | - | 100.51 | −5.74 | −5.45 | −5.17 |
| DU-145 | 109.40 | 9.04 | 101.28 | 122.63 | −5.73 | −5.46 | −5.19 |
| Breast Cancer | |||||||
| MCF7 | 111.19 | 22.84 | 96.38 | 98.77 | −5.65 | −5.25 | −4.24 |
| MDA-MB-231/ATCC | 107.13 | 13.85 | 85.06 | 119.48 | −5.70 | −5.41 | −5.12 |
| HS 578T | 65.01 | 21.84 | 106.83 | 125.43 | −5.55 | −5.07 | >−4.00 |
| BT-549 | 130.31 | 73.90 | 95.21 | 98.87 | −5.82 | −5.50 | −5.17 |
| T-47D | 108.41 | −35.57 | 73.92 | 85.37 | −5.73 | −5.32 | −4.18 |
| MDA-MB-468 | 91.37 | 54.38 | 81.60 | 96.15 | −5.77 | −5.45 | −5.13 |
| Mean | 103.88 | 29.51 | 96.94 | 105.13 | |||
| Mean MG_MID 7 | −5.66 | −5.3 | −4.78 |
| Compound | Mean Growth % 2 10−5 M | Growth % of the MOST Sensitive Cell Line | Cancer Type | Cell Line |
|---|---|---|---|---|
| 2b | 103.88 | −5.14 | Ovarian C. | IGROV1 |
| I | 80.85 | 29.76 | Leukemia | RPMI-8826 |
| II | 106.11 | 55.97 | NS Cell Lung C. | HOP-92 |
| III | 105.59 | 73.81 | NS Cell Lung C. | HOP-92 |
| 3b | 29.51 | −82.97 | Renal C. | UO-31 |
| IV | 20.78 | −85.59 | Renal C. | UO-31 |
| V | 64.11 | −88.95 | Renal C. | CAKI-1 |
| 4b | 96.94 | 70.40 | NS Cell Lung C. | NCI-H522 |
| 4c | 105.13 | 82.87 | Renal C. | UO-31 |
| VI | 48.46 | −43.11 | CNS C. | SF-295 |
| VII | 89.50 | 54.52 | Melanoma | UACC-62 |
| Number of Cell Lines Giving Positive log GI50 2, log TGI 3, log LC50 4 (≤−4.01) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cd | N | log GI50 | log TGI | log LC50 | ||||||
| N1 | Range 5 | MG_MID 6/Δ 7 | N2 | Range | MG_MID/Δ | N3 | Range | MG_MID/Δ | ||
| 3b | 58 | 58 | −4.86 to −6.69 | −5.495146 | 58 | −4.31 to −6.33 | −5.145631 | 45 | −4.03 to −5.82 | −4.596153846 |
| IV | 59 | 37 | −5.28 to −6.26 | −6.728395 | 7 | −4.52 to −5.51 | 4.39/1.12 | 3 | −5.17 to −5.26 | −3.317073171 |
| V | 59 | 58 | −4.23 to −8.00 | −5.097087 | 49 | −4.10 to −5.90 | −4.663366 | 35 | −4.07 to −5.42 | −4.631578947 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becan, L.; Pyra, A.; Rembiałkowska, N.; Bryndal, I. Synthesis, Structural Characterization and Anticancer Activity of New 5-Trifluoromethyl-2-thioxo-thiazolo[4,5-d]pyrimidine Derivatives. Pharmaceuticals 2022, 15, 92. https://doi.org/10.3390/ph15010092
Becan L, Pyra A, Rembiałkowska N, Bryndal I. Synthesis, Structural Characterization and Anticancer Activity of New 5-Trifluoromethyl-2-thioxo-thiazolo[4,5-d]pyrimidine Derivatives. Pharmaceuticals. 2022; 15(1):92. https://doi.org/10.3390/ph15010092
Chicago/Turabian StyleBecan, Lilianna, Anna Pyra, Nina Rembiałkowska, and Iwona Bryndal. 2022. "Synthesis, Structural Characterization and Anticancer Activity of New 5-Trifluoromethyl-2-thioxo-thiazolo[4,5-d]pyrimidine Derivatives" Pharmaceuticals 15, no. 1: 92. https://doi.org/10.3390/ph15010092