
High-Throughput Sequencing Reveals the Effect of Feeding on Overwintering Hirudo nipponia


Abstract
:1. Introduction
2. Materials and Methods
2.1. H. nipponia Material and Culture Conditions
2.2. Sample Collection
2.3. DNA Extraction and Metagenomic Library Preparation
2.4. Analysis of Metagenomic Data
2.5. RNA Extraction and cDNA Library Preparation for Transcriptome Sequencing
2.6. Analysis of Transcriptome Data
2.7. qRT-PCR Analysis
3. Results
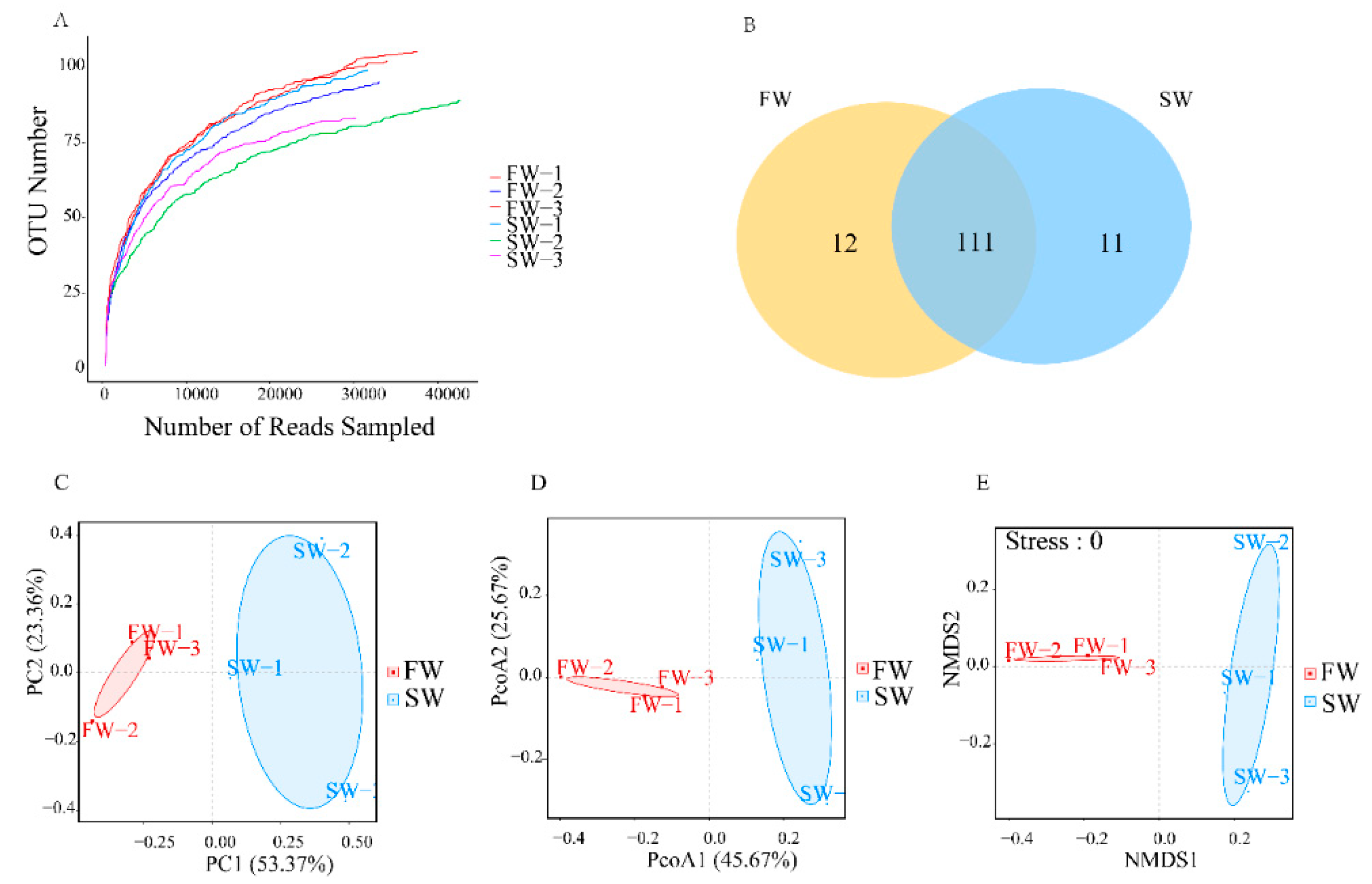
3.1. Microbial Diversity in the Digestive Tracts of H. nipponia
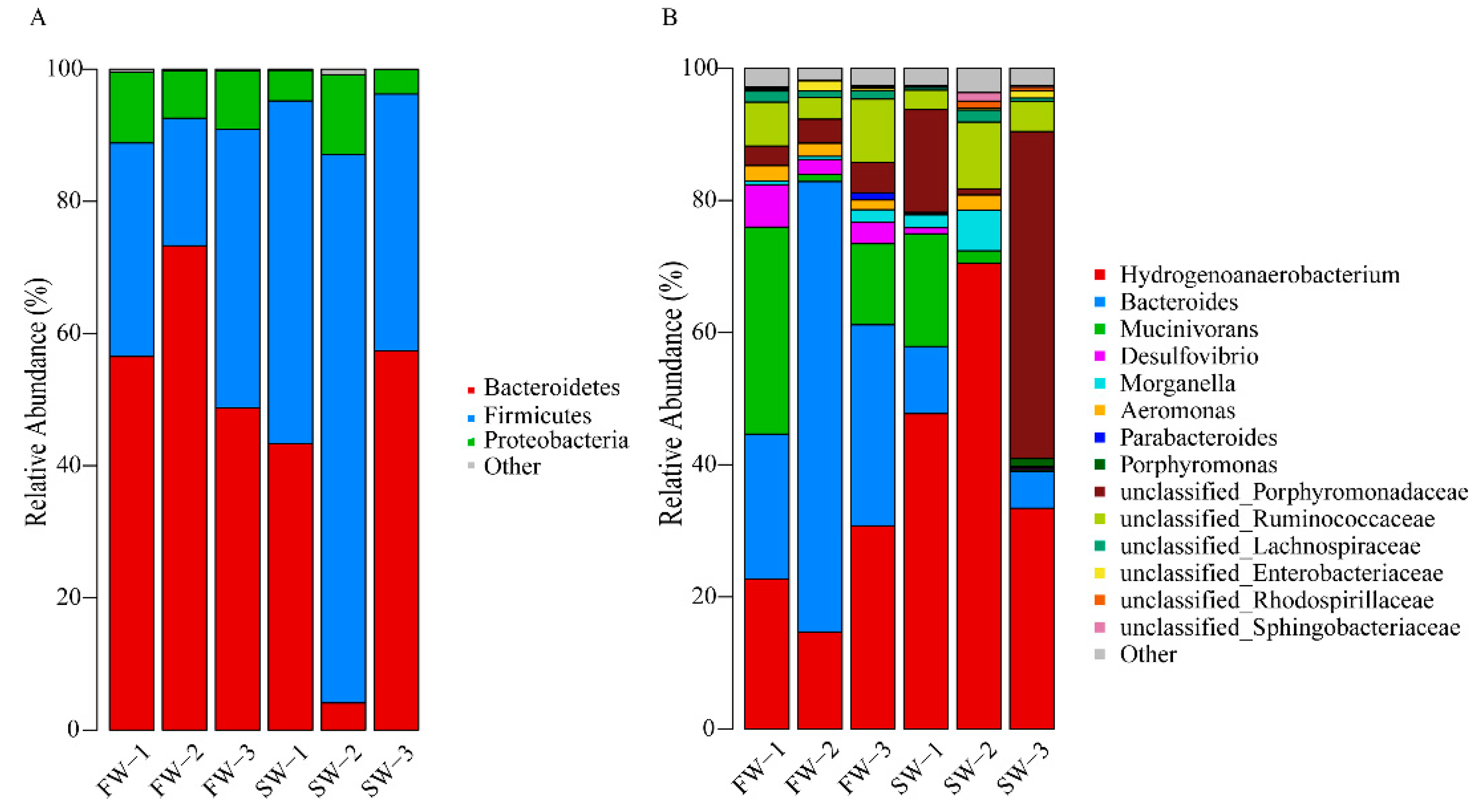
3.2. Changes in Microbial Abundance in the Digestive Tract of H. nipponia
3.3. Functional Prediction of Leeches
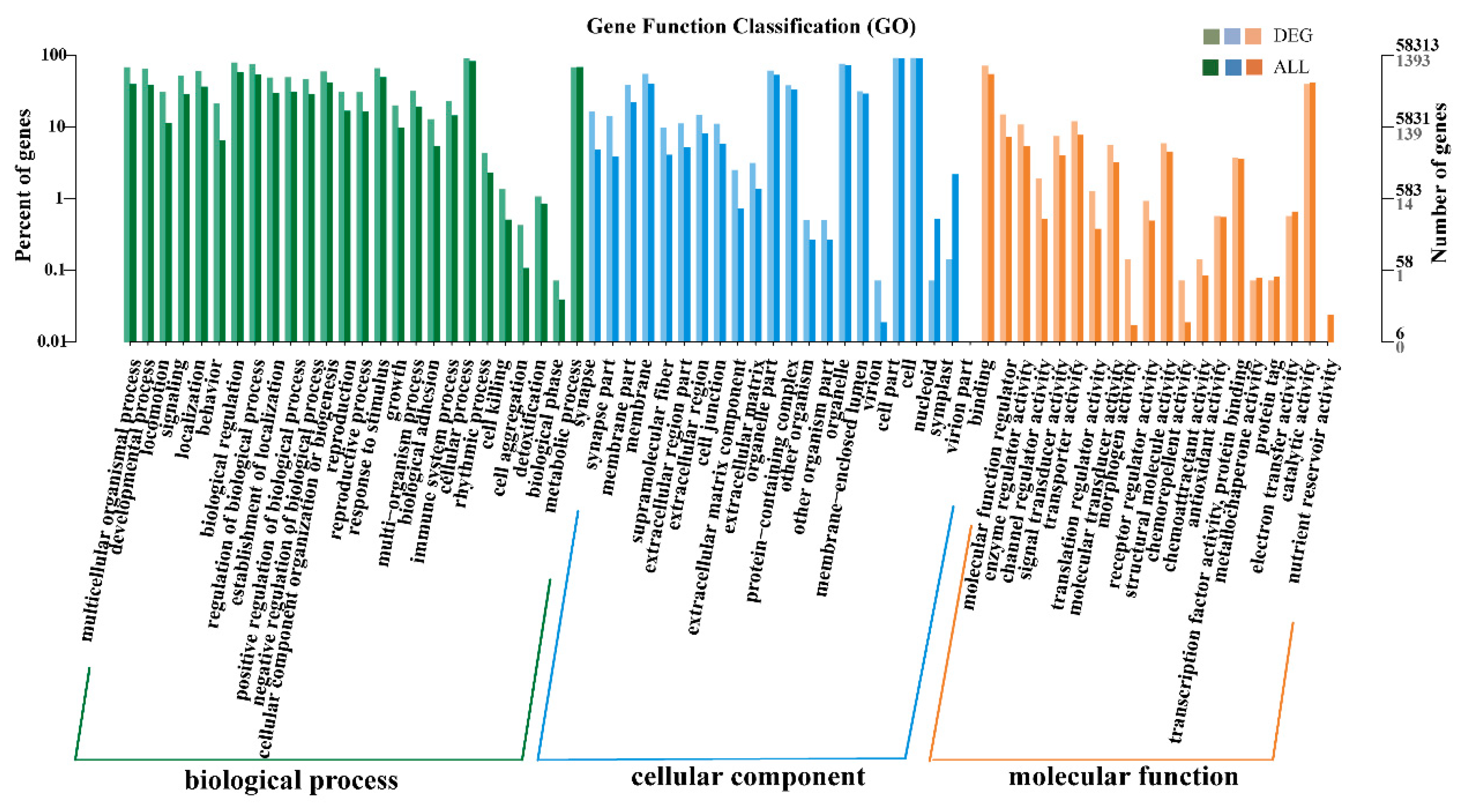
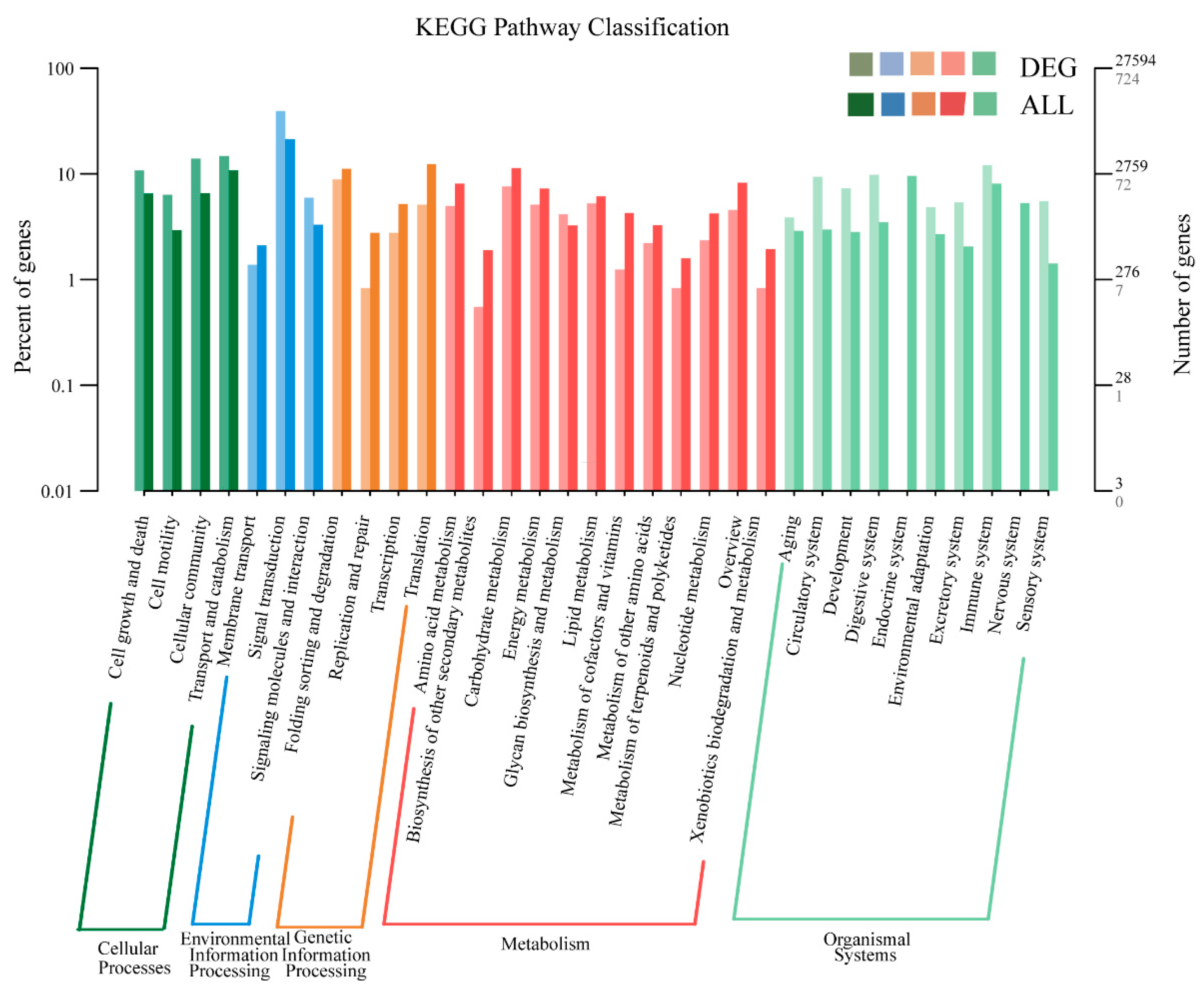
3.4. Identification of Differentially Expressed Genes (DEGs)
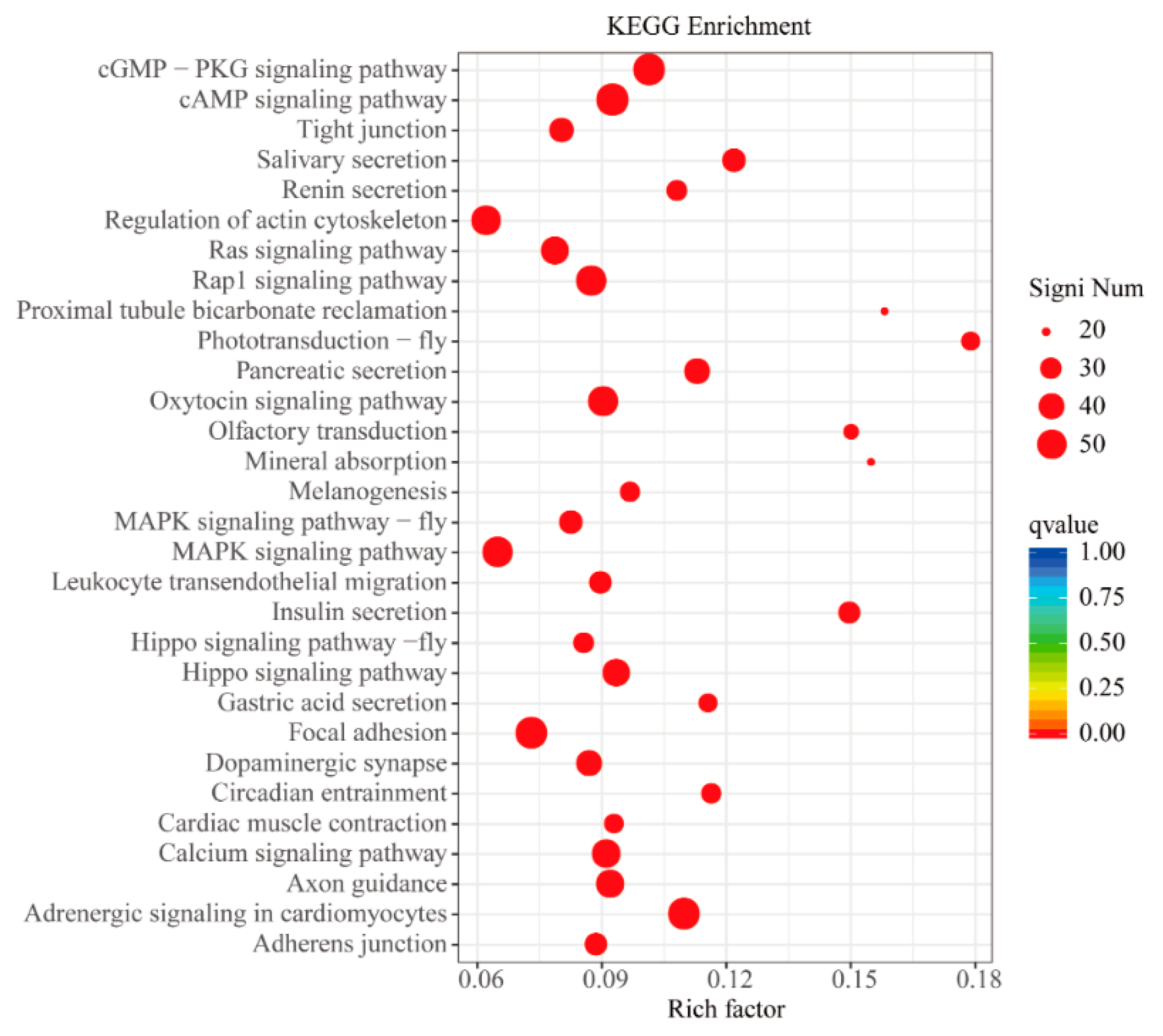
3.5. GO and KEGG Enrichment Analyses of DEGs
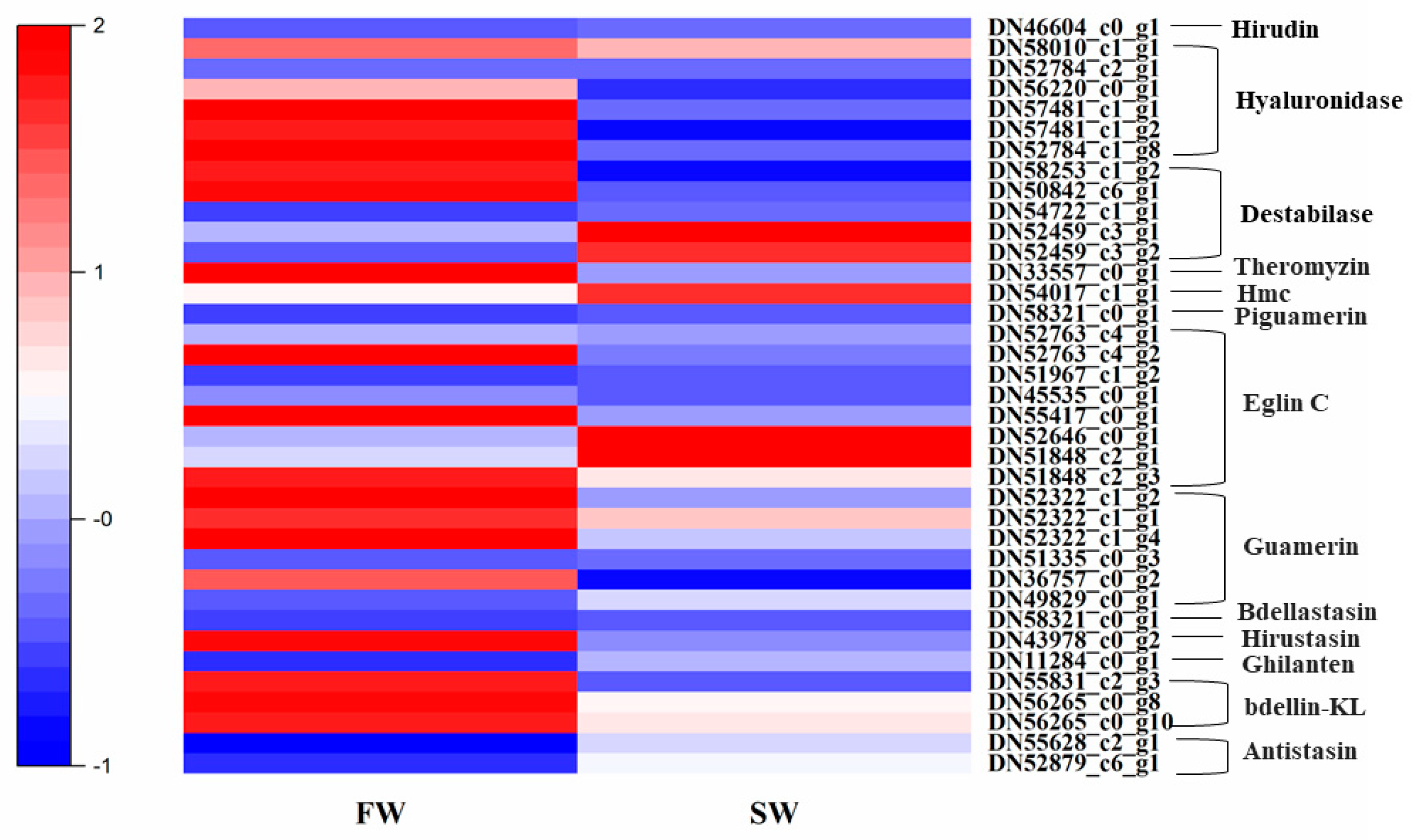
3.6. Expression Patterns of Genes Related to Anticoagulation and Antibacterial Activities
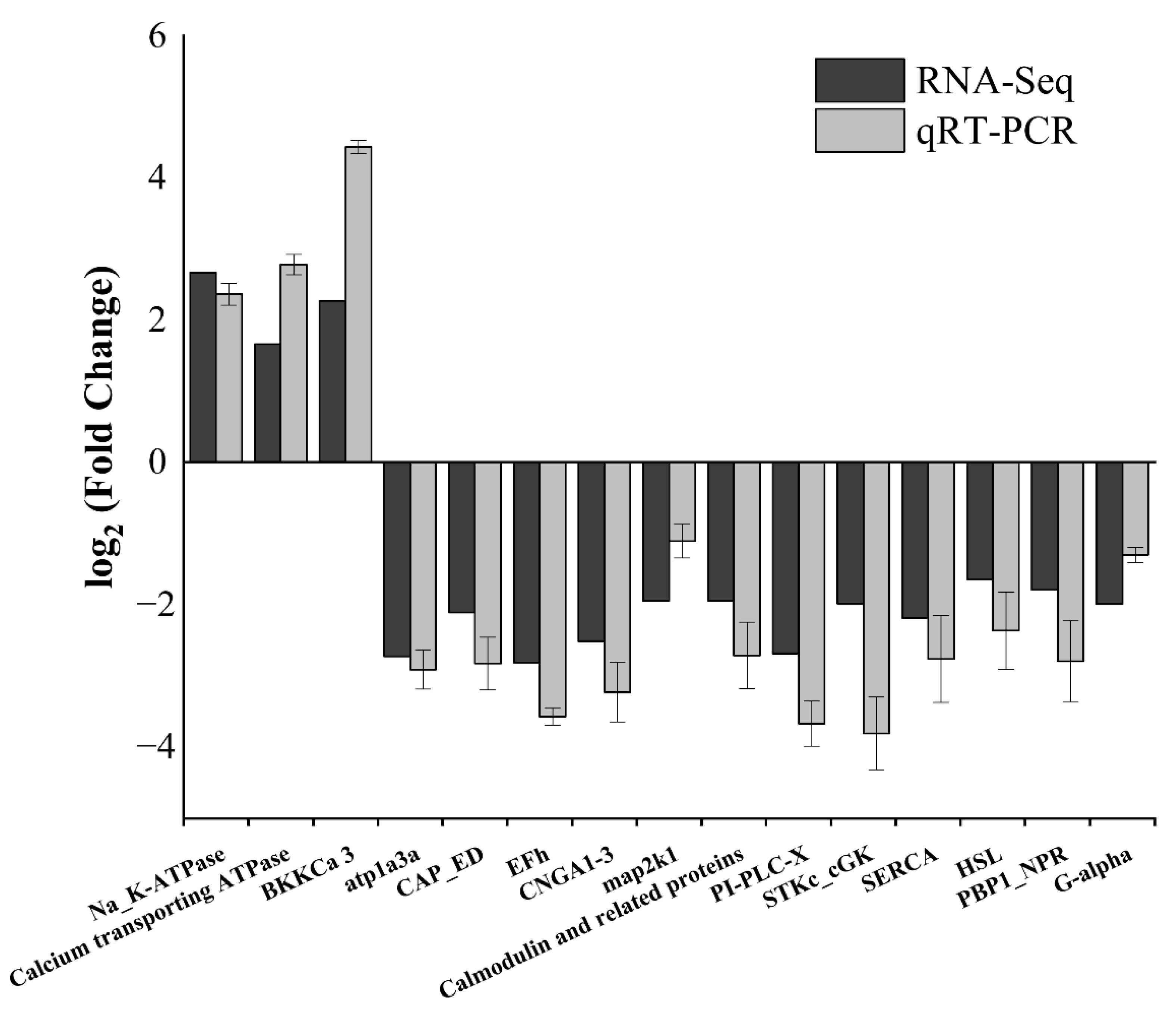
3.7. qRT-PCR Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, T. Annelida: Hirudinea. In Fauna Sinica; Science Press: Beijing, China, 1996; Volume 5, pp. 1–256. [Google Scholar]
- Zhang, T.; Yu, X.; Gong, Y.; Li, J.; Zhang, J. Effects of feed supplementation with Aeromonas hydrophila on the growth and survival of Hirudo nipponia. Hebei Fish. 2017, 6, 10–17. [Google Scholar] [CrossRef]
- Ding, Z.; Chen, K.; She, Y.; Chen, Y. Research Advances of Medicinal Leeches and Leech Polypeptides. Mod. Chin. Med. 2022, 24, 1–10. [Google Scholar] [CrossRef]
- Lu, Z.; He, Z.; Shi, P.; Li, Q.; You, H.; Chen, S. Anticoagulant activity analysis of Hirudo nipponica. Mod. Chin. Med. 2017, 19, 37–39. [Google Scholar] [CrossRef]
- Koh, C.Y.; Kini, R.M. Anticoagulants from hematophagous animals. Expert Rev. Hematol. 2008, 1, 135–139. [Google Scholar] [CrossRef]
- Chen, J.; Xie, X.; Zhang, H.; Li, G.; Yin, Y.; Cao, X.; Gao, Y.; Li, Y.; Zhang, Y.; Peng, F.; et al. Pharmacological Activities and Mechanisms of Hirudin and Its Derivatives—A Review. Front. Pharmacol. 2021, 12, 660757. [Google Scholar] [CrossRef]
- Scacheri, E.; Nitti, G.; Valsasina, B.; Orsini, G.; Visco, C.; Ferrera, M.; Sawyer, R.T.; Sarmientos, P. Novel hirudin variants from the leech Hirudinaria manillensis. Amino acid sequence, cDNA cloning and genomic organization. Eur. J. Biochem. 1993, 214, 295–304. [Google Scholar] [CrossRef]
- Dunwiddie, C.T.; Waxman, L.; Vlasuk, G.P.; Friedman, P.A. Purification and characterization of inhibitors of blood coagulation factor Xa from hematophagous organisms. Methods Enzymol. 1993, 223, 291–312. [Google Scholar] [CrossRef]
- Söllner, C.; Mentele, R.; Eckerskorn, C.; Fritz, H.; Sommerhoff, C.P. Isolation and characterization of hirustasin, an antista-sin-type serine-proteinase inhibitor from the medical leech Hirudo medicinalis. Eur. J. Biochem. 2010, 219, 937–943. [Google Scholar] [CrossRef]
- Jung, H.I.; Kim, S.I.; Ha, K.S.; Joe, C.O.; Kang, K.W. Isolation and characterization of guamerin, a new human leukocyte elastase inhibitor from Hirudo nipponia. J. Biol. Chem. 1995, 270, 13879–13884. [Google Scholar] [CrossRef]
- Gui, Y.; Dong, D.; Li, R. Bionic affinity purification and identification of a new anticoagulant protein from Hirudo nipponia. Chin. J. Biochem. Drugs 2008, 29, 145–148. [Google Scholar]
- Shi, P.; Zeng, L.; You, H.; He, Y.; Zhang, D.; Xing, K.; Chen, S. Effects of fasting and refeeding on Hirudo nipponia intestinal microflora diversity. Acta Hydrobiol Sin. 2019, 43, 537–544. [Google Scholar] [CrossRef]
- Aminov, R.; Frolov, A.; Aminova, A. The influence of the planting time and conditions on the reproductive properties of Hirudo verbana and Hirudo medicinalis. Ann. Parasitol. 2021, 67, 169–174. [Google Scholar] [CrossRef]
- Upadhyay, V.; Poroyko, V.; Kim, T.; Devkota, S.; Fu, S.; Liu, D.; Tumanov, A.V.; Koroleva, E.P.; Deng, L.; Nagler, C.; et al. Lymphotoxin regulates commensal responses to enable diet-induced obesity. Nat. Immunol. 2012, 13, 947–953. [Google Scholar] [CrossRef]
- Koppel, N.; Balskus, E.P. Exploring and understandingthe biochemical diversity of the human microbiota. Cell Chem. Biol. 2016, 23, 18–30. [Google Scholar] [CrossRef]
- Nelson, M.C.; Graf, J. Bacterial symbioses of the medicinal leech Hirudo verbana. Gut Microbes. 2012, 3, 322–331. [Google Scholar] [CrossRef]
- Graf, J.; Kikuchi, Y.; Rio, R.V.M. Leeches and their microbiota: Naturally simple symbiosis models. Trends Microbiol. 2006, 14, 365–371. [Google Scholar] [CrossRef]
- McClure, E.A.; Nelson, M.C.; Lin, A.; Graf, J. Macrobdella decora: Old World Leech Gut Microbial Community Structure Conserved in a New World Leech. Appl. Environ. Microbiol. 2021, 87, e02082-20. [Google Scholar] [CrossRef]
- Song, D.; Feng, Z. Leech; Science Press: Beijing, China, 1978. [Google Scholar]
- Ding, A. Gene cloning and expression of a partial sequence of Hirudomacin, an antimicrobial protein that is increased in leech (Hirudo nipponica Whitman) after a blood meal. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2019, 231, 75–86. [Google Scholar] [CrossRef]
- Lemke, E.; Pavlova, E.; Babenko, V.V.; Latsis, I.; Malakhova, M.; Lavrenova, V.; Bashkirov, P.; Belousov, D.; Klinov, D.; Lazarev, V. The Hirudo Medicinalis Microbiome Is a Source of New Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7141. [Google Scholar] [CrossRef]
- Lemke, S.; Vilcinskas, A. European Medicinal Leeches—New Roles in Modern Medicine. Biomedicines 2020, 8, 99. [Google Scholar] [CrossRef]
- Ma, M.; Tu, C.; Luo, J.; Lu, M.; Zhang, S.; Xu, L. Metabolic and immunological effects of gut microbiota in leaf beetles at the local and systemic levels. Integr Zool. 2021, 16, 313–323. [Google Scholar] [CrossRef]
- Luo, J.; Cheng, Y.; Guo, L.; Wang, A.; Lu, M.; Xu, L. Variation of gut microbiota caused by an imbalance diet is detrimental to bugs’ survival. Sci Total. Environ. 2021, 771, 144880. [Google Scholar] [CrossRef]
- Naggar, Y.A.; Singavarapu, B.; Paxton, R.J.; Wubet, T. Bees under interactive stressors: The novel insecticides flupyradifurone and sulfoxaflor along with the fungicide azoxystrobin disrupt the gut microbiota of honey bees and increase opportunistic bacterial pathogens. Sci Total Environ. 2022, 849, 157941. [Google Scholar] [CrossRef]
- Xu, L.; Deng, J.; Zhou, F.; Cheng, C.; Zhang, L.; Zhang, J.; Lu, M. Gut microbiota in an invasive bark beetle infected by a pathogenic fungus accelerates beetle mortality. J Pest Sci. 2019, 92, 343–351. [Google Scholar] [CrossRef]
- Heldmaier, G.; Ortmann, S.; Elvert, R. Natural hypometabolism during hibernation and daily torpor in mammals. Resp. Physiol. Neurobi. 2004, 141, 317–329. [Google Scholar] [CrossRef]
- Yu, M.; Liu, J.; Zhou, M.; Feng, X.; Cao, M. Primary investigation on habitat waters and biological characteristics of Hirudo nipponica. J. Chin. Med. Mater. 2019, 42, 2782–2785. [Google Scholar] [CrossRef]
- Sisa, C.; Turroni, S.; Amici, R.; Brigidi, P.; Candela, M.; Cerri, M. Potential role of the gut microbiota in synthetic torpor and therapeutic hypothermia. World J. Gastroentero. 2017, 23, 406–413. [Google Scholar] [CrossRef]
- Maltz, M.A.; Bomar, L.; Lapierre, P.; Morrison, H.G.; McClure, E.A.; Sogin, M.L.; Graf, J. Metagenomic analysis of the medicinal leech gut microbiota. Front. Microbiol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Zhu, Y.; Si, H.; Zhang, Y.; Xu, C. Research progress on interaction mechanism between animal hibernation behavior and gut microbiota. Chin. J. Anim. Nutr. 2021, 33, 3719–3725. [Google Scholar]
- Dittmer, J.; Brucker, R.M. When your host shuts down: Larval diapause impacts host-microbiome interactions in Nasonia vitripennis. Microbiome 2021, 9, 85. [Google Scholar] [CrossRef]
- Quail, M.A.; Kozarewa, I.; Smith, F.; Scally, A.; Stephens, P.J.; Durbin, R.; Swerdlow, H.; Turner, D.J. A large genome centre’s improvements to the Illumina sequencing system. Nat. Methods 2008, 5, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Reis-Filho, J.S. Next-generation sequencing. Breast Cancer Res. 2009, 11, S12. [Google Scholar] [CrossRef] [Green Version]
- Qin, N.; Li, D.; Yang, R. Next-generation sequencing technologies and the application in microbiology—A review. Acta Microbiol. Sin. 2011, 51, 445–457. [Google Scholar] [CrossRef]
- Song, P.; Ma, L.; Wang, Q.; Li, D.; Wei, Z. The advances of metagenome technology and its applications. Guizhou Agric. Sci. 2009, 37, 14–18. [Google Scholar]
- Zhao, Y.; Wu, J.; Yang, T.; Yao, M.; Pei, X.; Fu, L.; Liu, X. Advances in research on wildlife intestinal microbes based on metagenomics. Chin. J. Wildl. 2021, 42, 568–574. [Google Scholar] [CrossRef]
- Weng, F.C.H.; Yang, Y.J.; Wang, D. Functional analysis for gut microbes of the brown tree frog (Polypedates megacephalus) in artificial hibernation. BMC Genom. 2016, 17, 1024. [Google Scholar] [CrossRef]
- Xing, Y.; Guan, F.; Li, S.; Zhang, S.; Luo, Y. Comparative transcriptome analysis of salivary glands of Hirudo nipponia in response to starvation. J. Fish. China 2020, 44, 754–766. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, L.; Huang, Z.; Wang, D.; Hu, G. Transcription sequencing and its application on discovering the gene resources of wild soybean. Soybean Sci. 2013, 32, 845–851. [Google Scholar]
- Tang, H.; Huang, Y.; Lu, H.; Tian, D.; Zhang, Y. The application perspective of transcriptome sequencing on discovering the genes and variety breeding of bioenergy grass. Acta Agrestia Sin. 2016, 24, 731–737. [Google Scholar]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Babenko, V.V.; Podgorny, O.V.; Manuvera, V.A.; Kasianov, A.S.; Manolov, A.I.; Grafskaia, E.N.; Shirokov, D.A.; Kurdyumov, A.S.; Vinogradov, D.V.; Nikitina, A.S.; et al. Draft genome sequences of Hirudo medicinalis and salivary transcriptome of three closely related medicinal leeches. BMC Genom. 2020, 21, 331. [Google Scholar] [CrossRef]
- Zhang, S.; Lv, S.; Wang, J. The functional diversity of leech salivary gland secretions and its application. Chin. J. Biochem. Drugs 2017, 37, 316–319. [Google Scholar]
- Sears, C.L. A dynamic partnership: Celebrating our gut flora. Anaerobe 2005, 11, 247–251. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Scaldaferri, F.; Gerardi, V.; Lopetuso, L.R.; Zompo, F.D.; Mangiola, F.; Boškoski, I.; Bruno, G.; Petito, V.; Laterza, L.; Cammarota, G.; et al. Gut Microbial Flora, Prebiotics, and Probiotics in IBD: Their Current Usage and Utility. BioMed. Res. Int. 2013, 2013, 435268. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Shi, P.; Wei, J.; You, H.; Lu, Z.; Chen, S. Effects of acute temperature stress on intestinal microflora in Hirudo nipponia. Chin. J. Microecol. 2021, 33, 752–758. [Google Scholar] [CrossRef]
- Tran, N.T.; Xiong, F.; Hao, Y.T.; Zhang, J.; Wu, S.G.; Wang, G.T. Starvation influences the microbiota assembly and expression of immunity-related genes in the intestine of grass carp (Ctenopharyngodon idellus). Aquaculture 2018, 489, 121–129. [Google Scholar] [CrossRef]
- Kohl, K.D.; Amaya, J.; Passement, C.A.; Dearing, M.D.; McCue, M.D. Unique and shared responses of the gut microbiota to prolonged fasting: A comparative study across five classes of vertebrate hosts. FEMS Microbiol. Ecol. 2014, 90, 883–894. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, Y.; Wang, Z.; Zhou, Y.; Zhang, S.; Wang, P.; Xie, S.; Jiang, B. Changes of fecal flora and its correlation with inflammatory indicators in patients with inflammatory bowel disease. J. South Med. Univ. 2013, 33, 1474–1477. [Google Scholar] [CrossRef]
- Ikeda, T.; Aida, M.; Yoshida, Y.; Matsumoto, S.; Tanaka, M.; Nakayama, J.; Nagao, Y.; Nakata, R.; Oki, E.; Akahoshi, T.; et al. Alteration in faecal bile acids, gut microbial composition and diversity after laparoscopic sleeve gastrectomy. Br. J. Surg. 2020, 107, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Marden, J.N.; McClure, E.A.; Beka, L.; Graf, J. Host Matters: Medicinal Leech Digestive-Tract Symbionts and Their Pathogenic Potential. Front. Microbiol. 2016, 7, 1569. [Google Scholar] [CrossRef]
- Nelson, M.C.; Bomar, L.; Graf, J. Complete Genome Sequence of the Novel Leech Symbiont Mucinivorans hirudinis M3T. Genome Announc. 2015, 3, e01530-14. [Google Scholar] [CrossRef]
- Bomar, L.; Maltz, M.; Colston, S.; Graf, J. Directed Culturing of Microorganisms Using Metatranscriptomics. mBio 2011, 2, e00012-11. [Google Scholar] [CrossRef]
- Hubert, C.; Voordouw, G. Oil Field Souring Control by Nitrate-Reducing Sulfurospirillum spp. That Outcompete Sulfate-Reducing Bacteria for Organic Electron Donors. Appl. Environ. Microbiol. 2007, 73, 2644–2652. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shen, D. Ecological characters and application of sulfate-reducing bacteria. Chin. J. Appl. Environ. Biol. 1999, S1, 10–13. [Google Scholar]
- Chen, K.; Zhao, Y.; Feng, Y.; Liu, C.; Li, X.; Zhao, L.; Pang, X. One improved growth medium for sulphate-reducing bacteria isolated from gut. Chin. J. Microecol. 2013, 25, 1241–1244. [Google Scholar] [CrossRef]
- Laufer, A.S.; Siddall, M.E.; Graf, J. Characterization of the Digestive-Tract Microbiota of Hirudo orientalis, a European Medicinal Leech. Appl. Environ. Microbiol. 2008, 74, 6151–6154. [Google Scholar] [CrossRef]
- Neupane, S.; Modry, D.; Pafčo, B.; Zurek, L. Bacterial Community of the Digestive Tract of the European Medicinal Leech (Hirudo verbana) from the Danube River. Microb Ecol. 2019, 77, 1082–1090. [Google Scholar] [CrossRef]
- Siddall, M.E.; Worthen, P.L.; Johnson, M.; Graf, J. Novel Role for Aeromonas jandaei as a Digestive Tract Symbiont of the North American Medicinal Leech. Appl. Environ. Microbiol. 2007, 73, 655–658. [Google Scholar] [CrossRef]
- Qi, X.Z.; Tu, X.; Zha, J.W.; Huang, A.G.; Wang, G.X.; Ling, F. Immunosuppression-induced alterations in fish gut microbiota may increase the susceptibility to pathogens. Fish Shellfish Immunol. 2019, 88, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Yang, J.; Wang, L.; Duan, N.; Xiao, N. The Bacterial Pathogen Spectrum and Antibiotic Resistance of Infectious Diarrhea Outpatients in a Large Community in Beijing. Label. Immunoass. Clin. Med. 2019, 26, 777–780. [Google Scholar]
- Chopra, A.K.; Houston, C.W. Enterotoxins in Aeromonas-associated gastroenteritis. Microbes Infect. 1999, 1, 1129–1137. [Google Scholar] [CrossRef]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov.; sp. nov.; a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef]
- Kvist, S.; Manzano-Marín, A.; de Carle, D.; Trontelj, P.; Siddall, M.E. Draft genome of the European medicinal leech Hirudo medicinalis (Annelida, Clitellata, Hirudiniformes) with emphasis on anticoagulants. Sci. Rep. 2020, 10, 9885. [Google Scholar] [CrossRef] [PubMed]
- Min, G.S.; Sarkar, I.N.; Siddall, M.E. Salivary transcriptome of the North American medicinal leech, Macrobdella decora. J. Parasitol. 2010, 96, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Roters, F.J.; Zebe, E. Proteinases of the medicinal leech, Hirudo medicinalis: Purification and partial characterization of three enzymes from the digestive tract. Comp. Biochem. Physiol. B. 1992, 102, 627–634. [Google Scholar] [CrossRef]
- Baskova, I.P.; Zavalova, L.L. Proteinase Inhibitors from the Medicinal Leech Hirudo medicinalis. Biochemistry 2001, 66, 703–714. [Google Scholar] [CrossRef]
- Junger, W.G.; Hallström, S.; Redl, H.; Schlag, G. Inhibition of Human, Ovine, and Baboon Neutrophil Elastase with Eglin c and Secretory Leukocyte Proteinase Inhibitor. Biol. Chem. Hoppe-Seyler 2009, 373, 119–122. [Google Scholar] [CrossRef]
- Roters, F.J.; Zebe, E. Protease inhibitors in the alimentary tract of the medicinal leech Hirudo medicinalis: In vivo and in vitro studies. J. Comp. Physiol. B. 1992, 162, 85–92. [Google Scholar] [CrossRef]
- Zaidi, S.M.; Jameel, S.S.; Zaman, F.; Jilani, S.; Sultana, A.; Khan, S.A. A systematic overview of the medicinal importance of sanguivorous leeches. Altern. Med. Rev. 2011, 16, 59–65. [Google Scholar]
- Mast, A.E.; Enghild, J.J.; Pizzo, S.V.; Salvesen, G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase-complexed, and reactive site cleaved serpins: Comparison of alpha 1-proteinase inhibitor, alpha 1-antichymotrypsin, antithrombin III, alpha 2-antiplasmin, angiotensinogen, and ovalbumin. Biochemistry 1991, 30, 1723–1730. [Google Scholar] [CrossRef]
- Lu, Z.; Shi, P.; You, H.; Liu, Y.; Chen, S. Transcriptomic analysis of the salivary gland of medicinal leech Hirudo nipponia. PLoS ONE. 2018, 13, e0205875. [Google Scholar] [CrossRef] [PubMed]
- Brankamp, R.G.; Sreekrishna, K.; Smith, P.L.; Blankenship, D.T.; Cardin, A.D. Expression of a Synthetic Gene Encoding the Anticoagulant-Antimetastatic Protein Ghilanten by the Methylotropic YeastPichia pastoris. Protein Expr. Purif. 1995, 6, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chu, T.T.T.; Kim, D.Y.; Kim, D.R.; Nguyen, C.M.T.; Choi, J.; Lee, J.R.; Hahn, M.J.; Kim, K.K. The Crystal Structure of Guamerin in Complex with Chymotrypsin and the Development of an Elastase-specific Inhibitor. J. Mol Biol. 2008, 376, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.R.; Kang, K.W. Amino acid sequence of piguamerin, an antistasin-type protease inhibitor from the blood sucking leech Hirudo nipponia. Eur. J. Biochem. 1998, 254, 692–697. [Google Scholar] [CrossRef]
- Kvist, S.; Min, G.-S.; Siddall, M.E. Diversity and selective pressures of anticoagulants in three medicinal leeches (Hirudinida: Hirudinidae, Macrobdellidae). Ecol Evol. 2013, 3, 918–933. [Google Scholar] [CrossRef]
- Sobczak, N.; Kantyka, M. Hirudotherapy in veterinary medicine. Ann. Parasitol. 2014, 60, 89–92. [Google Scholar]
- Tasiemski, A.; Vandenbulcke, F.; Mitta, G.; Lemoine, J.; Lefebvre, C.; Sautière, P.E.; Salzet, M. Molecular Characterization of Two Novel Antibacterial Peptides Inducible upon Bacterial Challenge in an Annelid, the Leech Theromyzon tessulatum. J. Biol. Chem. 2004, 279, 30973–30982. [Google Scholar] [CrossRef]
- Chen, L.L.; Yudina, T.G.; Artamonova, I.I.; Baskova, I.P. Antibacterial Non-Glycosidase Activity of Invertebrate Destabilase-Lysozyme and of Its Helical Amphipathic Peptides. Chemotherapy 2006, 52, 158–160. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, M.; Wang, J.; Lv, L.; Ning, S. Study progresses on hirudin. J. Jilin Med. Univ. 2021, 42, 135–137. [Google Scholar] [CrossRef]
- Worthen, P.L.; Gode, C.J.; Graf, J. Culture-Independent Characterization of the Digestive-Tract Microbiota of the Medicinal Leech Reveals a Tripartite Symbiosis. Appl. Environ. Microbiol. 2006, 72, 4775–4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Chao1 | Ace | Shannon | Coverage |
|---|---|---|---|---|
| FW | 114.21 ± 6.83 | 120.82 ± 6.14 | 1.75 ± 0.31 | 0.99 ± 0.00 |
| SW | 105.86 ± 13.31 | 105.98 ± 11.24 | 1.48 ± 0.16 | 0.99 ± 0.00 |
| OTUs | FW | SW | FW vs. SW |
|---|---|---|---|
| Bacteroidetes | 59.52 ± 12.53 | 34.92 ± 27.61 | Up |
| Firmicutes | 31.25 ± 11.46 | 57.93 ± 22.70 | Down |
| Proteobacteria | 8.93 ± 1.78 | 6.78 ± 4.57 | Up |
| Hydrogenoanaerobacterium | 22.71 ± 8.04 | 50.54 ± 18.69 | down |
| Mucinivorans | 14.90 ± 15.30 | 6.40 ± 9.26 | up |
| Bacteroides | 40.17 ± 24.61 | 5.25 ± 5.06 | up |
| Desulfovibrio | 3.96 ± 2.16 | 0.38 ± 0.46 | up |
| Aeromonas | 1.93 ± 0.45 | 0.84 ± 1.23 | up |
| unclassified_Porphyromonadaceae | 3.72 ± 0.86 | 21.98 ± 24.95 | down |
| unclassified_Ruminococcaceae | 6.51 ± 3.13 | 5.87 ± 3.76 | up |
| CategoryL2 | FW | SW | t | p |
|---|---|---|---|---|
| Membrane Transport | 11.6526 ± 1.1476 | 14.451 ± 0.8439 | −3.403 | 0.027 |
| Carbohydrate Metabolism | 10.6905 ± 0.1332 | 11.0392 ± 0.1289 | −3.257 | 0.031 |
| Amino Acid Metabolism | 9.6914 ± 0.3057 | 8.9745 ± 0.1936 | 3.431 | 0.027 |
| Poorly Characterized | 5.1011 ± 0.0733 | 4.9643 ± 0.0246 | 3.063 | 0.038 |
| Metabolism of Cofactors and Vitamins | 4.2304 ± 0.1063 | 3.8005 ± 0.0944 | 3.704 | 0.021 |
| Nucleotide Metabolism | 4.1021 ± 0.0956 | 3.8637 ± 0.1097 | 2.838 | 0.047 |
| Cellular Processes and Signaling | 4.1819 ± 0.0610 | 4.3046 ± 0.0391 | −2.94 | 0.042 |
| Glycan Biosynthesis and Metabolism | 2.3785 ± 0.3398 | 1.4765 ± 0.2654 | 3.624 | 0.022 |
| Lipid Metabolism | 2.5681 ± 0.0394 | 2.4725 ± 0.0022 | 4.2 | 0.014 |
| Folding, Sorting and Degradation | 2.5454 ± 0.1745 | 2.169 ± 0.0985 | 3.253 | 0.031 |
| Genetic Information Processing | 2.5558 ± 0.0449 | 2.432 ± 0.0366 | 3.701 | 0.021 |
| Transcription | 2.5205 ± 0.1004 | 2.7929 ± 0.1074 | −3.21 | 0.033 |
| Cell Motility | 2.7142 ± 0.4425 | 3.6728 ± 0.2267 | −3.34 | 0.029 |
| Metabolism of Terpenoids and Polyketides | 1.6484 ± 0.0425 | 1.5329 ± 0.0235 | 0.914 | 0.043 |
| Xenobiotics Biodegradation and Metabolism | 1.8792 ± 0.0965 | 2.2023 ± 0.1472 | −3.18 | 0.034 |
| Metabolism of Other Amino Acids | 1.3821 ± 0.1269 | 1.0696 ± 0.0516 | 3.949 | 0.017 |
| Biosynthesis of Other Secondary Metabolites | 0.9477 ± 0.0719 | 0.7393 ± 0.1480 | 3.747 | 0.02 |
| Cell Growth and Death | 0.4773 ± 0.0180 | 0.4328 ± 0.0110 | 3.656 | 0.022 |
| Transport and Catabolism | 0.3407 ± 0.0835 | 0.1277 ± 0.0532 | 3.729 | 0.02 |
| Infectious Diseases | 0.3891 ± 0.0175 | 0.3525 ± 0.0072 | 3.358 | 0.028 |
| Signaling Molecules and Interaction | 0.2137 ± 0.0417 | 0.1411 ± 0.0155 | 2.83 | 0.047 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, H.; Cai, M.; Li, J.; Luo, Y. High-Throughput Sequencing Reveals the Effect of Feeding on Overwintering Hirudo nipponia. Diversity 2022, 14, 768. https://doi.org/10.3390/d14090768
Shen H, Cai M, Li J, Luo Y. High-Throughput Sequencing Reveals the Effect of Feeding on Overwintering Hirudo nipponia. Diversity. 2022; 14(9):768. https://doi.org/10.3390/d14090768
Chicago/Turabian StyleShen, Hongying, Meixiang Cai, Jiayi Li, and Yuanyuan Luo. 2022. "High-Throughput Sequencing Reveals the Effect of Feeding on Overwintering Hirudo nipponia" Diversity 14, no. 9: 768. https://doi.org/10.3390/d14090768