Comparative Phylogenomics, a Stepping Stone for Bird Biodiversity Studies


Abstract
:1. Introduction
2. Current Status of Bird Genome Sequencing
2.1. Comparison of Currently Available Sequencing Strategies
2.2. Towards Complete Genomic Representation of Birds
3. Insight into Phylogeny from Whole Genome Data
3.1. Centrality of the Phylogeny for Comparative Studies
3.2. Challenges to Phylogenetic Resolution
3.2.1. Incongruence as a Sign of Data Problems
3.2.2. Phylogenetic Incongruence as a Pointer to Evolutionary Processes
3.3. Outlook
4. Genomic Evolution
4.1. Genomic Architecture of Birds
4.2. Understanding Adaptation by Integrating Genomic Data with Phenotypes
4.3. Outlook
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| B10K | Bird 10,000 Genome Project |
| VGP | Vertebrate Genome Project |
| ILS | Incomplete Lineage Sorting |
| bp | base pairs |
| kb | kilo base pairs |
| Mb | mega base pairs |
| Gb | giga base pairs |
References
- Brusatte, S.L.; O’Connor, J.K.; Jarvis, E.D. The Origin and Diversification of Birds. Curr. Biol. 2015, 25, R888–R898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, C.J.; Wenny, D.G.; Marquis, R.J. Ecosystem Services Provided by Birds. Ann. N. Y. Acad. Sci. 2008, 1134, 25–60. [Google Scholar] [CrossRef] [PubMed]
- Pereira, H.; Cooper, H.D. Towards the global monitoring of biodiversity change. Trends Ecol. Evol. 2006, 21, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Butchart, S.; Walpole, M.; Collen, B.; Van Strien, A.; Scharlemann, J.; Almond, R.; Baillie, J.; Bomhard, B.; Brown, C.; Bruno, J.; et al. Global biodiversity: Indicators of recent declines. Science 2010, 328, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- International Chicken Genome Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef] [PubMed]
- Dalloul, R.A.; Long, J.A.; Zimin, A.V.; Aslam, L.; Beal, K.; Blomberg, L.A.; Bouffard, P.; Burt, D.W.; Crasta, O.; Crooijmans, R.P.M.A.; et al. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): Genome assembly and analysis. PLoS Biol. 2010, 8, e1000475. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.; Clayton, D.; Ellegren, H.; Arnold, A.; Hillier, L.; Künstner, A.; Searle, S.; White, S.; Vilella, A.; Fairley, S.; et al. The genome of a songbird. Nature 2010, 464, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Jarvis, E.D.; Gilbert, M.T.P. A flock of genomes. Science 2014, 346, 1308–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G. Bird sequencing project takes off. Nature 2015, 522, 34. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.L.; Harshman, J.; Huddleston, C.J.; Kingston, S.; et al. Why Do Phylogenomic Data Sets Yield Conflicting Trees? Data Type Influences the Avian Tree of Life more than Taxon Sampling. Syst. Biol. 2017, 66, 857–879. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fan, W.; Tian, G.; Zhu, H.; He, L.; Cai, J.; Huang, Q.; Cai, Q.; Li, B.; Bai, Y.; et al. The sequence and de novo assembly of the giant panda genome. Nature 2009, 463, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2009, 20, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genome 10K Community of Scientists. Genome 10K: A Proposal to Obtain Whole-Genome Sequence for 10,000 Vertebrate Species. J. Hered. 2009, 100, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Koepfli, K.P.; Paten, B.; O’Brien, S.J. The Genome 10K Project: A Way Forward. Annu. Rev. Anim. Biosci. 2015, 3, 57–111. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Li, B.; Li, C.; Gilbert, M.T.P.; Jarvis, E.D.; Wang, J. Comparative genomic data of the Avian Phylogenomics Project. GigaScience 2014, 3. [Google Scholar] [CrossRef]
- Peona, V.; Weissensteiner, M.H.; Suh, A. How complete are “complete” genome assemblies?—An avian perspective. Mol. Ecol. Resour. 2018. [Google Scholar] [CrossRef]
- Burt, D.W. Origin and evolution of avian microchromosomes. Cytogenet. Genome Res. 2002, 96, 97–112. [Google Scholar] [CrossRef]
- Wang, O.; Chin, R.; Cheng, X.; Wu, M.K.Y.; Mao, Q.; Tang, J.; Sun, Y.; Anderson, E.; Lam, H.K.; Chen, D.; et al. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing haplotyping, and de novo assembly. Genome Res. 2019, 29, 798–808. [Google Scholar] [CrossRef] [PubMed]
- van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A Method to Study the Three-dimensional Architecture of Genomes. J. Vis. Exp. 2010, 39. [Google Scholar] [CrossRef]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, C.; Zhou, Q.; Zhang, G. Improving the ostrich genome assembly using optical mapping data. GigaScience 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018, 19, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, O.; Batra, S.; Omer, A.; Nyquist, S.; Hoeger, M.; Durand, N.; Shamim, M.; Machol, I.; Lander, E.; Aiden, A.; et al. De novo assembly of the Aedes Aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Golloshi, R.; Sanders, J.T.; McCord, R.P. Iteratively improving Hi-C experiments one step at a time. Methods 2018, 142, 47–58. [Google Scholar] [CrossRef]
- Ghurye, J.; Pop, M.; Koren, S.; Bickhart, D.; Chin, C.S. Scaffolding of long read assemblies using long range contact information. BMC Genom. 2017, 18. [Google Scholar] [CrossRef]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, E.C.; Bahr, N.; Dowsett, R.; Pearson, D.; Remsen, V.; Roselaar, C.; Schodde, D. The Howard and Moore complete checklist of birds of the world. In The Howard and Moore Complete Checklist of Birds of the World; A & C Black: London, UK, 2014. [Google Scholar]
- Brown, J.W.; Wang, N.; Smith, S.A. The development of scientific consensus: Analyzing conflict and concordance among avian phylogenies. Mol. Phylogenet. Evol. 2017, 116, 69–77. [Google Scholar] [CrossRef]
- Dunn, C.W.; Zapata, F.; Munro, C.; Siebert, S.; Hejnol, A. Pairwise comparisons across species are problematic when analyzing functional genomic data. Proc. Natl. Acad. Sci. USA 2018, 115, E409–E417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, A. The phylogenomic forest of bird trees contains a hard polytomy at the root of Neoaves. Zool. Scr. 2016, 45, 50–62. [Google Scholar] [CrossRef]
- Feduccia, A. Explosive evolution in Tertiary birds and animals. Science 1995, 267, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Feduccia, A. ‘Big bang’ for tertiary birds? Trends Ecol. Evol. 2003, 18, 172–176. [Google Scholar] [CrossRef]
- Poe, S.; Chubb, A.L. Birds in a bush: Five genes indicate explosive evolution of avian orders. Evolution 2004, 58, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Ksepka, D.T.; Stidham, T.A.; Williamson, T.E. Early Paleocene landbird supports rapid phylogenetic and morphological diversification of crown birds after the K–Pg mass extinction. Proc. Natl. Acad. Sci. USA 2017, 114, 8047–8052. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.B.; Lockhart, P.J. Deciphering ancient rapid radiations. Trends Ecol. Evol. 2007, 22, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.; Battistuzzi, F.; Lentino, M.; Aguilar, R.; Kumar, S.; Escalante, A. Evolution of modern birds revealed by mitogenomics: Timing the radiation and origin of major orders. Mol. Biol. Evol. 2011, 28, 1927–1942. [Google Scholar] [CrossRef] [PubMed]
- Hackett, S.J.; Kimball, R.T.; Reddy, S.; Bowie, R.C.K.; Braun, E.L.; Braun, M.J.; Chojnowski, J.L.; Cox, W.A.; Han, K.L.; Harshman, J.; et al. A Phylogenomic Study of Birds Reveals Their Evolutionary History. Science 2008, 320, 1763–1768. [Google Scholar] [CrossRef]
- McCormack, J.E.; Harvey, M.G.; Faircloth, B.C.; Crawford, N.G.; Glenn, T.C.; Brumfield, R.T. A Phylogeny of Birds Based on Over 1,500 Loci Collected by Target Enrichment and High-Throughput Sequencing. PLoS ONE 2013, 8, e54848. [Google Scholar] [CrossRef]
- Prum, R.; Berv, J.; Dornburg, A.; Field, D.; Townsend, J.; Lemmon, E.; Lemmon, A. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Bravo, G.; Antonelli, A.; Bacon, C.; Bartoszek, K.; Blom, M.; Huynh, S.; Jones, G.; Knowles, L.; Lamichhaney, S.; Marcussen, T.; et al. Embracing heterogeneity: Coalescing the Tree of Life and the future of phylogenomics. PeerJ 2019, 7, e6399. [Google Scholar] [CrossRef] [PubMed]
- King, N.; Rokas, A. Embracing Uncertainty in Reconstructing Early Animal Evolution. Curr. Biol. 2017, 27, R1081–R1088. [Google Scholar] [CrossRef] [PubMed]
- Mirarab, S.; Bayzid, M.S.; Boussau, B.; Warnow, T. Statistical binning enables an accurate coalescent-based estimation of the avian tree. Science 2014, 346, 1250463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayzid, M.S.; Mirarab, S.; Boussau, B.; Warnow, T. Weighted Statistical Binning: Enabling Statistically Consistent Genome-Scale Phylogenetic Analyses. PLoS ONE 2015, 10, e0129183. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Aberer, A.J.; Stamatakis, A. ExaML version 3: A tool for phylogenomic analyses on supercomputers. Bioinformatics 2015, 31, 2577–2579. [Google Scholar] [CrossRef]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef]
- Paten, B.; Earl, D.; Nguyen, N.; Diekhans, M.; Zerbino, D.; Haussler, D. Cactus: Algorithms for genome multiple sequence alignment. Genome Res. 2011, 21, 1512–1528. [Google Scholar] [CrossRef] [Green Version]
- Green, R.; Braun, E.; Armstrong, J.; Earl, D.; Nguyen, N.; Hickey, G.; Vandewege, M.; St, J.J.; Capella-Gutiérrez, S.; Castoe, T.; et al. Three crocodilian genomes reveal ancestral patterns of evolution among archosaurs. Science 2014, 346, 1254449. [Google Scholar] [CrossRef]
- Kronenberg, Z.N.; Fiddes, I.T.; Gordon, D.; Murali, S.; Cantsilieris, S.; Meyerson, O.S.; Underwood, J.G.; Nelson, B.J.; Chaisson, M.J.P.; Dougherty, M.L.; et al. High-resolution comparative analysis of great ape genomes. Science 2018, 360, eaar6343. [Google Scholar] [CrossRef]
- Bergsten, J. A review of long-branch attraction. Cladistics 2005, 21, 163–193. [Google Scholar] [CrossRef]
- Brinkmann, H.; van der Giezen, M.; Zhou, Y.; de Raucourt, G.P.; Philippe, H. An Empirical Assessment of Long-Branch Attraction Artefacts in Deep Eukaryotic Phylogenomics. Syst. Biol. 2005, 54, 743–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roch, S.; Nute, M.; Warnow, T. Long-Branch Attraction in Species Tree Estimation: Inconsistency of Partitioned Likelihood and Topology-Based Summary Methods. Syst. Biol. 2018, 68, 281–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollock, D.D.; Zwickl, D.J.; McGuire, J.A.; Hillis, D.M. Increased Taxon Sampling Is Advantageous for Phylogenetic Inference. Syst. Biol. 2002, 51, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwickl, D.J.; Hillis, D.M. Increased Taxon Sampling Greatly Reduces Phylogenetic Error. Syst. Biol. 2002, 51, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M.; Pollock, D.D.; McGuire, J.A.; Zwickl, D.J. Is Sparse Taxon Sampling a Problem for Phylogenetic Inference? Syst. Biol. 2003, 52, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Heath, T.A.; Zwickl, D.J.; Kim, J.; Hillis, D.M. Taxon Sampling Affects Inferences of Macroevolutionary Processes from Phylogenetic Trees. Syst. Biol. 2008, 57, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, A.R.; Sarkar, I.N. The impact of taxon sampling on phylogenetic inference: A review of two decades of controversy. Brief. Bioinform. 2011, 13, 122–134. [Google Scholar] [CrossRef]
- Betancur-R, R.; Arcila, D.; Vari, R.P.; Hughes, L.C.; Oliveira, C.; Sabaj, M.H.; Ortí, G. Phylogenomic incongruence, hypothesis testing, and taxonomic sampling: The monophyly of characiform fishes. Evolution 2019, 73, 329–345. [Google Scholar] [CrossRef]
- Tamashiro, R.A.; White, N.D.; Braun, M.J.; Faircloth, B.C.; Braun, E.L.; Kimball, R.T. What are the roles of taxon sampling and model fit in tests of cyto-nuclear discordance using avian mitogenomic data? Mol. Phylogenet. Evol. 2019, 130, 132–142. [Google Scholar] [CrossRef]
- Kelchner, S.A.; Thomas, M.A. Model use in phylogenetics: Nine key questions. Trends Ecol. Evol. 2007, 22, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Tarver, J.E.; dos Reis, M.; Mirarab, S.; Moran, R.J.; Parker, S.; O’Reilly, J.E.; King, B.L.; O’Connell, M.J.; Asher, R.J.; Warnow, T.; et al. The Interrelationships of Placental Mammals and the Limits of Phylogenetic Inference. Genome Biol. Evol. 2016, 8, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Philippe, H.; Delsuc, F.; Brinkmann, H.; Lartillot, N. Phylogenomics. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 541–562. [Google Scholar] [CrossRef]
- Rodríguez-Ezpeleta, N.; Brinkmann, H.; Roure, B.; Lartillot, N.; Lang, B.F.; Philippe, H. Detecting and Overcoming Systematic Errors in Genome-Scale Phylogenies. Syst. Biol. 2007, 56, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbiza, L.; Patricio, M.; Dopazo, H.; Posada, D. Genome-Wide Heterogeneity of Nucleotide Substitution Model Fit. Genome Biol. Evol. 2011, 3, 896–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naser-Khdour, S.; Minh, B.Q.; Zhang, W.; Stone, E.; Lanfear, R. The Prevalence and Impact of Model Violations in Phylogenetics Analysis. bioRxiv 2018. [Google Scholar] [CrossRef]
- Weber, C.C.; Boussau, B.; Romiguier, J.; Jarvis, E.D.; Ellegren, H. Evidence for GC-biased gene conversion as a driver of between-lineage differences in avian base composition. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian Mixture Model for Across-Site Heterogeneities in the Amino-Acid Replacement Process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef]
- Lartillot, N.; Brinkmann, H.; Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 2007, 7, S4. [Google Scholar] [CrossRef]
- Maddison, W.P. Gene Trees in Species Trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Degnan, J.H.; DeGiorgio, M.; Bryant, D.; Rosenberg, N.A. Properties of Consensus Methods for Inferring Species Trees from Gene Trees. Syst. Biol. 2009, 58, 35–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakhleh, L. Computational approaches to species phylogeny inference and gene tree reconciliation. Trends Ecol. Evol. 2013, 28, 719–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, C.; Kubatko, L.S. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: A model. Theor. Popul. Biol. 2009, 75, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Degnan, J.H.; Nakhleh, L. The Probability of a Gene Tree Topology within a Phylogenetic Network with Applications to Hybridization Detection. PLoS Genet. 2012, 8, e1002660. [Google Scholar] [CrossRef] [PubMed]
- Lamichhaney, S.; Han, F.; Webster, M.T.; Andersson, L.; Grant, B.R.; Grant, P.R. Rapid hybrid speciation in Darwin’s finches. Science 2017, 359, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Ottenburghs, J.; Ydenberg, R.C.; Hooft, P.V.; Wieren, S.E.V.; Prins, H.H. The Avian Hybrids Project: Gathering the scientific literature on avian hybridization. Ibis 2015, 157, 892–894. [Google Scholar] [CrossRef]
- Ottenburghs, J.; Kraus, R.H.S.; van Hooft, P.; van Wieren, S.E.; Ydenberg, R.C.; Prins, H.H.T. Avian introgression in the genomic era. Avian Res. 2017, 8. [Google Scholar] [CrossRef]
- Degnan, J.H.; Rosenberg, N.A. Discordance of Species Trees with Their Most Likely Gene Trees. PLoS Genet. 2006, 2, e68. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P.; Knowles, L.L. Inferring Phylogeny Despite Incomplete Lineage Sorting. Syst. Biol. 2006, 55, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Mailund, T.; Munch, K.; Schierup, M.H. Lineage Sorting in Apes. Annu. Rev. Genet. 2014, 48, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.C. Microevolutionary processes generate phylogenomic discordance at ancient divergences. Evolution 2013, 67, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Suh, A.; Smeds, L.; Ellegren, H. The Dynamics of Incomplete Lineage Sorting across the Ancient Adaptive Radiation of Neoavian Birds. PLoS Biol. 2015, 13, e1002224. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, A.; Sackton, T.B.; Grayson, P.; Clamp, M.; Baker, A.J.; Edwards, S.V. Whole-Genome Analyses Resolve the Phylogeny of Flightless Birds (Palaeognathae) in the Presence of an Empirical Anomaly Zone. Syst. Biol. 2019. [Google Scholar] [CrossRef]
- Rokas, A.; Carroll, S.B. Bushes in the Tree of Life. PLoS Biol. 2006, 4, e352. [Google Scholar] [CrossRef] [PubMed]
- Sayyari, E.; Mirarab, S. Testing for Polytomies in Phylogenetic Species Trees Using Quartet Frequencies. Genes 2018, 9, 132. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Kimball, R.T.; Braun, E.L. Introns outperform exons in analyses of basal avian phylogeny using clathrin heavy chain genes. Gene 2008, 410, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Suh, A.; Paus, M.; Kiefmann, M.; Churakov, G.; Franke, F.A.; Brosius, J.; Kriegs, J.O.; Schmitz, J. Mesozoic retroposons reveal parrots as the closest living relatives of passerine birds. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef]
- Edwards, S.V.; Cloutier, A.; Baker, A.J. Conserved Nonexonic Elements: A Novel Class of Marker for Phylogenomics. Syst. Biol. 2017, 66, 1028–1044. [Google Scholar] [CrossRef]
- Edwards, S.V. Natural selection and phylogenetic analysis. Proc. Natl. Acad. Sci. USA 2009, 106, 8799–8800. [Google Scholar] [CrossRef] [Green Version]
- Claramunt, S.; Cracraft, J. A new time tree reveals Earth history’s imprint on the evolution of modern birds. Sci. Adv. 2015, 1, e1501005. [Google Scholar] [CrossRef]
- Moyle, R.G.; Oliveros, C.H.; Andersen, M.J.; Hosner, P.A.; Benz, B.W.; Manthey, J.D.; Travers, S.L.; Brown, R.M.; Faircloth, B.C. Tectonic collision and uplift of Wallacea triggered the global songbird radiation. Nat. Commun. 2016, 7, 12709. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, C.H.; Field, D.J.; Ksepka, D.T.; Barker, F.K.; Aleixo, A.; Andersen, M.J.; Alström, P.; Benz, B.W.; Braun, E.L.; Braun, M.J.; et al. Earth history and the passerine superradiation. Proc. Natl. Acad. Sci. USA 2019, 116, 7916–7925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Guang, A.; Zapata, F.; Howison, M.; Lawrence, C.E.; Dunn, C.W. An Integrated Perspective on Phylogenetic Workflows. Trends Ecol. Evol. 2016, 31, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Kozlov, A.M.; Stamatakis, A. ParGenes: A tool for massively parallel model selection and phylogenetic tree inference on thousands of genes. Bioinformatics 2018, 35, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Elworth, R.A.L.; Ogilvie, H.A.; Zhu, J.; Nakhleh, L. Advances in Computational Methods for Phylogenetic Networks in the Presence of Hybridization. In Bioinformatics and Phylogenetics; Springer Nature Switzerland: Cham, Switzerland, 2019; pp. 317–360. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.Y.; Liang, D.; Zhang, P. Selecting Question-Specific Genes to Reduce Incongruence in Phylogenomics: A Case Study of Jawed Vertebrate Backbone Phylogeny. Syst. Biol. 2015, 64, 1104–1120. [Google Scholar] [CrossRef] [Green Version]
- Botero-Castro, F.; Figuet, E.; Tilak, M.K.; Nabholz, B.; Galtier, N. Avian Genomes Revisited: Hidden Genes Uncovered and the Rates versus Traits Paradox in Birds. Mol. Biol. Evol. 2017, 34, 3123–3131. [Google Scholar] [CrossRef]
- Gregory, T.; Nicol, J.; Tamm, H.; Kullman, B.; Kullman, K.; Leitch, I.; Murray, B.; Kapraun, D.; Greilhuber, J.; Bennett, M. Eukaryotic genome size databases. Nucleic Acids Res. 2007, 35, D332–D338. [Google Scholar] [CrossRef]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapusta, A.; Suh, A. Evolution of bird genomes-a transposon’s-eye view. Ann. N. Y. Acad. Sci. 2016, 1389, 164–185. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R. A birds’s-eye view of the C-value enigma: Genome size, cell size, and metabolic rate in the class Aves. Evolution 2002, 56, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R.; Andrews, C.B.; McGuire, J.A.; Witt, C.C. The smallest avian genomes are found in hummingbirds. Proc. R. Soc. B Biol. Sci. 2009, 276, 3753–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and Diversity of Transposable Elements in Vertebrate Genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeds, L.; Warmuth, V.; Bolivar, P.; Uebbing, S.; Burri, R.; Suh, A.; Nater, A.; Bureš, S.; Garamszegi, L.Z.; Hogner, S.; et al. Evolutionary analysis of the female-specific avian W chromosome. Nat. Commun. 2015, 6, 7330. [Google Scholar] [CrossRef] [Green Version]
- Ellegren, H. Evolutionary stasis: The stable chromosomes of birds. Trends Ecol. Evol. 2010, 25, 283–291. [Google Scholar] [CrossRef]
- O’Connor, R.E.; Kiazim, L.; Skinner, B.; Fonseka, G.; Joseph, S.; Jennings, R.; Larkin, D.M.; Griffin, D.K. Patterns of microchromosome organization remain highly conserved throughout avian evolution. Chromosoma 2018, 128, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G. The bird’s-eye view on chromosome evolution. Genome Biol. 2018, 19. [Google Scholar] [CrossRef]
- Kretschmer, R.; Ferguson-Smith, M.; de Oliveira, E. Karyotype Evolution in Birds: From Conventional Staining to Chromosome Painting. Genes 2018, 9, 181. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Yang, W.; An, N.; Zhang, P.; Zhang, G.; Zhou, Q. Temporal genomic evolution of bird sex chromosomes. BMC Evol. Biol. 2014, 14, 250. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, J.; Bachtrog, D.; An, N.; Huang, Q.; Jarvis, E.D.; Gilbert, M.T.P.; Zhang, G. Complex evolutionary trajectories of sex chromosomes across bird taxa. Science 2014, 346, 1246338. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Auer, G.; Peona, V.; Suh, A.; Deng, Y.; Feng, S.; Zhang, G.; Blom, M.P.; Christidis, L.; Prost, S.; et al. Dynamic evolutionary history and gene content of sex chromosomes across diverse songbirds. Nat. Ecol. Evol. 2019, 3, 834. [Google Scholar] [CrossRef] [PubMed]
- Pala, I.; Naurin, S.; Stervander, M.; Hasselquist, D.; Bensch, S.; Hansson, B. Evidence of a neo-sex chromosome in birds. Heredity 2011, 108, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meredith, R.W.; Zhang, G.; Gilbert, M.T.P.; Jarvis, E.D.; Springer, M.S. Evidence for a single loss of mineralized teeth in the common avian ancestor. Science 2014, 346, 1254390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louchart, A.; Viriot, L. From snout to beak: The loss of teeth in birds. Trends Ecol. Evol. 2011, 26, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Lamichhaney, S.; Berglund, J.; Almén, M.S.; Maqbool, K.; Grabherr, M.; Martinez-Barrio, A.; Promerová, M.; Rubin, C.J.; Wang, C.; Zamani, N.; et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 2015, 518, 371. [Google Scholar] [CrossRef] [PubMed]
- Lamichhaney, S.; Han, F.; Berglund, J.; Wang, C.; Almén, M.; Webster, M.; Grant, B.; Grant, P.; Andersson, L. A beak size locus in Darwin’s finches facilitated character displacement during a drought. Science 2016, 352, 470–474. [Google Scholar] [CrossRef]
- Ng, C.S.; Li, W.H. Genetic and Molecular Basis of Feather Diversity in Birds. Genome Biol. Evol. 2018, 10, 2572–2586. [Google Scholar] [CrossRef]
- Greenwold, M.J.; Bao, W.; Jarvis, E.D.; Hu, H.; Li, C.; Gilbert, M.T.P.; Zhang, G.; Sawyer, R.H. Dynamic evolution of the alpha and beta keratins has accompanied integument diversification and the adaptation of birds into novel lifestyles. BMC Evol. Biol. 2014, 14. [Google Scholar] [CrossRef]
- Borges, R.; Khan, I.; Johnson, W.; Gilbert, M.; Zhang, G.; Jarvis, E.; O’Brien, S.; Antunes, A. Gene loss, adaptive evolution and the co-evolution of plumage coloration genes with opsins in birds. BMC Genom. 2015, 16, 751. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, Y.; Li, J.; Kong, L.; Hu, H.; Pan, H.; Xu, L.; Deng, Y.; Li, Q.; Jin, L.; et al. Two Antarctic penguin genomes reveal insights into their evolutionary history and molecular changes related to the Antarctic environment. GigaScience 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Le Duc, D.; Renaud, G.; Krishnan, A.; Almén, M.S.; Huynen, L.; Prohaska, S.J.; Ongyerth, M.; Bitarello, B.D.; Schiöth, H.B.; Hofreiter, M.; et al. Kiwi genome provides insights into evolution of a nocturnal lifestyle. Genome Biol. 2015, 16, 147. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, M.D.; Kronenberg, Z.; Li, C.; Domyan, E.T.; Pan, H.; Campbell, M.; Tan, H.; Huff, C.D.; Hu, H.; Vickrey, A.I.; et al. Genomic Diversity and Evolution of the Head Crest in the Rock Pigeon. Science 2013, 339, 1063–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfenning, A.; Hara, E.; Whitney, O.; Rivas, M.; Wang, R.; Roulhac, P.; Howard, J.; Wirthlin, M.; Lovell, P.; Ganapathy, G.; et al. Convergent transcriptional specializations in the brains of humans and song-learning birds. Science 2014, 346, 1256846. [Google Scholar] [CrossRef] [PubMed]
- Burga, A.; Wang, W.; Ben-David, E.; Wolf, P.C.; Ramey, A.M.; Verdugo, C.; Lyons, K.; Parker, P.G.; Kruglyak, L. A genetic signature of the evolution of loss of flight in the Galapagos cormorant. Science 2017, 356, eaal3345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackton, T.B.; Grayson, P.; Cloutier, A.; Hu, Z.; Liu, J.S.; Wheeler, N.E.; Gardner, P.P.; Clarke, J.A.; Baker, A.J.; Clamp, M.; et al. Convergent regulatory evolution and loss of flight in paleognathous birds. Science 2019, 364, 74–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, T.F.; Fischer, C.R.; Wu, P.; Jiang, T.X.; Xie, K.T.; Kuo, J.; Doctorov, E.; Zehnder, A.; Khosla, C.; Chuong, C.M.; et al. Genetic Mapping and Biochemical Basis of Yellow Feather Pigmentation in Budgerigars. Cell 2017, 171, 427–439.e21. [Google Scholar] [CrossRef] [Green Version]
- Projecto-Garcia, J.; Natarajan, C.; Moriyama, H.; Weber, R.E.; Fago, A.; Cheviron, Z.A.; Dudley, R.; McGuire, J.A.; Witt, C.C.; Storz, J.F. Repeated elevational transitions in hemoglobin function during the evolution of Andean hummingbirds. Proc. Natl. Acad. Sci. USA 2013, 110, 20669–20674. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, C.; Hoffmann, F.G.; Weber, R.E.; Fago, A.; Witt, C.C.; Storz, J.F. Predictable convergence in hemoglobin function has unpredictable molecular underpinnings. Science 2016, 354, 336–339. [Google Scholar] [CrossRef] [Green Version]
- Eöry, L.; Gilbert, M.T.P.; Li, C.; Li, B.; Archibald, A.; Aken, B.L.; Zhang, G.; Jarvis, E.; Flicek, P.; Burt, D.W. Avianbase: A community resource for bird genomics. Genome Biol. 2015, 16, 21. [Google Scholar] [CrossRef] [PubMed]




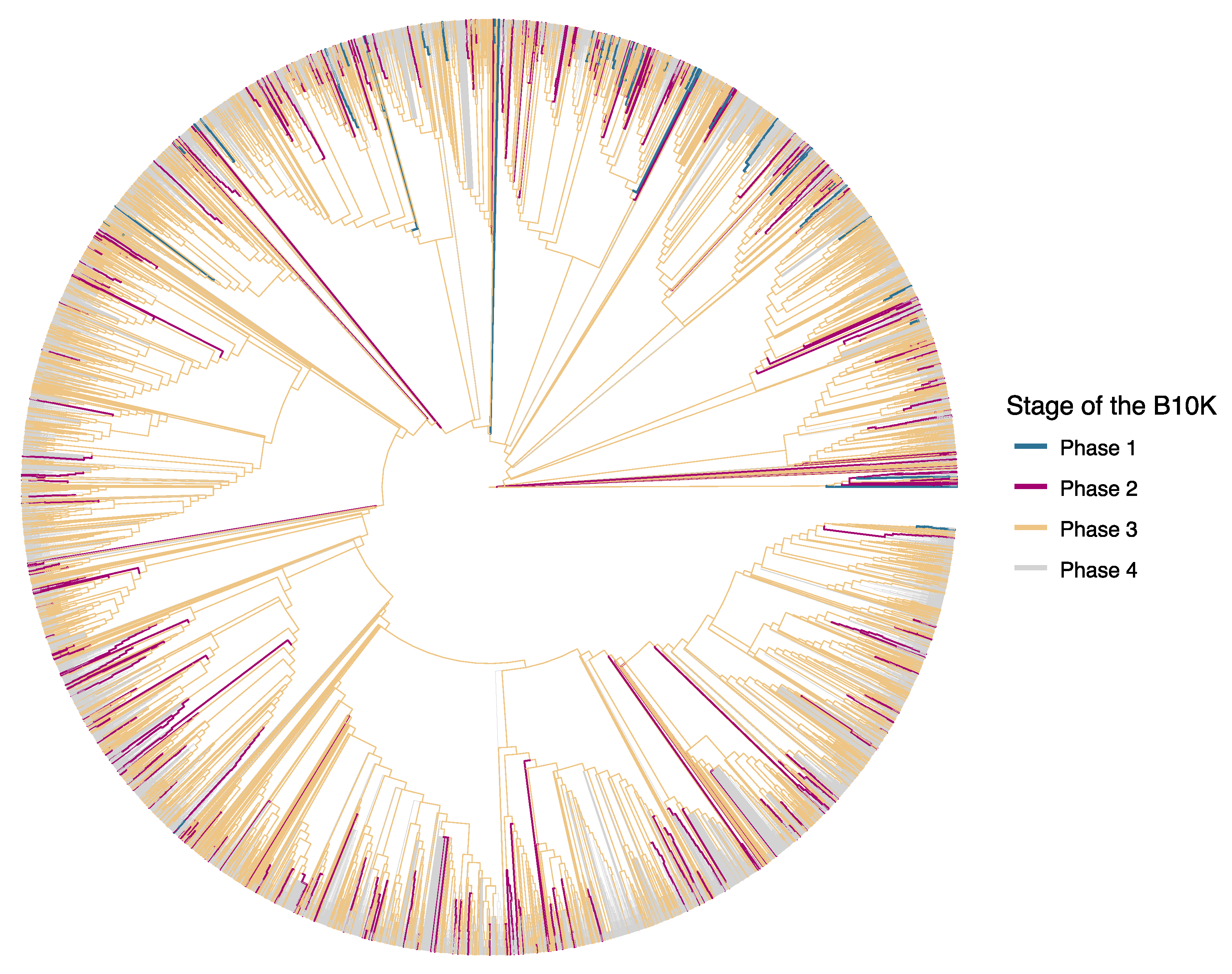{kind=link}
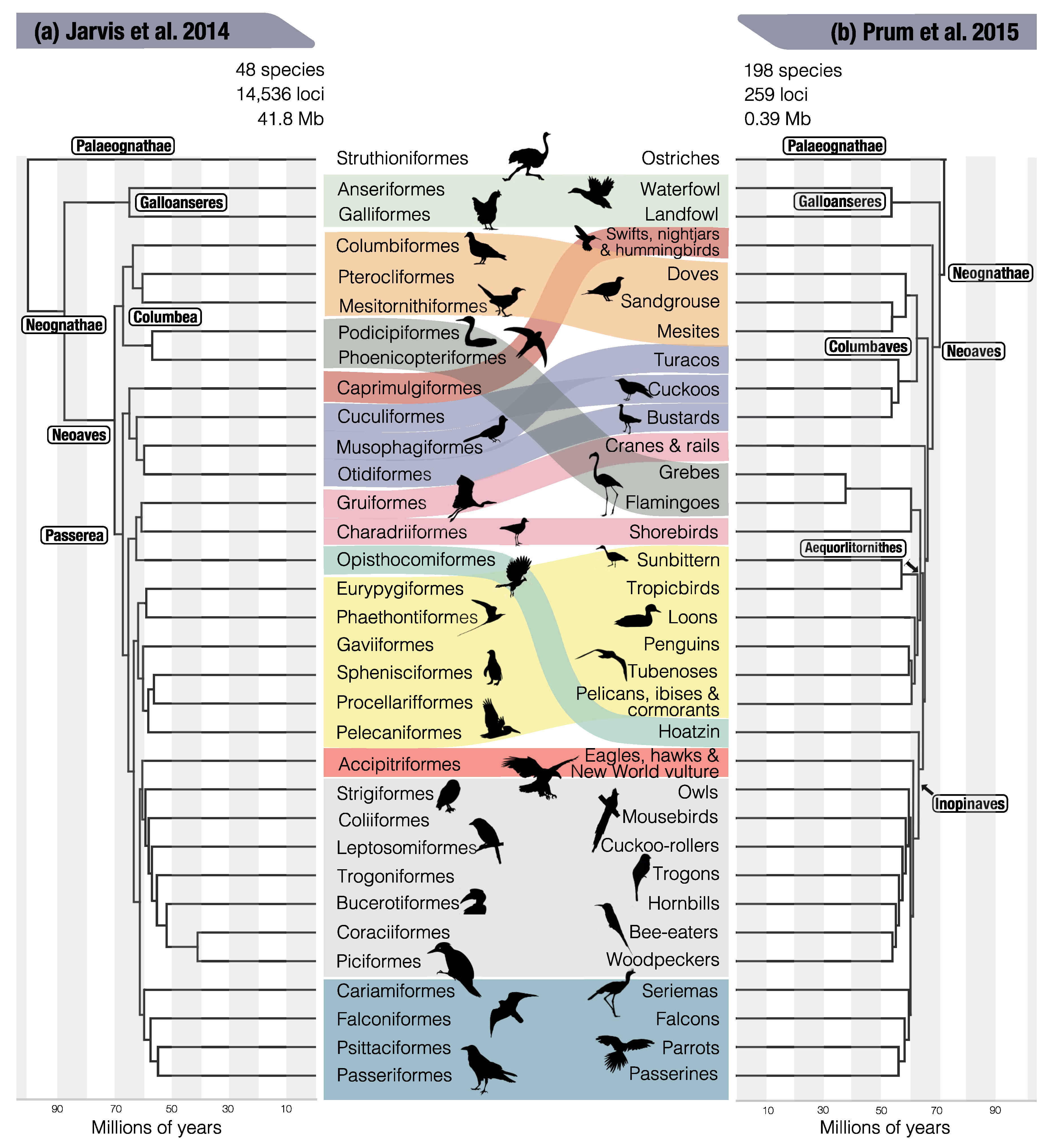{kind=link}
{kind=link}
| Short Read Sequencing | Mixed Sequencing | ||||||
|---|---|---|---|---|---|---|---|
| Assembly strategy | 1 WGS+1 MatePair library | Gradient insert size libraries (200 bp–20 kb) | 10X Genomics | stLFR | 1 WGS+Hi-C libraries | VGP strategy (PacBio+10X+Bionano+Hi-C) | |
| Input DNA | Quantity | ≥10 g | ≥125 g | ≥300 ng | ≥300 ng | ≥2 g DNA + 0.2 g tissue | ≥10 g DNA + 0.2 g tissue |
| Quality | >2 kb | >20 kb | >40 kb | >40 kb | >2 kb | >250 kb | |
| Sequencing coverage | 30–60× | 100× | 60–100× | 60–100× | 200× | 400× | |
| Assembly quality | Contig N50 | 10–40 kb | 20–80 kb | 100 kb | 100 kb | 10–40 kb | 10 Mb |
| Scaffold N50 | 30–800 kb | 0.2–10 Mb | 10 Mb | 10 Mb | 100 Mb (chr-level) | 100 Mb (chr-level) | |
| Complete-ness (e.g., BUSCO) | 90–95% | 93–98% | 93–98% | 93–98% | 90–95% | 95–99% | |
| Sequencing cost | $2200 | $7000 | $2000 | $1000 | $3000 | $20,000 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiller, J.; Zhang, G. Comparative Phylogenomics, a Stepping Stone for Bird Biodiversity Studies. Diversity 2019, 11, 115. https://doi.org/10.3390/d11070115
Stiller J, Zhang G. Comparative Phylogenomics, a Stepping Stone for Bird Biodiversity Studies. Diversity. 2019; 11(7):115. https://doi.org/10.3390/d11070115
Chicago/Turabian StyleStiller, Josefin, and Guojie Zhang. 2019. "Comparative Phylogenomics, a Stepping Stone for Bird Biodiversity Studies" Diversity 11, no. 7: 115. https://doi.org/10.3390/d11070115