
Red-Backed Shrike Lanius collurio Whole-Genome Sequencing Reveals Population Genetic Admixture



Abstract
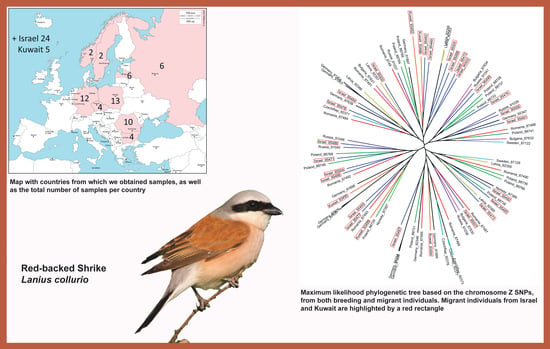
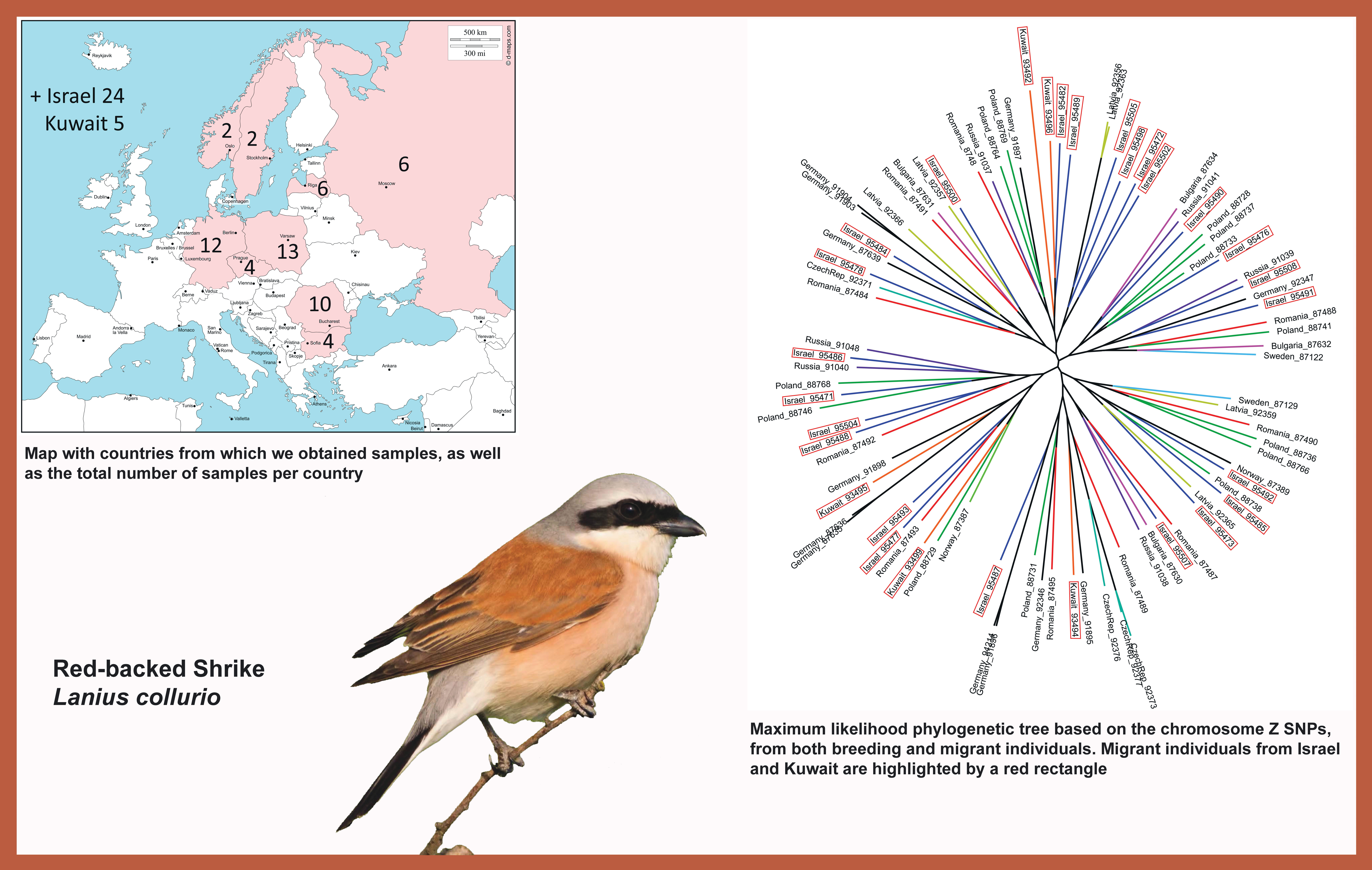
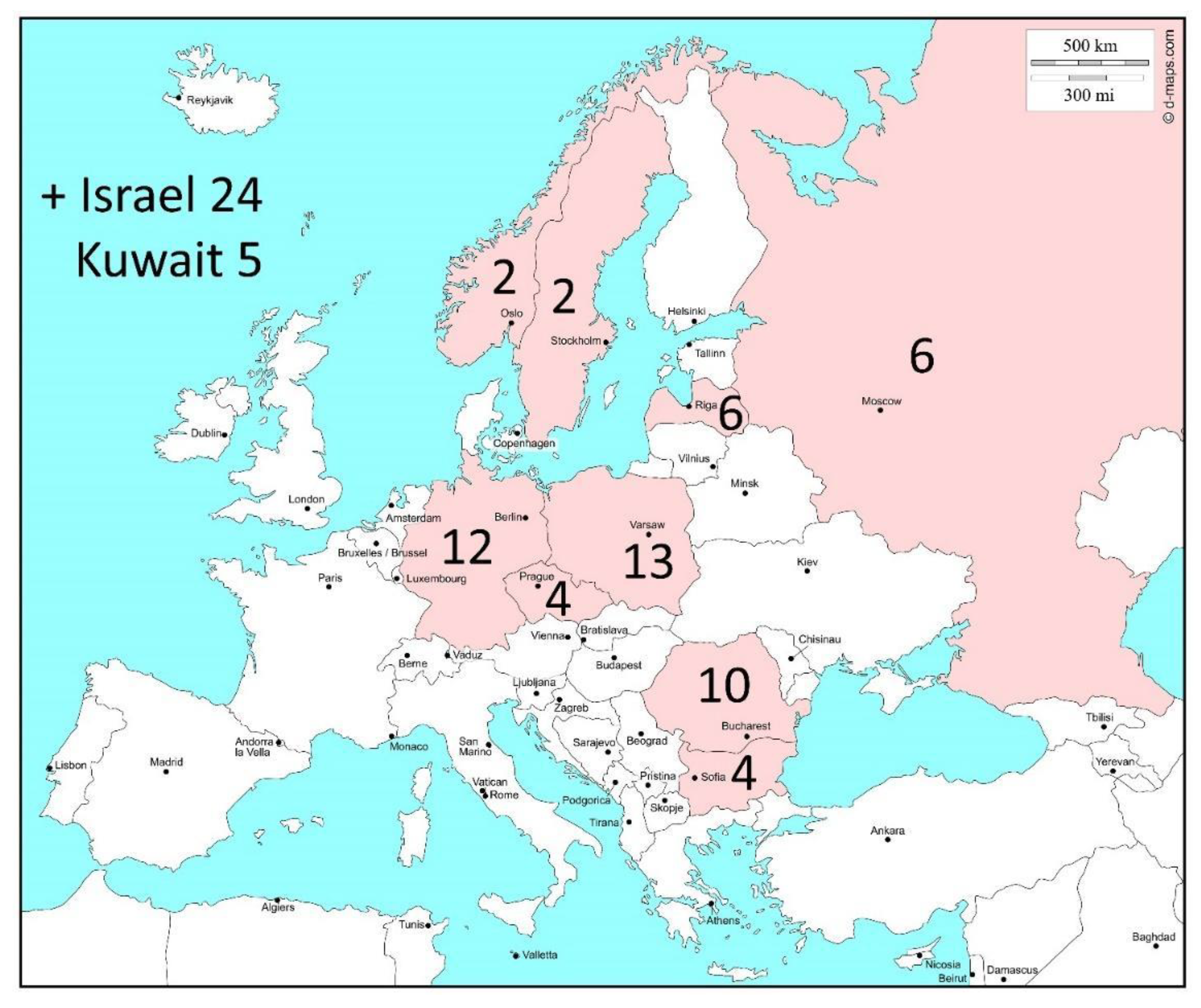
:
1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, and Sequencing
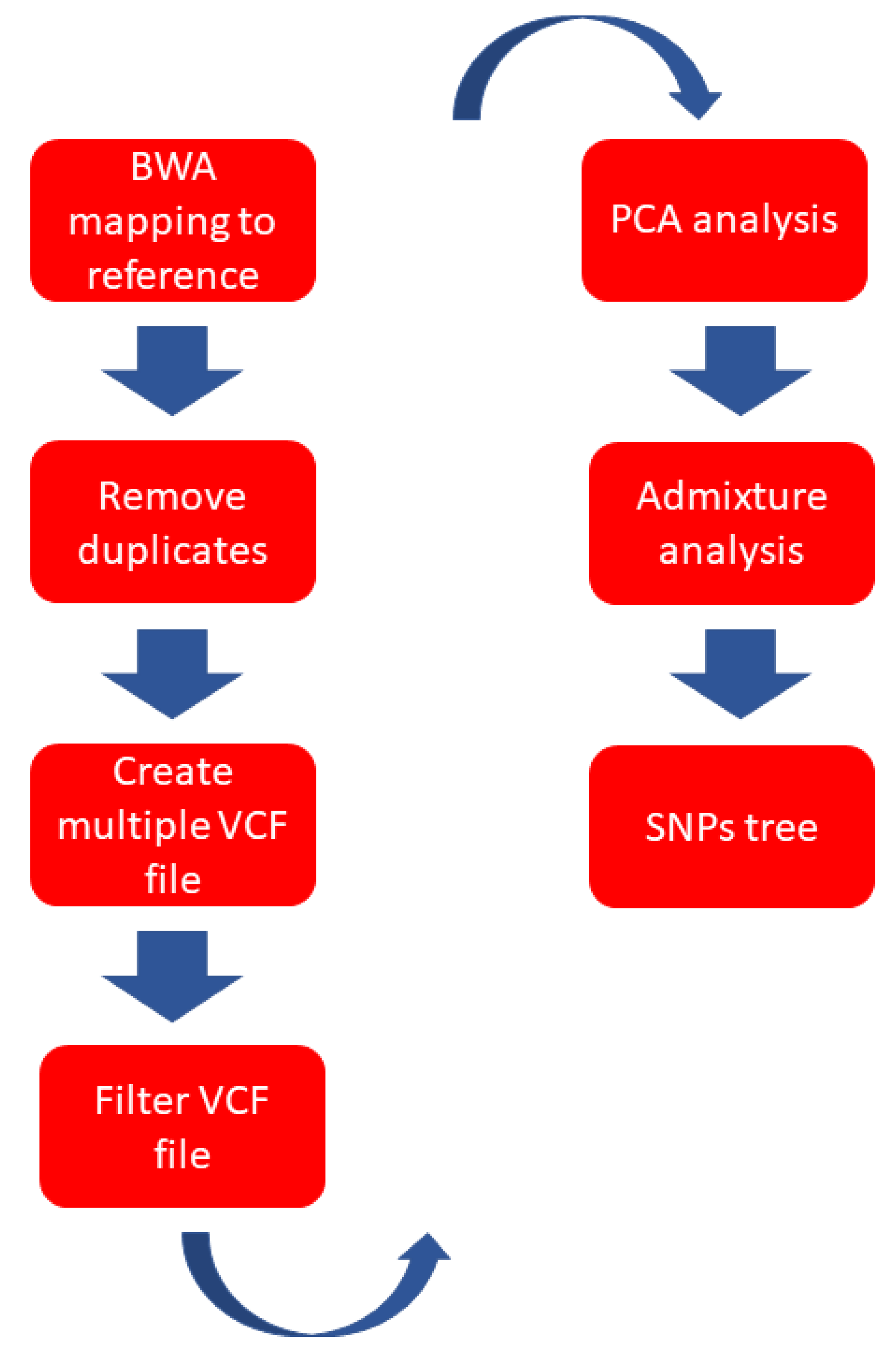
2.2. NGS Data Analysis
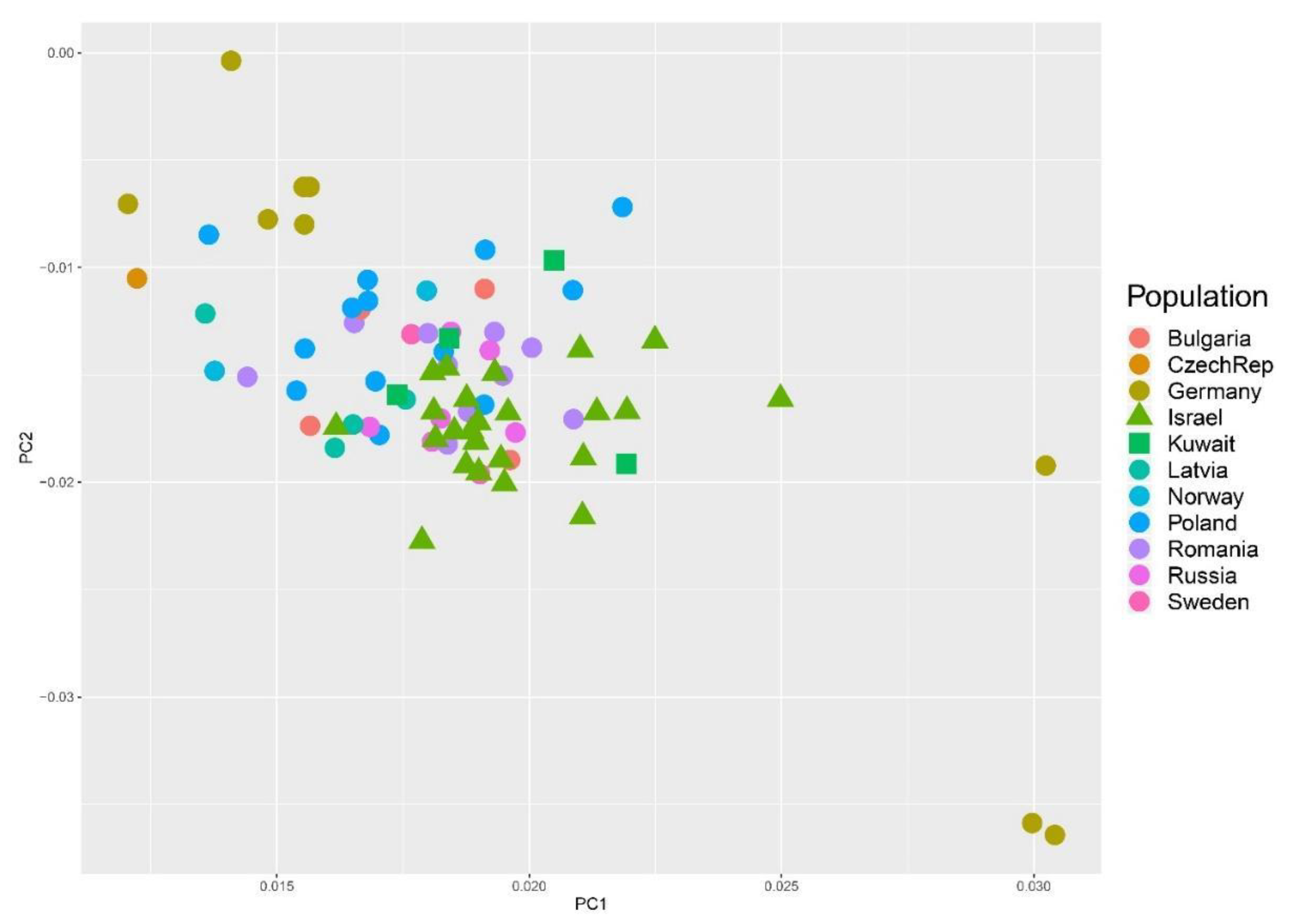
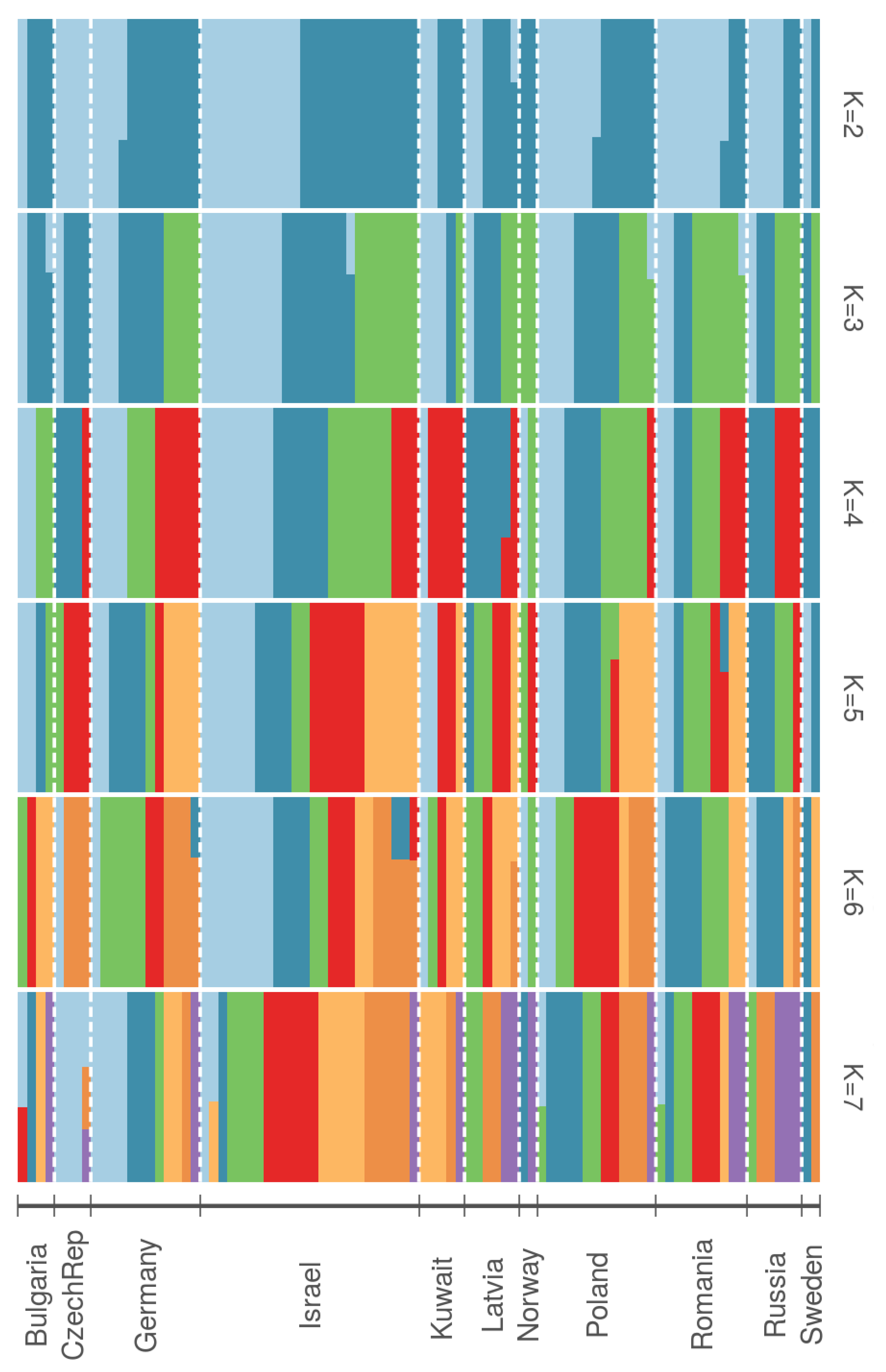
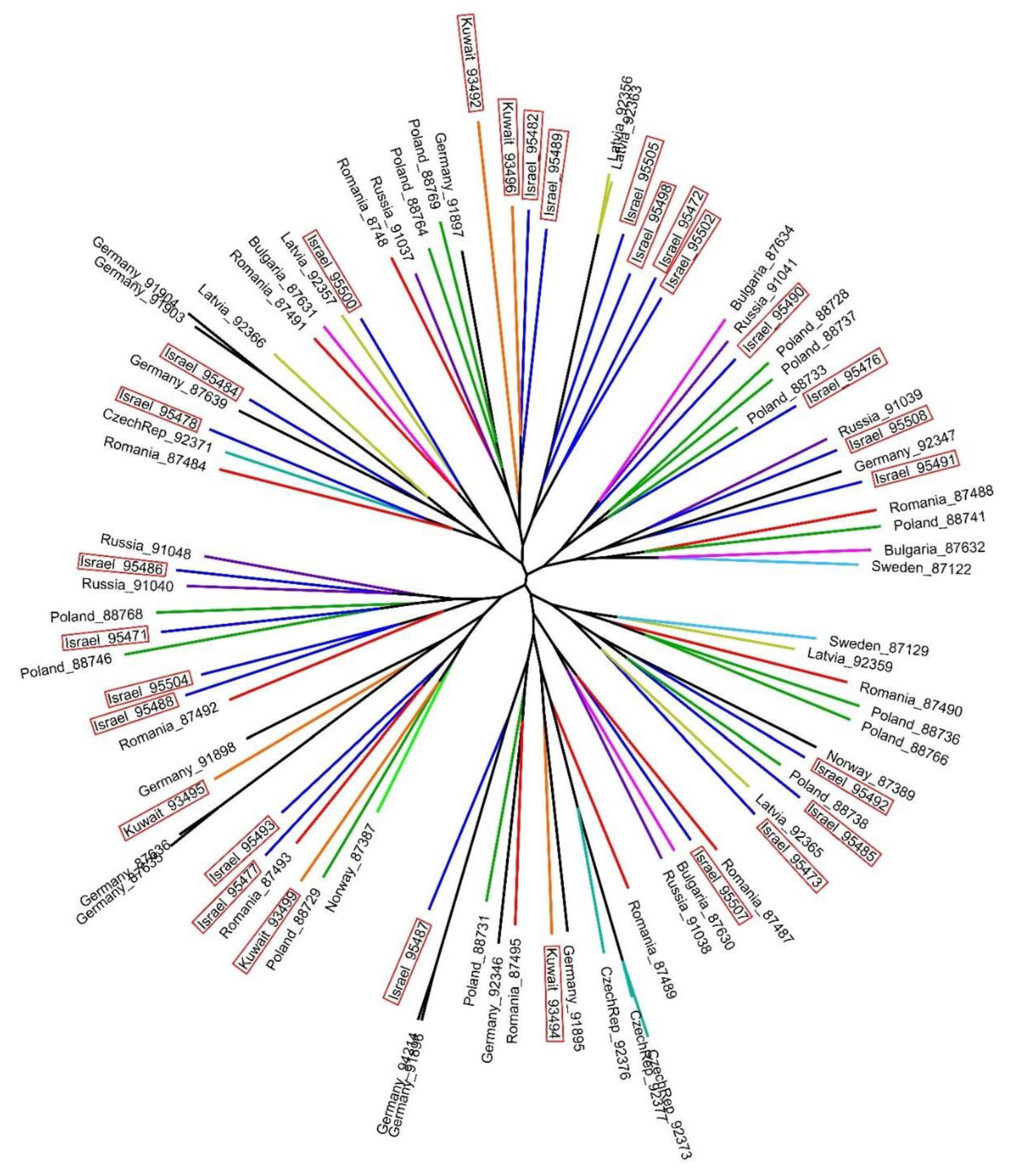
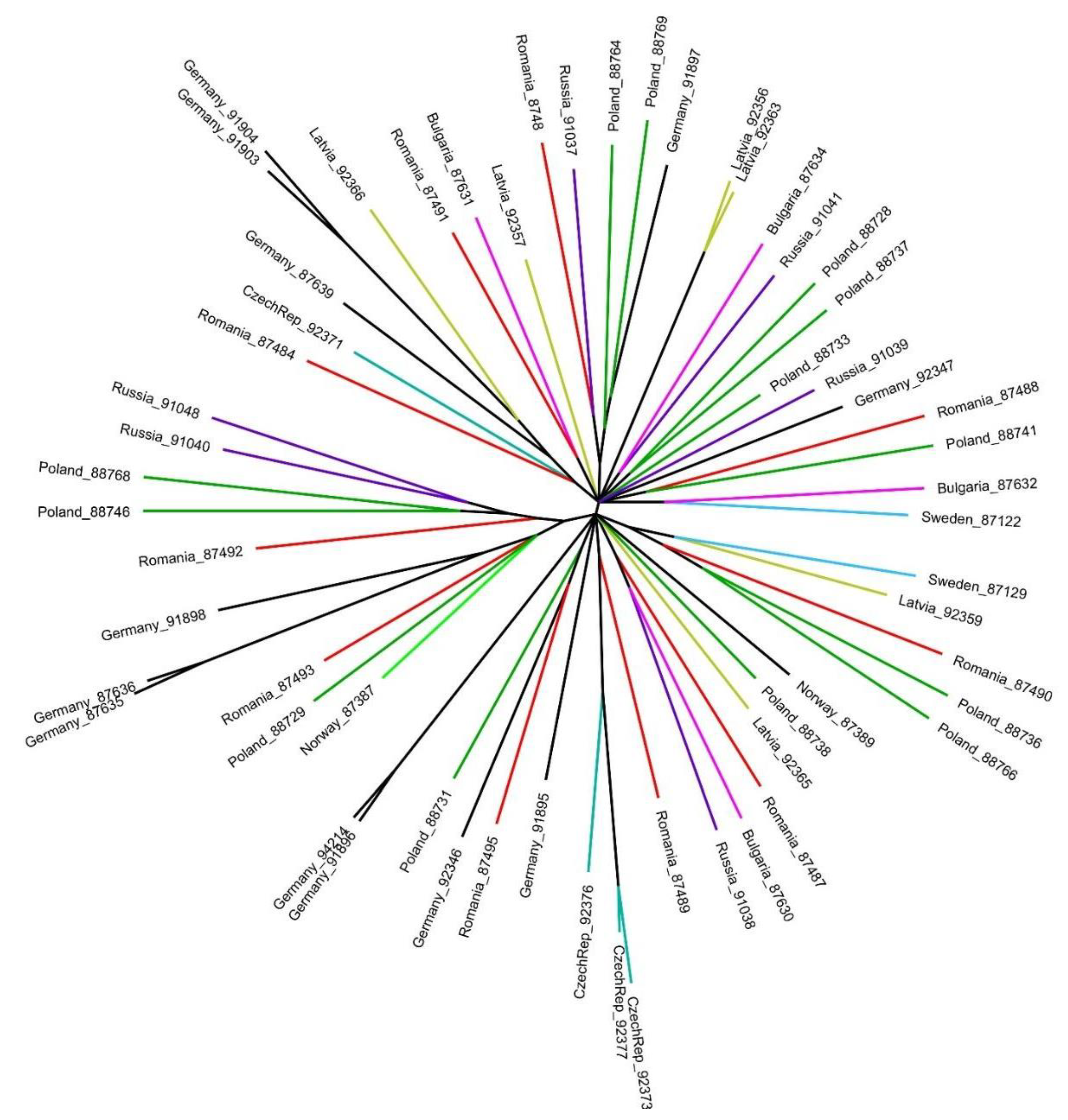
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.A. The Correlation between Relatives on the Supposition of Mendelian Inheritance, Trans. R. Soc. Edinb. 1919, 52, 399–433. [Google Scholar] [CrossRef] [Green Version]
- Haldane, J.B.S. A Mathematical Theory of Natural and Artificial Selection. Parts I—IX. Math. Proc. Camb. Philos. Soc. 1932, 23, 26–28. [Google Scholar]
- Wright, S. Evolution in Mendelian Populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Charlesworth, D. Population Genetics from 1966 to 2016. Heredity 2017, 118, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heather, J.M.; Chain, B. The Sequence of Sequencers: The History of Sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajora, O.P. (Ed.) Population Genomics; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Ellegren, H. Genome Sequencing and Population Genomics in Non-Model Organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 1997. [Google Scholar]
- Zhang, G. Bird Sequencing Project Takes Off. Nature 2015, 522, 34. [Google Scholar] [CrossRef]
- Stiller, J.; Zhang, G. Comparative Phylogenomics, a Stepping Stone for Bird Biodiversity Studies. Diversity 2019, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Wink, M. A Historical Perspective of Avian Genomics. In Avian Genomics in Ecology and Evolution; Kraus, R.H., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 7–19. [Google Scholar]
- Kraus, R.H.S.; Wink, M. Avian Genomics: Fledging into the Wild! J. Ornithol. 2015, 156, 851–865. [Google Scholar] [CrossRef] [Green Version]
- Wink, M. DNA Analyses Have Revolutionized Studies on the Taxonomy and Evolution in Birds. In Birds—Challenges and Opportunities for Business, Conservation and Research; Mikkola, H., Ed.; IntechOpen: London, UK, 2021. [Google Scholar]
- Kimball, R.T.; Oliveros, C.H.; Wang, N.; White, N.D.; Barker, F.K.; Field, D.J.; Ksepka, D.T.; Chesser, R.T.; Moyle, R.G.; Braun, M.J.; et al. A Phylogenomic Supertree of Birds. Diversity 2019, 11, 109. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T. Whole-Genome Analyses Resolve Early Branches in the Tree of Life of Modern Birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A Comprehensive Phylogeny of Birds (Aves) Using Targeted Next-Generation DNA Sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Hillier, L.W.; Miller, W.; Birney, E.; Warren, W.; Hardison, R.C.; Ponting, C.P.; Bork, P.; Burt, D.W.; Groenen, M.A.M.; Delany, M.E.; et al. Sequence and Comparative Analysis of the Chicken Genome Provide Unique Perspectives on Vertebrate Evolution. Nature 2004, 432, 695–716. [Google Scholar]
- Dalloul, R.A.; Long, J.A.; Zimin, A.V.; Aslam, L.; Beal, K.; Blomberg, L.A.; Bouffard, P.; Burt, D.W.; Crasta, O.; Crooijmans, R.P.M.A.; et al. Multi-Platform Next-Generation Sequencing of the Domestic Turkey (Meleagris Gallopavo): Genome Assembly and Analysis. PLoS Biol. 2010, 8, e1000475. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Dixon, A.; Batbayar, N.; Bragin, E.; Ayas, Z.; Deutschova, L.; Chavko, J.; Domashevsky, S.; Dorosencu, A.; Bagyura, J.; et al. Exonic versus Intronic SNPs: Contrasting Roles in Revealing the Population Genetic Differentiation of a Widespread Bird Species. Heredity 2015, 114, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Spurgin, L.G.; Bosse, M.; Adriaensen, F.; Albayrak, T.; Barboutis, C.; Belda, E.; Bushuev, A.; Cecere, J.G.; Charmantier, A.; Cichon, M.; et al. The Great Tit HapMap Project: A Continental-Scale Analysis of Genomic Variation in a Songbird. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Calderón, L.; Campagna, L.; Wilke, T.; Lormee, H.; Eraud, C.; Dunn, J.C.; Rocha, G.; Zehtindjiev, P.; Bakaloudis, D.E.; Metzger, B.; et al. Genomic Evidence of Demographic Fluctuations and Lack of Genetic Structure across Flyways in a Long Distance Migrant, the European Turtle Dove. BMC Evol. Biol. 2016, 16, 237. [Google Scholar] [CrossRef] [Green Version]
- von Rönn, J.A.C.; Shafer, A.B.A.; Wolf, J.B.W. Disruptive Selection without Genome-Wide Evolution across a Migratory Divide. Mol. Ecol. 2016, 25, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Vijay, N.; Bossu, C.M.; Poelstra, J.W.; Weissensteiner, M.H.; Suh, A.; Kryukov, A.P.; Wolf, J.B.W. Evolution of Heterogeneous Genome Differentiation across Multiple Contact Zones in a Crow Species Complex. Nat. Commun. 2016, 7, 13195. [Google Scholar] [CrossRef] [Green Version]
- Gill, F.; Donsker, D.; Rasmussen, P. IOC World Bird List V12.1. Available online: https://www.worldbirdnames.org/ioc-lists/crossref/ (accessed on 4 February 2022).
- Fuchs, J.; Alström, P.; Yosef, R.; Olsson, U. Miocene Diversification of an Open-habitat Predatorial Passerine Radiation, the Shrikes (Aves: Passeriformes: Laniidae). Zool. Scr. 2019, 48, 571–588. [Google Scholar] [CrossRef]
- Kvist, L.; Giralt, D.; Valera, F.; Hoi, H.; Kristin, A.; Darchiashvili, G.; Lovaszi, P. Population Decline Is Accompanied by Loss of Genetic Diversity in the Lesser Grey Shrike Lanius Minor. Ibis 2011, 153, 98–109. [Google Scholar] [CrossRef]
- Gonzalez, J.; Wink, M.; Garcia-del-Rey, E.; Castro, G.D. Evidence from DNA Nucleotide Sequences and ISSR Profiles Indicates Paraphyly in Subspecies of the Southern Grey Shrike (Lanius meridionalis). J. Ornithol. 2008, 149, 495–506. [Google Scholar] [CrossRef]
- Padilla, D.P.; Spurgin, L.G.; Fairfield, E.A.; Illera, J.C.; Richardson, D.S. Population History, Gene Flow, and Bottlenecks in Island Populations of a Secondary Seed Disperser, the Southern Grey Shrike (Lanius meridionalis koenigi). Ecol. Evol. 2015, 5, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundy, N.I.; Winchell, C.S.; Woodruff, D.S. Genetic Differences between the Endangered San Clemente Island Loggerhead Shrike Lanius ludovicianus mearnsi and Two Neighbouring Subspecies Demonstrated by MtDNA Control Region and Cytochrome b Sequence Variation. Mol. Ecol. 1997, 6, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, I.C.; Ashley, M.V. Genetic Analysis of the Endemic Island Loggerhead Shrike, Lanius ludovicianus anthonyi. Conserv. Genet. 2011, 12, 1485–1493. [Google Scholar] [CrossRef]
- Panov, E.N. The True Shrikes (Laniidae) of the World: Ecology, Behavior and Evolution; Pensoft: Sofia, Bulgaria; Moscow, Russia, 2011. [Google Scholar]
- Harris, T.; Franklin, K. Shrikes and Bush-Shrikes: Including Wood-Shrikes, Helmet-Shrikes, Flycatcher-Shrikes, Philentomas, Batises and Wattle-Eyes; Helm: London, UK, 2000. [Google Scholar]
- Olsson, U.; Alström, P.; Svensson, L.; Aliabadian, M.; Sundberg, P. The Lanius excubitor (Aves, Passeriformes) Conundrum-Taxonomic Dilemma When Molecular and Non-Molecular Data Tell Different Stories. Mol. Phylogenet. Evol. 2010, 55, 347–357. [Google Scholar] [CrossRef]
- del Hoyo, J.; Elliott, A.; Christie, D.A. (Eds.) Handbook of the Birds of the World. Vol. 13. Penduline-Tits to Shrikes; Lynx Edicions: Barcelona, Spain, 2008. [Google Scholar]
- Cramp, S.; Perrins, C.M. (Eds.) The Birds of the Western Palearctic; Oxford University Press: Oxford, UK, 1993; Volume 7. [Google Scholar]
- Pârâu, L.G.; Frias-Soler, R.C.; Wink, M. High Genetic Diversity among Breeding Red-Backed Shrikes Lanius collurio in the Western Palearctic. Diversity 2019, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012; Volume 1. [Google Scholar]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. Computing 2021, 1, 12–21. [Google Scholar]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, E.M. Vcf2phylip v2.0: Convert a VCF Matrix into Several Matrix Formats for Phylogenetic Analysis. Available online: https://zenodo.org/record/2540861 (accessed on 21 January 2020).
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A. FigTree v1. 3.1. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 13 December 2019).
- Oyler-McCance, S.J.; Cornman, R.S.; Jones, K.L.; Fike, J.A. Z Chromosome Divergence, Polymorphism and Relative Effective Population Size in a Genus of Lekking Birds. Heredity 2015, 115, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Ellegren, H.; Smeds, L.; Burri, R.; Olason, P.I.; Backström, N.; Kawakami, T.; Künstner, A.; Mäkinen, H.; Nadachowska-Brzyska, K.; Qvarnström, A.; et al. The Genomic Landscape of Species Divergence in Ficedula Flycatchers. Nature 2012, 491, 756–760. [Google Scholar] [CrossRef]
- Kloskowski, J.; Trembaczowski, A.; Filipiuk, M. Stable Isotope Tracing of Links between Marine Wintering and Freshwater Breeding Habitats of Red-Necked Grebes. J. Ornithol. 2019, 160, 593–605. [Google Scholar] [CrossRef]
- Guillemain, M.; Bacon, L.; Kardynal, K.J.; Olivier, A.; Podhrazsky, M.; Musil, P.; Hobson, K.A. Geographic Origin of Migratory Birds Based on Stable Isotope Analysis: The Case of the Greylag Goose (Anser snser) Wintering in Camargue, Southern France. Eur. J. Wildl. Res. 2019, 65, 67. [Google Scholar] [CrossRef] [Green Version]
- Jiguet, F.; Kardynal, K.J.; Piha, M.; Seimola, T.; Copete, J.L.; Czajkowski, M.A.; Dombrovski, V.; Efrat, R.; Minkevicius, S.; Raković, M.; et al. Stable Isotopes Reveal the Common Winter Moult of Central Rectrices in a Long-Distance Migrant Songbird. J. Ornithol. 2019, 160, 1077–1085. [Google Scholar] [CrossRef]
- Franzoi, A.; Bontempo, L.; Kardynal, K.J.; Camin, F.; Pedrini, P.; Hobson, K.A. Natal Origins and Timing of Migration of Two Passerine Species through the Southern Alps: Inferences from Multiple Stable Isotopes (δ 2 H, δ 13 C, δ 15 N, δ 34 S) and Ringing Data. Ibis 2020, 162, 293–306. [Google Scholar] [CrossRef]
- Pârâu, L.G.; Wink, M. Common Patterns in the Molecular Phylogeography of Western Palearctic Birds: A Comprehensive Review. J. Ornithol. 2021, 162, 937–959. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Zhang, D.; Braun, M.S.; Hotz-Wagenblatt, A.; Pärt, T.; Arlt, D.; Schmaljohann, H.; Bairlein, F.; Lei, F.; Wink, M. Can Mitogenomes of the Northern Wheatear (Oenanthe oenanthe) Reconstruct Its Phylogeography and Reveal the Origin of Migrant Birds? Sci. Rep. 2020, 10, 9290. [Google Scholar] [CrossRef]
- Tryjanowski, P.; Goławski, A.; Kuźniak, S.; Mokwa, T.; Antczak, M. Disperse or Stay? Exceptionally High Breeding-Site Infidelity in the Red-Backed Shrike Lanius collurio. Ardea 2007, 95, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Ottenburghs, J.; Lavretsky, P.; Peters, J.L.; Kawakami, T.; Kraus, R.H.S. Population Genomics and Phylogeography. In Avian Genomics in Ecology and Evolution; Kraus, R.H.S., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 237–265. [Google Scholar]
- Foote, A.D.; Martin, M.D.; Louis, M.; Pacheco, G.; Robertson, K.M.; Sinding, M.-H.S.; Amaral, A.R.; Baird, R.W.; Baker, C.S.; Ballance, L.; et al. Killer Whale Genomes Reveal a Complex History of Recurrent Admixture and Vicariance. Mol. Ecol. 2019, 28, 3427–3444. [Google Scholar] [CrossRef] [PubMed]
- Skovrind, M.; Olsen, M.T.; Vieira, F.G.; Pacheco, G.; Carl, H.; Gilbert, M.T.P.; Møller, P.R. Genomic Population Structure of Freshwater-Resident and Anadromous Ide (Leuciscus idus) in North-Western Europe. Ecol. Evol. 2016, 6, 1064–1074. [Google Scholar] [CrossRef]
- Krebs, J.E.; Goldstein, E.S.; Kilpatrick, S.T. Lewin’s Genes XII; Jones & Bartlett Learning: Burlington, MA, USA, 2018. [Google Scholar]
- Kelleher, J.; Wong, Y.; Wohns, A.W.; Fadil, C.; Albers, P.K.; McVean, G. Inferring Whole-Genome Histories in Large Population Datasets. Nat. Genet. 2019, 51, 1330–1338. [Google Scholar] [CrossRef]
- Excoffier, L.; Heckel, G. Computer Programs for Population Genetics Data Analysis: A Survival Guide. Nat. Rev. Genet. 2006, 7, 745–758. [Google Scholar] [CrossRef]
- Stamatakis, A. Population and Evolutionary Genetic Inferences in the Whole-Genome Era: Software Challenges. In Population Genomics; Rajora, O.P., Ed.; Springer Nature: Cham, Switzerland, 2019; pp. 161–175. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Reads | Raw Bases | Clean Reads | Clean Bases | Read Length | Q30 Percent | |
|---|---|---|---|---|---|---|
| Average | 64,015,481 | 19,204,644,286 | 63,883,753 | 19,165,126,023 | 150;150 | 94.00;91.22 |
| Bulgaria | CzechRep | Germany | Israel | Kuwait | Latvia | Norway | Poland | Romania | Russia | Sweden | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bulgaria | |||||||||||
| CzechRep | 0.102 | ||||||||||
| Germany | 0.074 | 0.095 | |||||||||
| Israel | 0.093 | 0.113 | 0.085 | ||||||||
| Kuwait | 0.072 | 0.093 | 0.064 | 0.083 | |||||||
| Latvia | 0.082 | 0.103 | 0.074 | 0.093 | 0.072 | ||||||
| Norway | 0.064 | 0.085 | 0.056 | 0.075 | 0.054 | 0.064 | |||||
| Poland | 0.067 | 0.087 | 0.059 | 0.078 | 0.057 | 0.067 | 0.049 | ||||
| Romania | 0.067 | 0.087 | 0.059 | 0.077 | 0.057 | 0.067 | 0.048 | 0.052 | |||
| Russia | 0.077 | 0.097 | 0.069 | 0.087 | 0.067 | 0.077 | 0.059 | 0.062 | 0.061 | ||
| Sweden | 0.069 | 0.090 | 0.061 | 0.080 | 0.059 | 0.069 | 0.051 | 0.054 | 0.053 | 0.063 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pârâu, L.G.; Wang, E.; Wink, M. Red-Backed Shrike Lanius collurio Whole-Genome Sequencing Reveals Population Genetic Admixture. Diversity 2022, 14, 216. https://doi.org/10.3390/d14030216
Pârâu LG, Wang E, Wink M. Red-Backed Shrike Lanius collurio Whole-Genome Sequencing Reveals Population Genetic Admixture. Diversity. 2022; 14(3):216. https://doi.org/10.3390/d14030216
Chicago/Turabian StylePârâu, Liviu G., Erjia Wang, and Michael Wink. 2022. "Red-Backed Shrike Lanius collurio Whole-Genome Sequencing Reveals Population Genetic Admixture" Diversity 14, no. 3: 216. https://doi.org/10.3390/d14030216