
Conservation Significance of the Rare and Endangered Tree Species, Trigonobalanus doichangensis (Fagaceae)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Genotyping-by-Sequencing
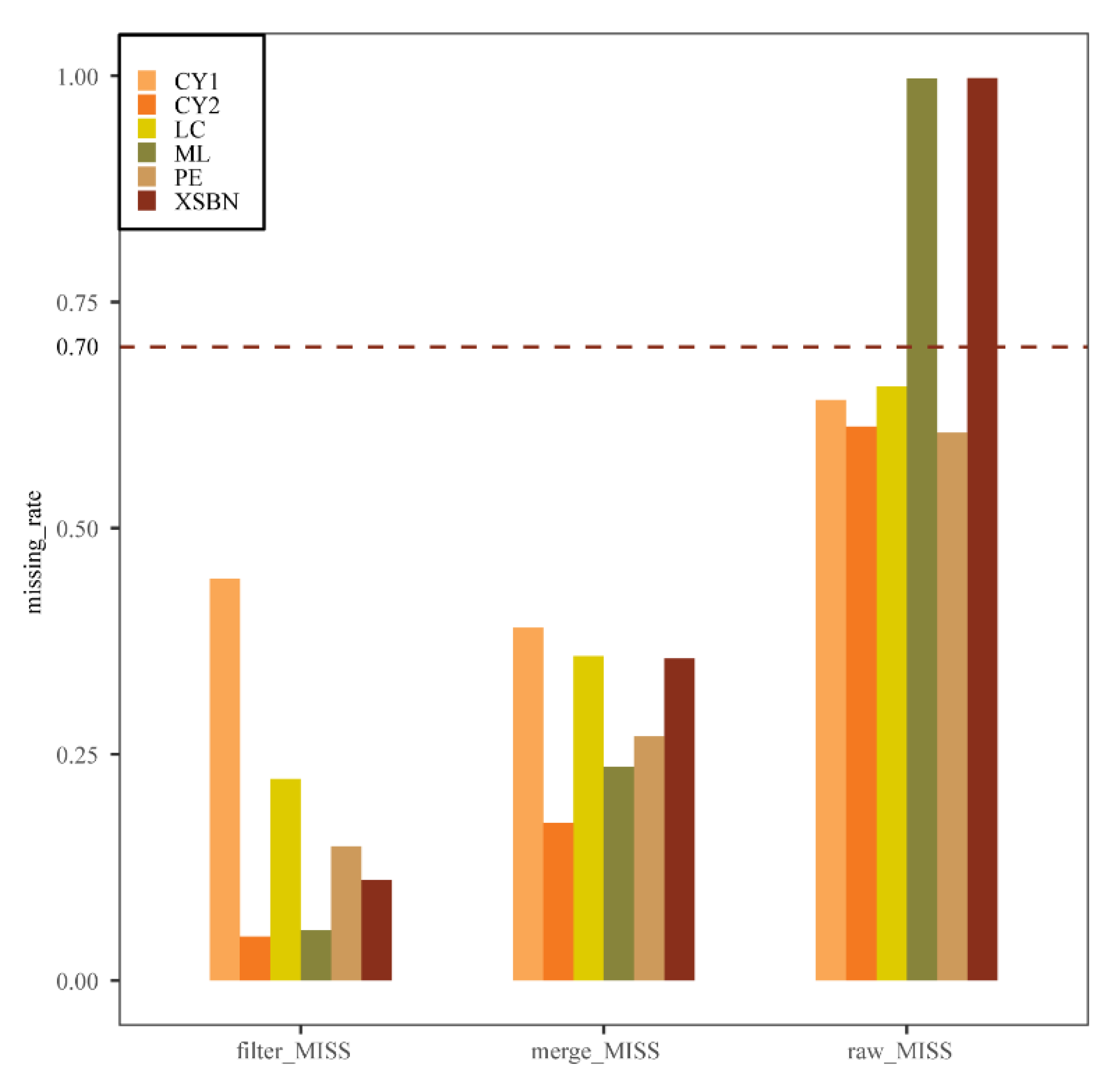
2.2. SNP Calling and Quality Filter
2.3. Calculation of Genetic Diversity Parameter and AMOVA
2.4. Isolation by Distance
2.5. Population Structure and Gene Flow
3. Results
3.1. Genotyping-by-Sequencing and Quality Control
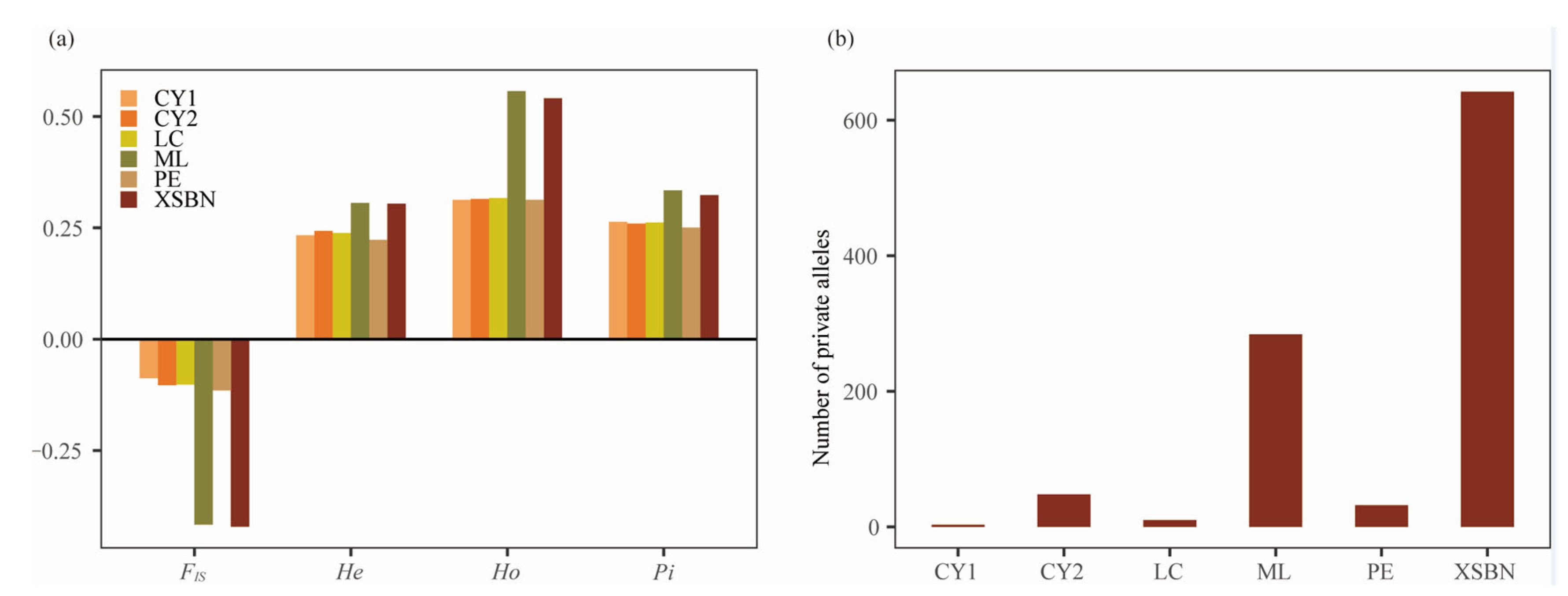
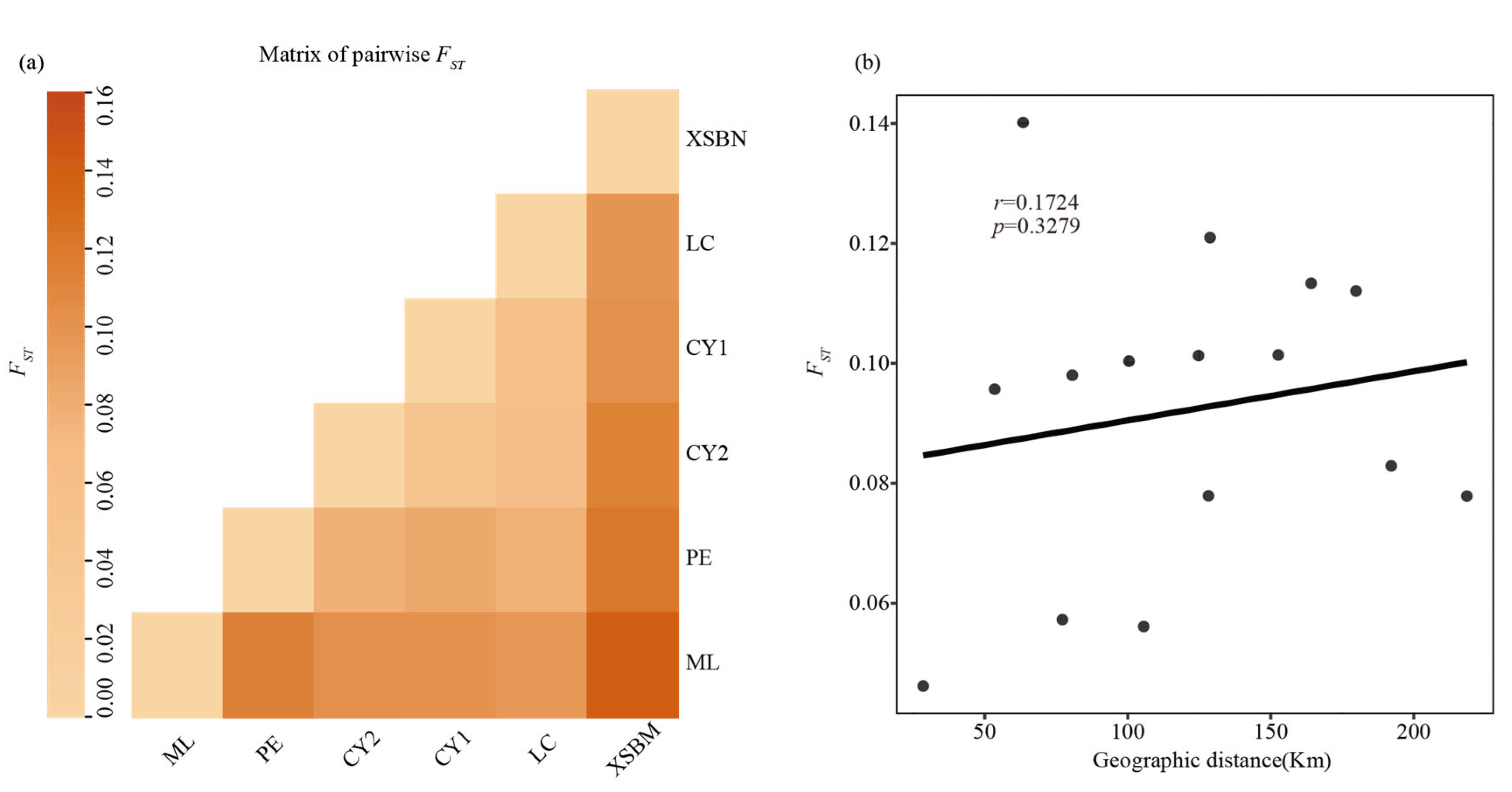
3.2. Genetic Diversity and Genetic Differentiation
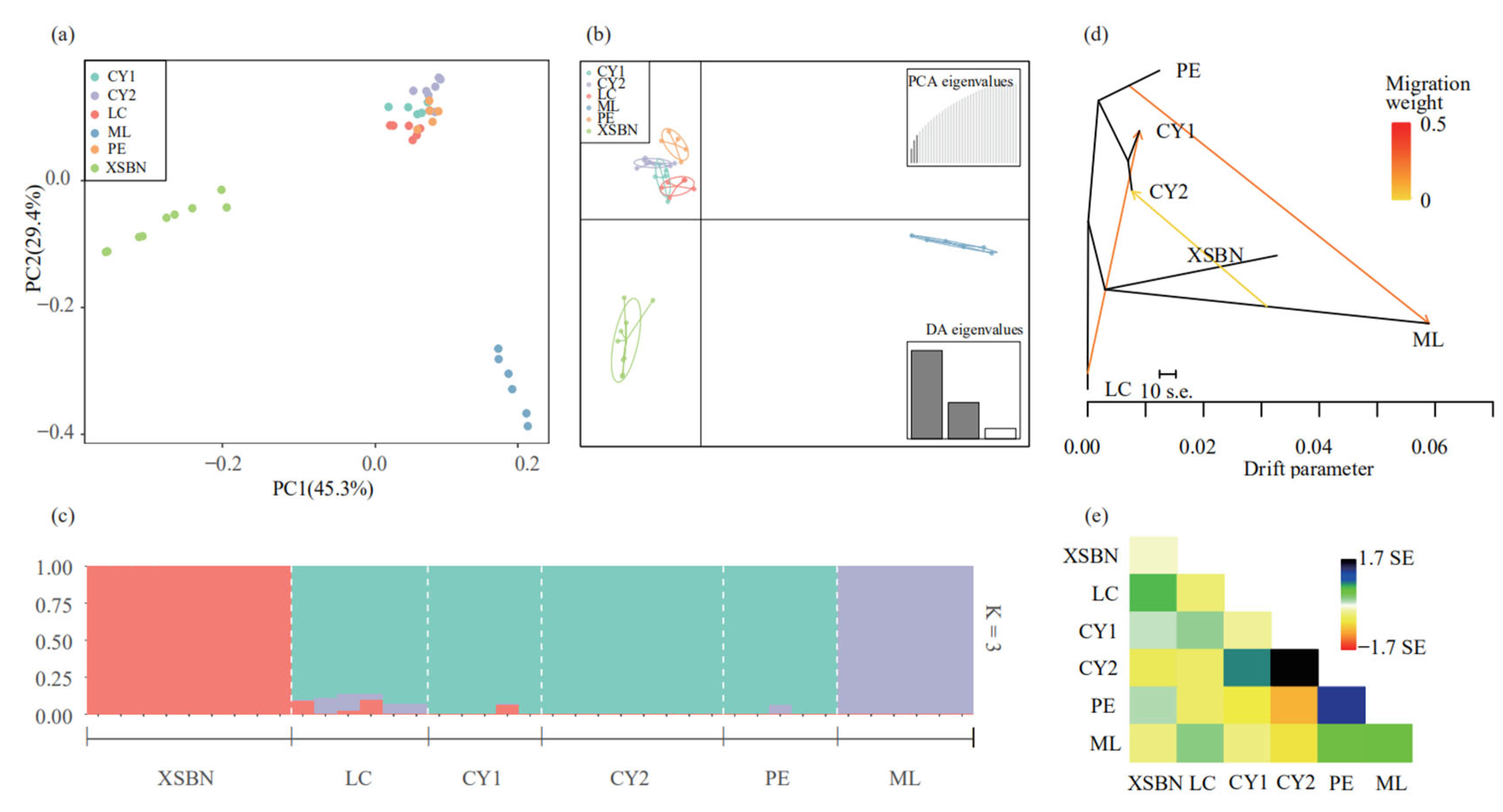
3.3. Genetic Structure of T. doichangensis
3.4. Gene Flow Analysis
4. Discussion
4.1. Genetic Diversity
4.2. Genetic Differentiation, Genetic Structure and Gene Flow
4.3. Conservation Significance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meng, H.H.; Gao, X.Y.; Song, Y.G.; Cao, G.L.; Li, J. Biodiversity arks in the Anthropocene. Reg. Sustain. 2021, 2, 109–115. [Google Scholar] [CrossRef]
- Folke, C.; Polasky, S.; Rockström, J.; Galaz, V.; Westley, F.; Lamont, M.; Scheffer, M.; Österblom, H.; Carpenter, S.R.; Chapin, F.S.; et al. Our future in the Anthropocene biosphere. Ambio 2021, 50, 834–869. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, M.; Ma, T.; Huang, M.; Huang, G.; Zhou, W.; Ping, X.; Lu, Y.; Wei, F. Integrated index-based assessment reveals long-term conservation progress in implementation of Convention on Biological Diversity. Sci. Adv. 2022, 8, eabj8093. [Google Scholar] [CrossRef]
- López-Pujol, J.; Zhang, F.M.; Ge, S. Plant biodiversity in China: Richly varied, endangered, and in need of conservation. Biodivers. Conserv. 2006, 15, 3983–4026. [Google Scholar] [CrossRef]
- Mi, X.; Feng, G.; Hu, Y.; Zhang, J.; Chen, L.; Corlett, R.T.; Hughes, A.C.; Pimm, S.; Schmid, B.; Shi, S.; et al. The global significance of biodiversity science in China: An overview. Natl. Sci. Rev. 2021, 8, nwab032. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.N.; Zhao, L.N. Evaluating the threat status of higher plants in China. Biodivers. Sci. 2017, 25, 689–695. [Google Scholar] [CrossRef]
- López-Bao, J.V.; Chapron, G.; Treves, A. The Achilles heel of participatory conservation. Biol. Conserv. 2017, 212, 139–143. [Google Scholar] [CrossRef]
- Meng, H.H.; Zhou, S.S.; Li, L.; Tan, Y.H.; Li, J.W.; Li, J. Conflict between biodiversity conservation and economic growth: Insight into rare plants in tropical China. Biodivers. Conserv. 2019, 28, 523–537. [Google Scholar] [CrossRef]
- Yang, W.Z.; Xiang, Z.Y.; Zhang, S.S.; Kang, H.M.; Shi, F.Q. Plant species with extremely small populations (PSESP) and their significance in China’s national plant conservation strategy. Biodivers. Sci. 2015, 23, 419–425. [Google Scholar] [CrossRef]
- Crane, P. Conserving our global botanical heritage: The PSESP plant conservation program. Plant Divers. 2020, 42, 319–322. [Google Scholar] [CrossRef]
- Sun, W.B.; Ma, Y.P.; Blackmore, S. How a new conservation action concept has accelerated plant conservation in China. Trends Plant Sci. 2019, 24, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.L.; Wang, H.C.; Yu, Z.X.; Schinnerl, J.; Tang, R.; Geng, Y.P.; Chen, G. Genetic diversity and structure of the endemic and endangered species Aristolochia delavayi growing along the Jinsha River. Plant Divers. 2021, 43, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, S.L.; Pei, S.Y.; Ning, M.M.; Tang, S.Q. Genetic diversity and population structure of Camellia huana (Theaceae), a limestone species with narrow geographic range, based on chloroplast DNA sequence and microsatellite markers. Plant Divers. 2020, 42, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.Y.; Ji, Y.H.; Liu, J.; Gong, X. Genetic characterization of the entire range of Cycas panzhihuaensis (Cycadaceae). Plant Divers. 2020, 42, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.N.; Xiao, J.H.; Ci, X.Q.; Conran, J.G.; Li, J. Genetic diversity of Horsfieldia tetratepala (Myristicaceae), an endangered Plant Species with Extremely Small Populations to China: Implications for its conservation. Plant Syst. Evol. 2021, 307, 50. [Google Scholar] [CrossRef]
- Zhang, X.J.; Liu, X.F.; Liu, D.T.; Cao, Y.R.; Li, Z.H.; Ma, Y.P.; Ma, H. Genetic diversity and structure of Rhododendron meddianum, a plant species with extremely small populations. Plant Divers. 2021, 43, 472–479. [Google Scholar] [CrossRef]
- Zhang, X.M.; Qin, H.T.; Xie, W.J.; Ma, Y.P.; Sun, W.B. Comparative population genetic analyses suggest hybrid origin of Rhododendron pubicostatum, an endangered plant species with extremely small populations endemic to Yunnan, China. Plant Divers. 2020, 42, 312–318. [Google Scholar] [CrossRef]
- Sun, W.B.; Han, C.Y. Researches and conservation for plant species with extremely small populations (PSESP). Biodivers. Sci. 2015, 23, 426–429. [Google Scholar] [CrossRef]
- Phillips, R.D.; Dixon, K.W.; Peakall, R. Low population genetic differentiation in the Orchidaceae: Implications for the diversification of the family. Mol. Ecol. 2012, 21, 5208–5220. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Jia, H.X.; Wang, Z.B.; Sun, P.; Cao, D.M.; Hu, J.J. Genetic diversity and population structure of Populus yunnanensis. Biodivers. Sci. 2019, 27, 355–365. [Google Scholar]
- Xiao, J.H.; Ding, X.; Li, L.; Ma, H.; Ci, X.Q.; van der Merwe, M.; Conran, J.G.; Li, J. Miocene diversification of a golden-thread nanmu tree species (Phoebe zhennan, Lauraceae) around the Sichuan Basin shaped by the East Asian monsoon. Ecol. Evol. 2020, 10, 10543–10557. [Google Scholar] [CrossRef] [PubMed]
- Waqar, Z.; Moraes, R.C.S.; Benchimol, M.; Morante-Filho, J.C.; Mariano-Neto, E.; Gaiotto, F.A. Gene flow and genetic structure reveal reduced diversity between generations of a tropical tree, Manilkara multifida Penn., in atlantic forest fragments. Genes 2021, 12, 2025. [Google Scholar] [CrossRef] [PubMed]
- Pekkala, N.; Knott, K.E.; Kotiaho, J.S.; Nissinen, K.; Puurtinen, M. The effect of inbreeding rate on fitness, inbreeding depression and heterosis over a range of inbreeding coefficients. Evol. Appl. 2014, 7, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.P.; Sloop, C.M.; Davis, H.G.; Cushman, J. Population genetic diversity and structure of two rare vernal pool grasses in central California. Conserv. Genet. 2012, 13, 117–130. [Google Scholar] [CrossRef]
- Wu, F.Q.; Shen, S.K.; Zhang, X.J.; Wang, Y.H.; Sun, W.B. Genetic diversity and population structure of an extremely endangered species: The world′s largest Rhododendron. AoB Plants 2015, 7, plu082. [Google Scholar] [CrossRef]
- Ci, X.Q.; Chen, J.Q.; Li, Q.M.; Li, J. AFLP and ISSR analysis reveals high genetic variation and inter-population differentiation in fragmented populations of the endangered Litsea szemaois (Lauraceae) from Southwest China. Plant Syst. Evol. 2008, 273, 237–246. [Google Scholar] [CrossRef]
- Narum, S.R.; Buerkle, C.A.; Davey, J.W.; Miller, M.R.; Hohenlohe, P.A. Genotyping-by-sequencing in ecological and conservation Genomics. Mol. Ecol. 2013, 22, 2841–2847. [Google Scholar] [CrossRef]
- Deschamps, S.; Llaca, V.; May, G.D. Genotyping-by-sequencing in plants. Biology 2012, 1, 460–483. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Shekhovtsov, S.V.; Shekhovtsova, I.N.; Kosterin, O.E. Genotyping-by-Sequencing Analysis Shows That Siberian Lindens Are Nested within Tilia cordata Mill. Diversity 2022, 14, 256. [Google Scholar] [CrossRef]
- Forman, L. Trigonobalanus, a new genus of Fagaceae, with notes on the classification of the family. Kew Bull. 1964, 17, 381–396. [Google Scholar] [CrossRef]
- Nixon, K.C.; Crepet, W.L. Trigonobalanus (Fagaceae): Taxonomic status and phylogenetic relationships. Am. J. Bot. 1989, 76, 828–841. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Wang, C.J.; Wu, C.Y.; Li, H.W. Trigonobalanus Forman-A new recorded genus of Fagaceae in China. Acta Bot. Yunnan 1981, 3, 213–215. [Google Scholar]
- Zheng, Y.L.; Sun, W.B.; Zhou, Y.; Coombs, D. Variation in seed and seedling traits among natural populations of Trigonobalanus doichangensis (A. Camus) Forman (Fagaceae), a rare and endangered plant in southwest China. New For. 2009, 37, 285–294. [Google Scholar] [CrossRef]
- Sun, W.B.; Zhou, Y.; Han, C.Y.; Zeng, C.X.; Shi, X.D.; Xiang, Q.B.; Coombes, A. Status and conservation of Trigonobalanus doichangensis (Fagaceae). Biodivers. Conserv. 2006, 15, 1303–1318. [Google Scholar] [CrossRef]
- Wang, S.; Xie, Y. China Species Red List; High Education Press: Beijing, China, 2004. [Google Scholar]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Staton, M.; Addo-Quaye, C.; Cannon, N.; Yu, J.; Zhebentyayeva, T.; Huff, M.; Islam-Faridi, N.; Fan, S.; Georgi, L.L.; Nelson, C.D.; et al. A reference genome assembly and adaptive trait analysis of Castanea mollissima ‘Vanuxem,’a source of resistance to chestnut blight in restoration breeding. Tree Genet. Genomes 2020, 16, 1392. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Lischer, H.E.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, R.J.; Karney, C.; Williams, E.; Vennes, C. Package “geosphere”: Spherical Trigonometry. 2021. Available online: https://cran.r-project.org/web/packages/geosphere/geosphere.pdf (accessed on 10 March 2022).
- Dray, S.; Dufour, A.B.; Chessel, D. The ade4 package-II: Two-table and K-table methods. R News 2007, 7, 47–52. [Google Scholar]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ezpeleta, N.; Bradbury, I.R.; Mendibil, I.; Álvarez, P.; Cotano, U.; Irigoien, X. Population structure of Atlantic mackerel inferred from RAD-seq-derived SNP markers: Effects of sequence clustering parameters and hierarchical SNP selection. Mol. Ecol. Resour. 2016, 16, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Duan, X.Z.; Zhao, D.S.; Sun, J.T.; Hong, X.Y. Genome-wide single nucleotide polymorphisms are robust in resolving fine-scale population genetic structure of the small brown planthopper, Laodelphax striatellus (Fallén) (Hemiptera: Delphacidae). J. Econ. Entomol. 2019, 112, 2362–2368. [Google Scholar] [CrossRef]
- Dierickx, E.G.; Shultz, A.J.; Sato, F.; Hiraoka, T.; Edwards, S.V. Morphological and genomic comparisons of Hawaiian and Japanese Black-footed Albatrosses (Phoebastria nigripes) using double digest RADseq: Implications for conservation. Evol. Appl. 2015, 8, 662–678. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, D.; Wariss, H.M.; Zhang, R.; Tao, L.; Milne, R.I.; Sun, W. Demographic history and identification of threats revealed by population genomic analysis provide insights into conservation for an endangered maple. Mol. Ecol. 2022, 31, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Fitak, R.R. OptM: Estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 2021, 6, bpab017. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Patterson, N.; Barbieri, C.; Berthold, F.; Gerlach, L.; Güldemann, T.; Kure, B.; Mpoloka, S.W.; Nakagawa, H.; Naumann, C.; et al. The genetic prehistory of southern Africa. Nat. Commun. 2012, 3, 1143. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Huang, H.W. Gene flow dynamics and related adaptive evolution in plant populations. Chin. Bull. Bot. 2009, 44, 351. [Google Scholar]
- Laikre, L.; Allendorf, F.W.; Aroner, L.C.; Baker, C.S.; Gregovich, D.P.; Hansen, M.M.; Jackson, J.A.; Kendall, K.C.; McKELVEY, K.; Neel, M.C.; et al. Neglect of genetic diversity in implementation of the convention on biological diversity. Conserv. Biol. 2010, 24, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, Y.G.; Zhang, C.Y.; Wang, T.R.; Su, T.H.; Huang, P.H.; Meng, H.H.; Li, J. Latitudinal diversity gradient in the changing world: Retrospectives and perspectives. Diversity 2022, 14, 334. [Google Scholar] [CrossRef]
- Setoguchi, H.; Mitsui, Y.; Ikeda, H.; Nomura, N.; Tamura, A. Genetic structure of the critically endangered plant Tricyrtis ishiiana (Convallariaceae) in relict populations of Japan. Conserv. Genet. 2011, 12, 491–501. [Google Scholar] [CrossRef]
- Ramanatha, R.V.; Hodgkin, T. Genetic diversity and conservation and utilization of plant genetic resources. Plant Cell Tissue Organ Cult. 2002, 68, 1–19. [Google Scholar] [CrossRef]
- Zhou, Y. Preliminary study on geographic populations of Trigonobalanus doichangensis and its effect on seed germination and seedling. Seed 2003, 4, 3–4. [Google Scholar]
- Sun, W.B.; Zhou, Y.; Zhao, J.C.; Chen, G. Current distribution, population attributes and biological characters of Trigonobalanus doichangensis in relation to its conservation. Acta Ecol. Sin. 2004, 24, 352–358. [Google Scholar]
- Aoki, K.; Ueno, S.; Kamijo, T.; Setoguchi, H.; Murakami, N.; Kato, M.; Tsumura, Y. Genetic differentiation and genetic diversity of Castanopsis (Fagaceae), the dominant tree species in Japanese broadleaved evergreen forests, revealed by analysis of EST-associated microsatellites. PLoS ONE 2014, 9, e87429. [Google Scholar] [CrossRef] [PubMed]
- Valencia, C.L.; Piñero, D.; Mussali, G.P.; Valencia-Ávalos, S.; Tovar-Sánchez, E. Effect of a red oak species gradient on genetic structure and diversity of Quercus castanea (Fagaceae) in Mexico. Tree Genet. Genomes 2014, 10, 641–652. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Plant Population Genetics, Breeding, and Genetic Resources; Sinauer: Sunderland, MA, USA, 1990. [Google Scholar]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef]
- Gamba, D.; Muchhala, N. Global patterns of population genetic differentiation in seed plants. Mol. Ecol. 2020, 29, 3413–3428. [Google Scholar] [CrossRef]
- Li, S.; Gan, X.H.; Han, H.Y.; Zhang, X.M.; Tian, Z.Q. Low within-population genetic diversity and high genetic differentiation among populations of the endangered plant Tetracentron sinense Oliver revealed by inter-simple sequence repeat analysis. Ann. For. Sci. 2018, 75, 74. [Google Scholar] [CrossRef]
- Souza, F.L.; Cunha, A.F.; Oliveira, M.A.; Pereira, G.A.; Reis, S.F.d. Estimating dispersal and gene flow in the neotropical freshwater turtle Hydromedusa maximiliani (Chelidae) by combining ecological and genetic methods. Genet. Mol. Biol. 2002, 25, 151–155. [Google Scholar] [CrossRef]
- Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Sun, W.B.; Han, C.Y.; Gao, L.M.; Wilson, C.A. Genetic diversity of the rare Asian plant, Trigonobalanus doichangensis (Fagaceae). Aust. J. Bot. 2007, 55, 10–17. [Google Scholar] [CrossRef]
- Fu, L.G. China Plant Red Data Book; Science Press: Beijing, China, 1992; Volume 1, pp. 302–303. [Google Scholar]
- Meng, H.H.; Zhou, S.S.; Jiang, X.L.; Gugger, P.F.; Li, L.; Tan, Y.H.; Li, J. Are mountaintops climate refugia for plants under global warming? A lesson from high-mountain oaks in tropical rainforest. Alp. Bot. 2019, 129, 175–183. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Populations | No. | Locations | Longitude | Latitude | Elevation (m) |
|---|---|---|---|---|---|---|
| 1 | XSBN | 9 | Padianliangzi, Xishuangbanna, Yunnan | 100°04′58″ | 22°02′32″ | 2038 |
| 2 | LC | 6 | Donghuixiang, Lancang, Yunnan | 99°48′50″ | 22°43′20″ | 1400 |
| 3 | CY1 | 5 | Chengbian, Cangyuan, Yunnan | 99°14′28″ | 23°10′18″ | 1482 |
| 4 | CY2 | 8 | Nanbancun, Cangyuan, Yunnan | 99°01′04″ | 23°19′27″ | 1482 |
| 5 | PE | 5 | Dazhaishuiku, Puer, Yunnan | 101°03′43″ | 22°45′44″ | 1564 |
| 6 | ML | 6 | Dengzhanzhai, Menglian, Yunnan | 99°32′30″ | 22°18′47″ | 1153 |
| Populations | PA | Ho | He | Pi | FIS |
|---|---|---|---|---|---|
| XSBN | 642 | 0.5410 | 0.3044 | 0.3235 | −0.4213 |
| LC | 10 | 0.3169 | 0.2383 | 0.2621 | −0.1022 |
| CY1 | 3 | 0.3129 | 0.2335 | 0.2637 | −0.0879 |
| CY2 | 48 | 0.3150 | 0.2430 | 0.2598 | −0.1033 |
| PE | 32 | 0.3129 | 0.2232 | 0.2504 | −0.1151 |
| ML | 284 | 0.5571 | 0.3059 | 0.3343 | −0.4166 |
| Populations | XSBN | LC | CY1 | CY2 | PE | ML |
|---|---|---|---|---|---|---|
| XSBN | - | 80.5984 | 152.6067 | 179.8030 | 128.7604 | 63.4062 |
| LC | 0.0980 | - | 77.1407 | 105.5585 | 128.2083 | 53.4627 |
| CY1 | 0.1014 | 0.0573 | - | 28.4602 | 192.1101 | 100.4373 |
| CY2 | 0.1121 | 0.0561 | 0.0462 | - | 218.5402 | 124.7333 |
| PE | 0.1210 | 0.0779 | 0.0829 | 0.0779 | - | 164.1128 |
| ML | 0.1402 | 0.0957 | 0.1004 | 0.1013 | 0.1133 | - |
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation |
|---|---|---|---|---|
| Among groups | 2 | 3210.467 | 49.1535 | 9.97 |
| Among populations within groups | 3 | 1617.129 | 8.8010 | 1.79 |
| Within populations | 72 | 31312.635 | 434.8977 | 88.24 |
| Total | 77 | 36140.231 | 492.8522 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, L.; Le, X.-G.; Zhou, S.-S.; Zhang, C.-Y.; Tan, Y.-H.; Ren, Q.; Meng, H.-H.; Cun, Y.; Li, J. Conservation Significance of the Rare and Endangered Tree Species, Trigonobalanus doichangensis (Fagaceae). Diversity 2022, 14, 666. https://doi.org/10.3390/d14080666
Hu L, Le X-G, Zhou S-S, Zhang C-Y, Tan Y-H, Ren Q, Meng H-H, Cun Y, Li J. Conservation Significance of the Rare and Endangered Tree Species, Trigonobalanus doichangensis (Fagaceae). Diversity. 2022; 14(8):666. https://doi.org/10.3390/d14080666
Chicago/Turabian StyleHu, Ling, Xin-Gui Le, Shi-Shun Zhou, Can-Yu Zhang, Yun-Hong Tan, Qiang Ren, Hong-Hu Meng, Yupeng Cun, and Jie Li. 2022. "Conservation Significance of the Rare and Endangered Tree Species, Trigonobalanus doichangensis (Fagaceae)" Diversity 14, no. 8: 666. https://doi.org/10.3390/d14080666