

Modulating the Activity of the Human Organic Cation Transporter 2 Emerges as a Potential Strategy to Mitigate Unwanted Toxicities Associated with Cisplatin Chemotherapy
 , , and
, , and 
Abstract
:1. Introduction
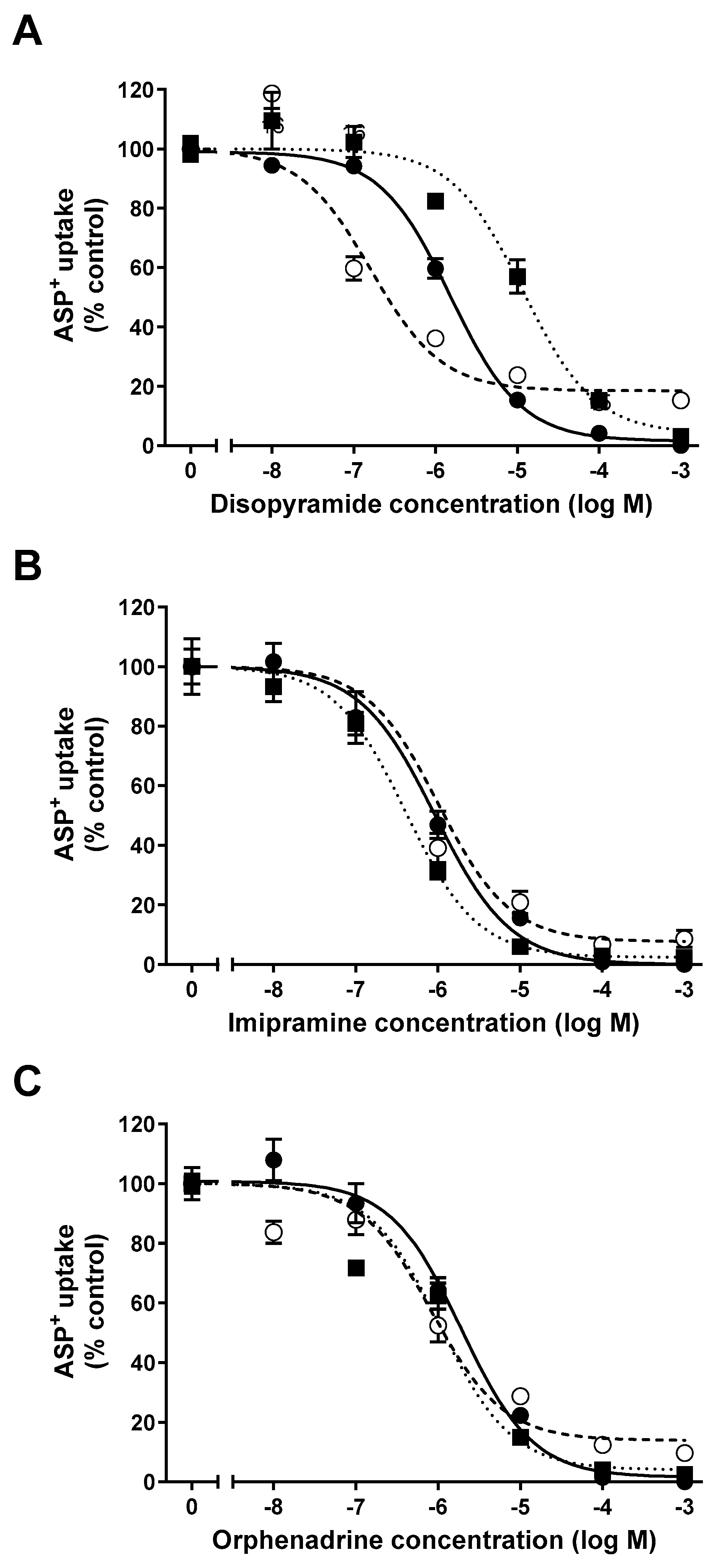
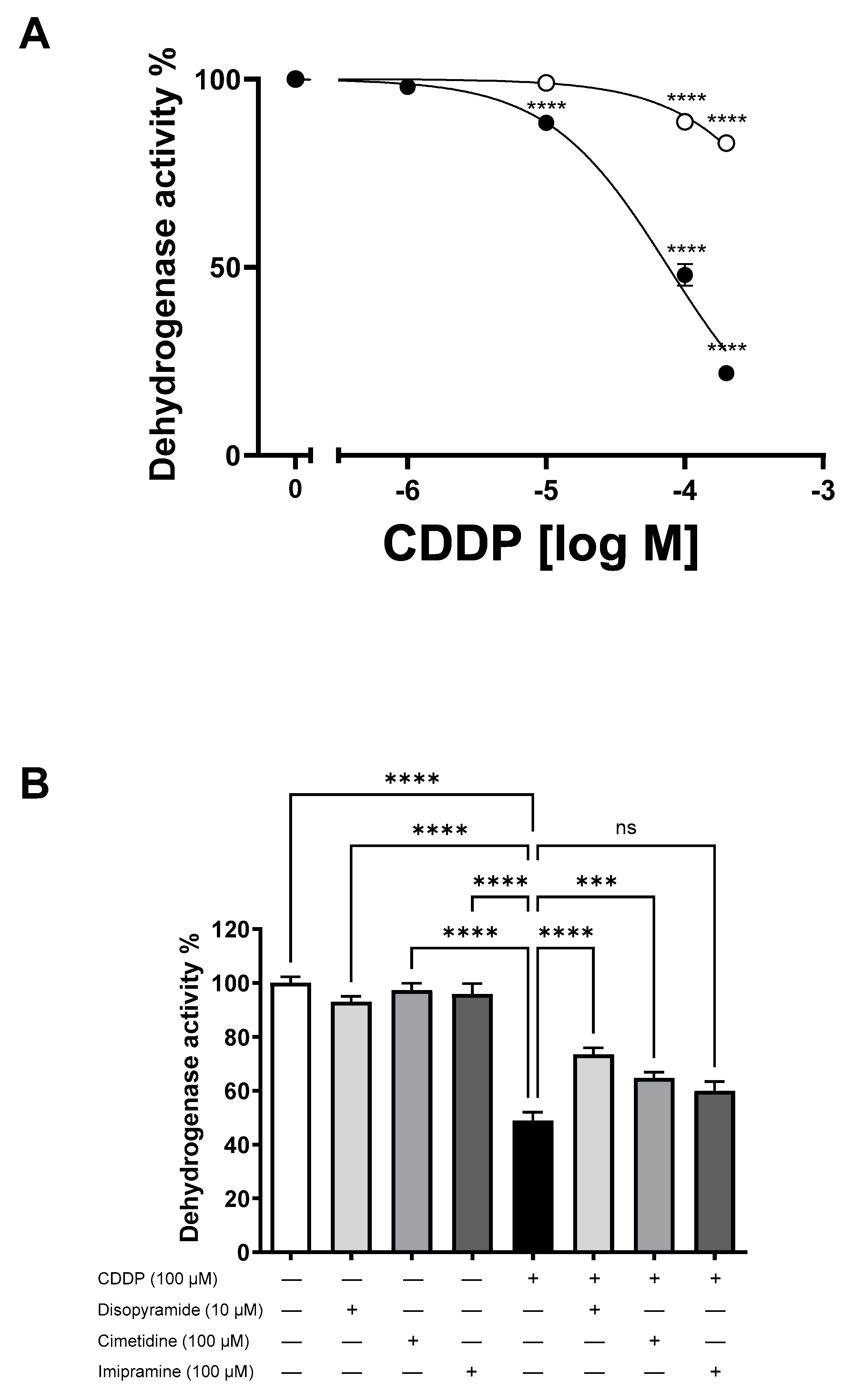
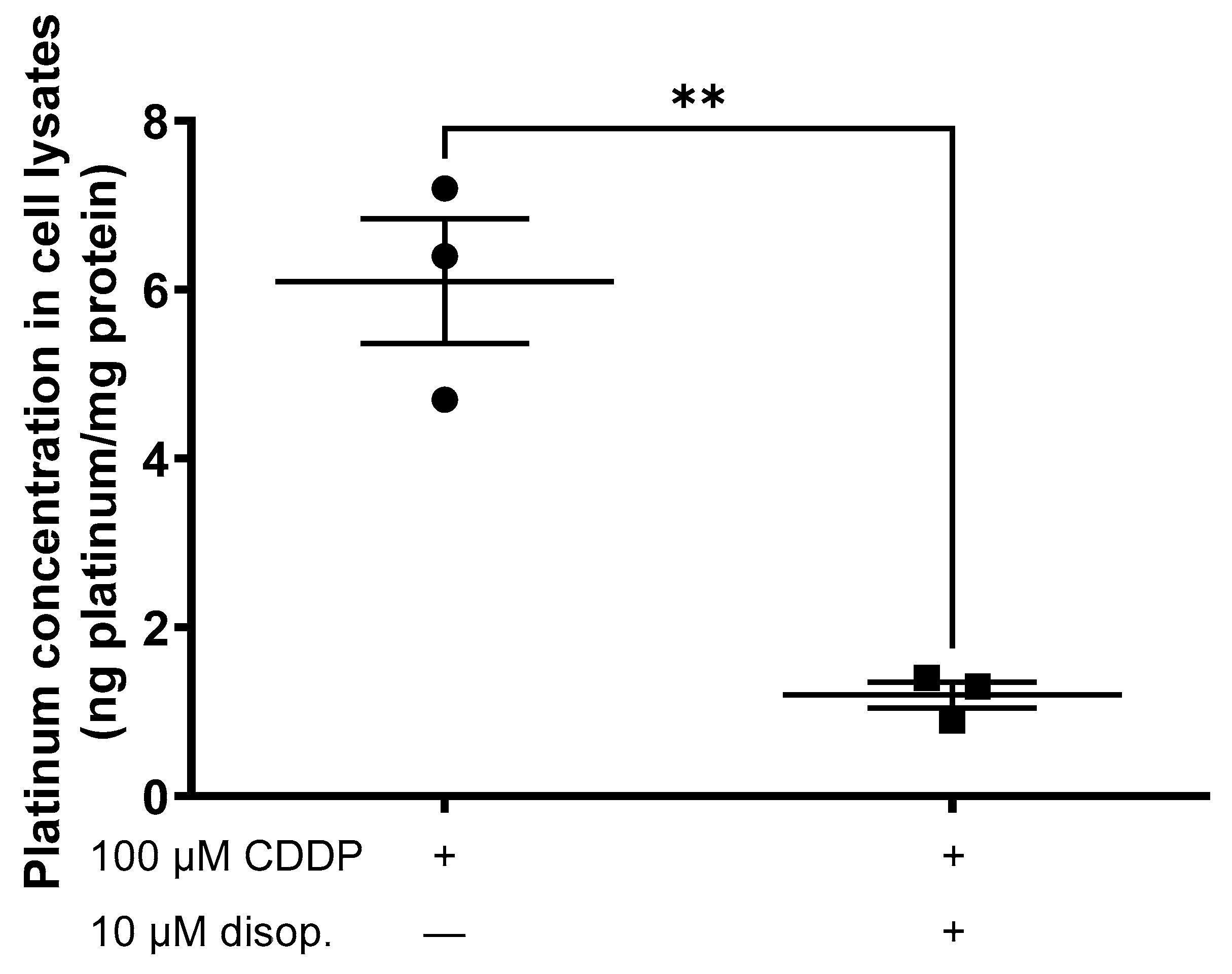
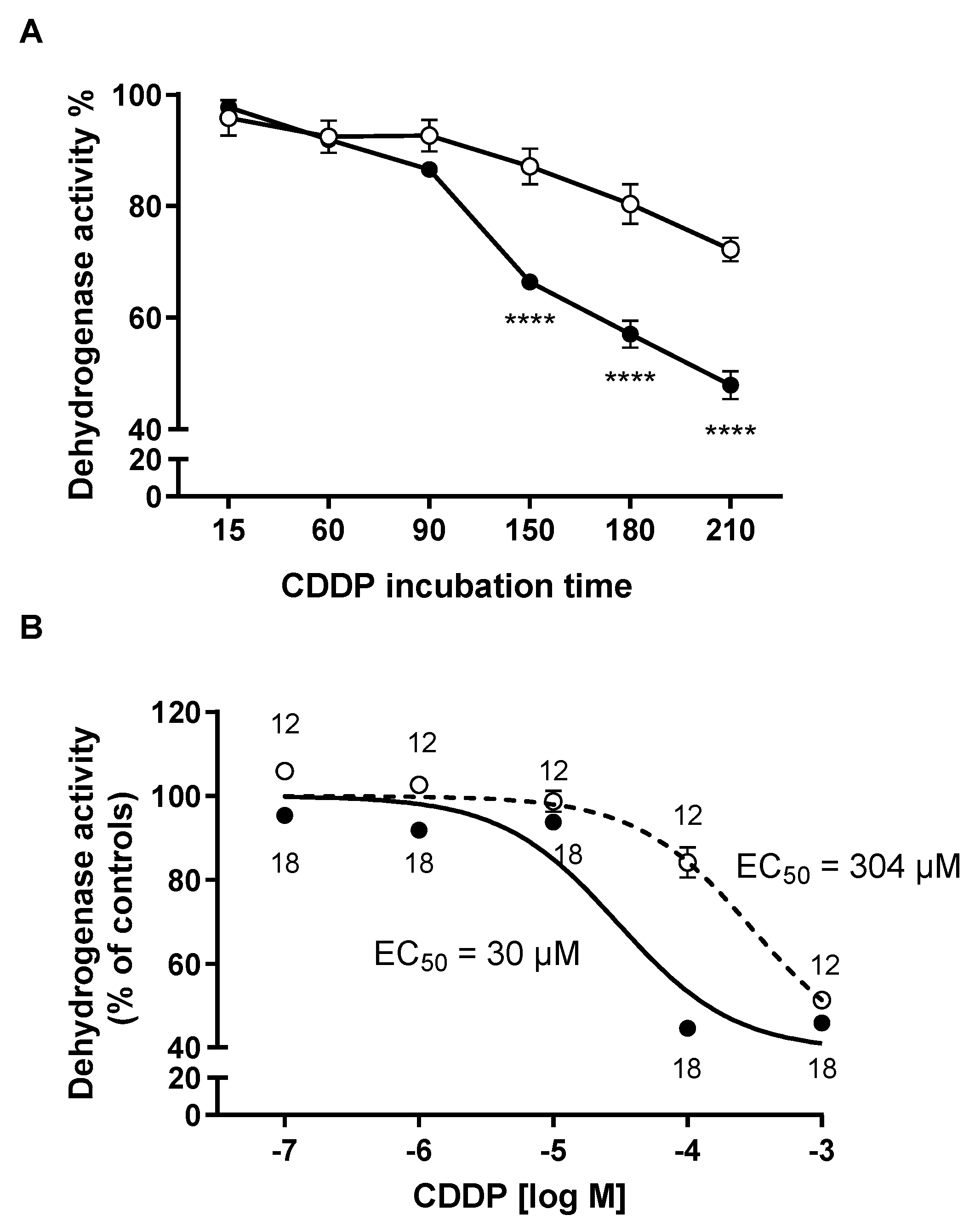
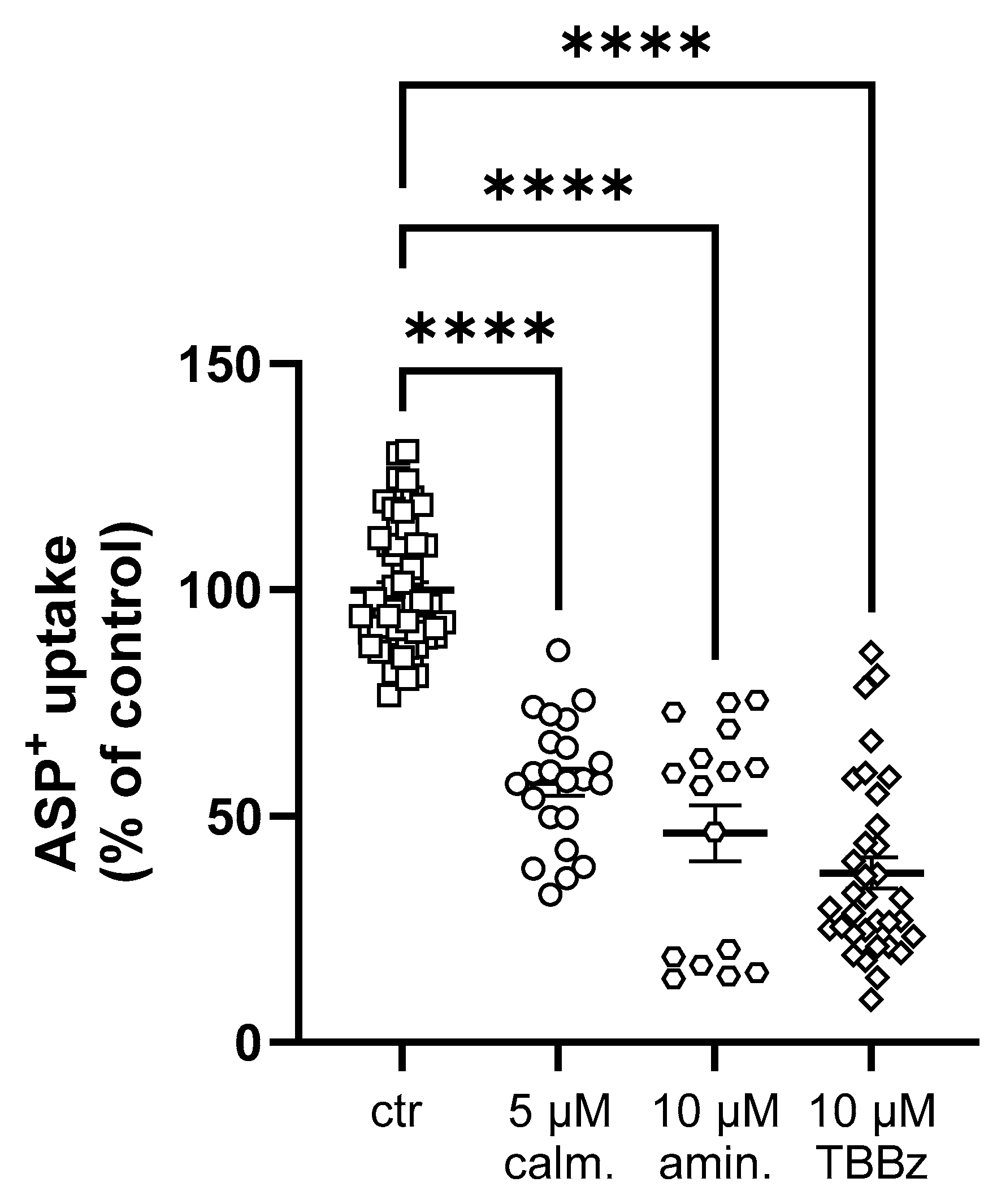
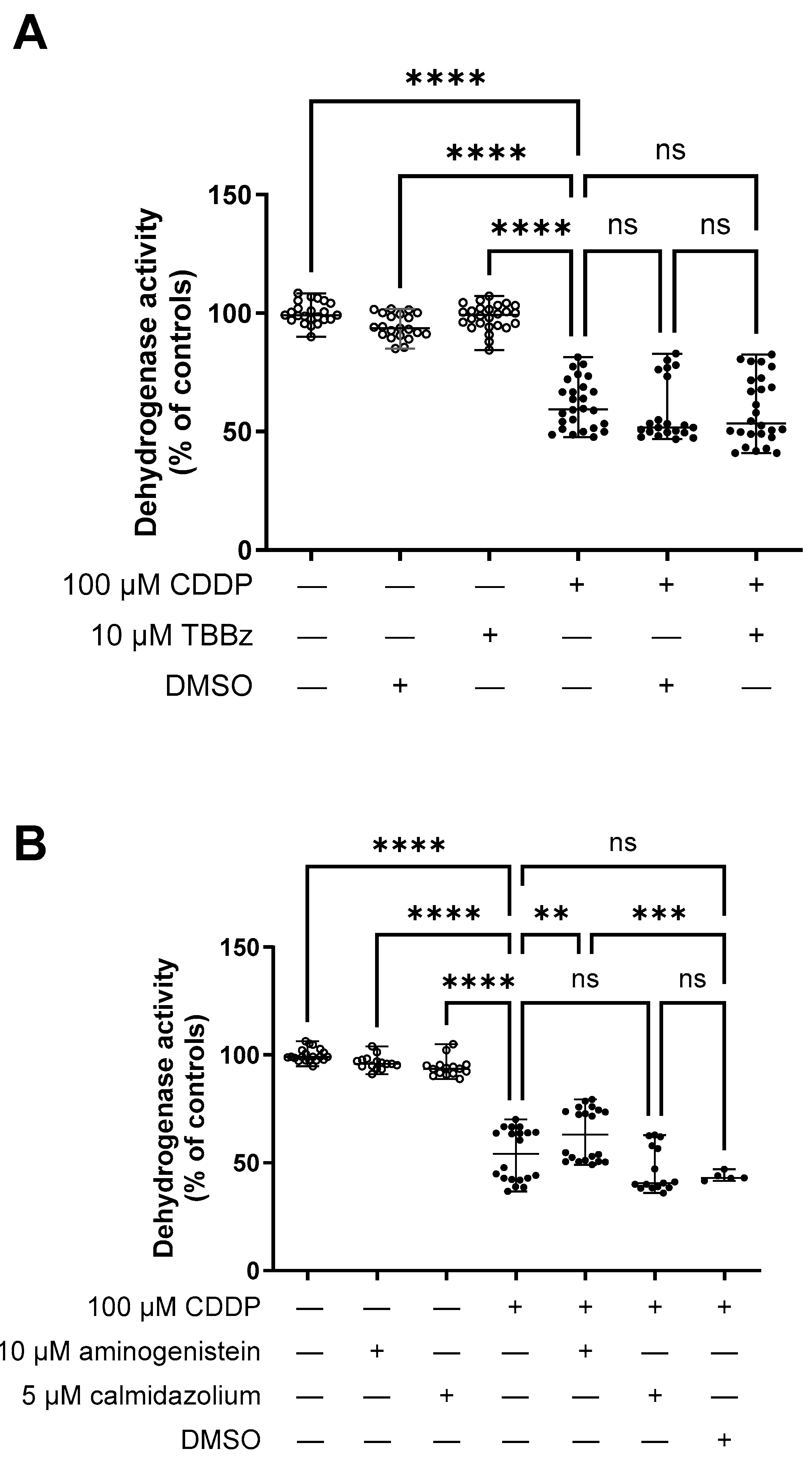
2. Results
Interaction of Selected Drugs with OCT Transport
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Evaluation of OCT Function
4.3. CDDP Toxicity Test
4.4. Measurement of Intracellular CDDP Amount
4.5. Chemicals
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bai, Y.; Aodeng, G.; Ga, L.; Hai, W.; Ai, J. Research Progress of Metal Anticancer Drugs. Pharmaceutics 2023, 15, 2750. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Ahmad, S. Kinetic Aspects of Platinum Anticancer Agents. Polyhedron 2017, 138, 109–124. [Google Scholar] [CrossRef]
- Ratanaphan, A. A DNA Repair Protein BRCA1 as a Potentially Molecular Target for the Anticancer Platinum Drug Cisplatin. In DNA Repair; Kruman, I., Ed.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Minerva; Bhat, A.; Verma, S.; Chander, G.; Jamwal, R.S.; Sharma, B.; Bhat, A.; Katyal, T.; Kumar, R.; Shah, R. Cisplatin-Based Combination Therapy for Cancer. J. Cancer Res. Ther. 2023, 19, 530–536. [Google Scholar] [CrossRef]
- Ruano, L.; Cárdenas, G.; Nogueira, J.J. The Permeation Mechanism of Cisplatin Through a Dioleoylphosphocholine Bilayer. Chem. Phys. Chem. 2021, 22, 1251–1261. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the Anticancer Drug Cisplatin Mediated by the Copper Transporter Ctr1 in Yeast and Mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstädt, H.; Koepsell, H.; Piechota, H.-J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin Nephrotoxicity Is Critically Mediated via the Human Organic Cation Transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Tang, X.; Peterson, D.R.; Kilari, D.; Chow, C.-W.; Fujimoto, J.; Kalhor, N.; Swisher, S.G.; Stewart, D.J.; Wistuba, I.I.; et al. Copper Transporter CTR1 Expression and Tissue Platinum Concentration in Non-Small Cell Lung Cancer. Lung Cancer 2014, 85, 88–93. [Google Scholar] [CrossRef]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstädt, H.; Lanvers-Kaminsky, C.; am Zehnhoff-Dinnesen, A.; Schinkel, A.H.; et al. Organic Cation Transporter 2 Mediates Cisplatin-Induced Oto- and Nephrotoxicity and Is a Target for Protective Interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Masuda, S.; Nishihara, K.; Yano, I.; Katsura, T.; Inui, K. Association between Tubular Toxicity of Cisplatin and Expression of Organic Cation Transporter ROCT2 (Slc22a2) in the Rat. Biochem. Pharmacol. 2005, 70, 1823–1831. [Google Scholar] [CrossRef]
- Sprowl, J.A.; Lancaster, C.S.; Pabla, N.; Hermann, E.; Kosloske, A.M.; Gibson, A.A.; Li, L.; Zeeh, D.; Schlatter, E.; Janke, L.J.; et al. Cisplatin-Induced Renal Injury Is Independently Mediated by OCT2 and P53. Clin. Cancer Res. 2014, 20, 4026–4035. [Google Scholar] [CrossRef]
- Sprowl, J.A.; van Doorn, L.; Hu, S.; van Gerven, L.; de Bruijn, P.; Li, L.; Gibson, A.A.; Mathijssen, R.H.; Sparreboom, A. Conjunctive Therapy of Cisplatin with the OCT2 Inhibitor Cimetidine: Influence on Antitumor Efficacy and Systemic Clearance. Clin. Pharmacol. Ther. 2013, 94, 585–592. [Google Scholar] [CrossRef]
- Basit, A.; Radi, Z.; Vaidya, V.S.; Karasu, M.; Prasad, B. Kidney Cortical Transporter Expression across Species Using Quantitative Proteomics. Drug Metab. Dispos. 2019, 47, 802–808. [Google Scholar] [CrossRef]
- Zhou, S.; Zeng, S.; Shu, Y. Drug-Drug Interactions at Organic Cation Transporter 1. Front. Pharmacol. 2021, 12, 628705. [Google Scholar] [CrossRef]
- Bönisch, H. Substrates and Inhibitors of Organic Cation Transporters (OCTs) and Plasma Membrane Monoamine Transporter (PMAT) and Therapeutic Implications. Handb. Exp. Pharmacol. 2021, 266, 119–167. [Google Scholar]
- Hanada, K.; Odaka, K.; Kudo, A.; Ogata, H. Effects of Disopyramide and Verapamil on Renal Disposition and Nephrotoxicity of Cisplatin in Rats. Pharm. Res. 1999, 16, 1589–1595. [Google Scholar] [CrossRef]
- Jahchan, N.S.; Dudley, J.T.; Mazur, P.K.; Flores, N.; Yang, D.; Palmerton, A.; Zmoos, A.-F.; Vaka, D.; Tran, K.Q.T.; Zhou, M.; et al. A Drug Repositioning Approach Identifies Tricyclic Antidepressants as Inhibitors of Small Cell Lung Cancer and Other Neuroendocrine Tumors. Cancer Discov. 2013, 3, 1364–1377. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Nakao, Y.; Masuda, S.; Katsura, T.; Kamba, T.; Ogawa, O.; Inui, K.-I. Precise Comparison of Protein Localization among OCT, OAT, and MATE in Human Kidney. J. Pharm. Sci. 2013, 102, 3302–3308. [Google Scholar] [CrossRef] [PubMed]
- Caceres, P.S.; Gravotta, D.; Zager, P.J.; Dephoure, N.; Rodriguez-Boulan, E. Quantitative Proteomics of MDCK Cells Identify Unrecognized Roles of Clathrin Adaptor AP-1 in Polarized Distribution of Surface Proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 11796–11805. [Google Scholar] [CrossRef] [PubMed]
- Koepp, T.N.; Tokaj, A.; Nedvetsky, P.I.; Conchon Costa, A.C.; Snieder, B.; Schröter, R.; Ciarimboli, G. Properties of Transport Mediated by the Human Organic Cation Transporter 2 Studied in a Polarized Three-Dimensional Epithelial Cell Culture Model. Int. J. Mol. Sci. 2021, 22, 9658. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci. Ther. 2019, 11, 97. [Google Scholar] [PubMed]
- Einhorn, L.H. Curing Metastatic Testicular Cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 4592–4595. [Google Scholar] [CrossRef]
- Mwenda, V.; Githuku, J.; Gathecha, G.; Wambugu, B.M.; Roka, Z.G.; Ong’or, W.O. Prevalence and Factors Associated with Chronic Kidney Disease among Medical Inpatients at the Kenyatta National Hospital, Kenya, 2018: A Cross-Sectional Study. Pan. Afr. Med. J. 2019, 33, 321. [Google Scholar] [CrossRef] [PubMed]
- Isiiko, J.; Atwiine, B.; Oloro, J. Prevalence and Risk Factors of Nephrotoxicity Among Adult Cancer Patients at Mbarara Regional Referral Hospital. Cancer Manag. Res. 2021, 13, 7677–7684. [Google Scholar] [CrossRef] [PubMed]
- Mohri, J.; Katada, C.; Ueda, M.; Sugawara, M.; Yamashita, K.; Moriya, H.; Komori, S.; Hayakawa, K.; Koizumi, W.; Atsuda, K. Predisposing Factors for Chemotherapy-Induced Nephrotoxicity in Patients with Advanced Esophageal Cancer Who Received Combination Chemotherapy with Docetaxel, Cisplatin, and 5-Fluorouracil. J. Transl. Int. Med. 2018, 6, 32–37. [Google Scholar] [CrossRef]
- Duan, Z.; Cai, G.; Li, J.; Chen, X. Cisplatin-Induced Renal Toxicity in Elderly People. Ther. Adv. Med. Oncol. 2020, 12, 175883592092343. [Google Scholar] [CrossRef]
- Schofield, J.; Harcus, M.; Pizer, B.; Jorgensen, A.; McWilliam, S. Long-Term Cisplatin Nephrotoxicity after Childhood Cancer: A Systematic Review and Meta-Analysis. Pediatr. Nephrol. 2023, 39, 399–710. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin Nephrotoxicity: Mechanisms and Renoprotective Strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef]
- Daugaard, G. Cisplatin Nephrotoxicity: Experimental and Clinical Studies. Dan. Med. Bull. 1990, 37, 1–12. [Google Scholar]
- Rybak, L.P.; Ramkumar, V. Ototoxicity. Kidney Int. 2007, 72, 931–935. [Google Scholar] [CrossRef]
- Rybak, L.; Mukherjea, D.; Ramkumar, V. Mechanisms of Cisplatin-Induced Ototoxicity and Prevention. Semin. Hear. 2019, 40, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Skinner, R. Best Practice in Assessing Ototoxicity in Children with Cancer. Eur. J. Cancer 2004, 40, 2352–2354. [Google Scholar] [CrossRef] [PubMed]
- Filipski, K.K.; Loos, W.J.; Verweij, J.; Sparreboom, A. Interaction of Cisplatin with the Human Organic Cation Transporter 2. Clin. Cancer Res. 2008, 14, 3875–3880. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.H.; Dantzler, W.H. Molecular and Cellular Physiology of Renal Organic Cation and Anion Transport. Physiol. Rev. 2004, 84, 987–1049. [Google Scholar] [CrossRef]
- Alnouti, Y.; Petrick, J.S.; Klaassen, C.D. Tissue Distribution and Ontogeny of Organic Cation Transporters in Mice. Drug Metab. Dispos. 2006, 34, 477–482. [Google Scholar] [CrossRef]
- Safaei, R.; Howell, S.B. Copper Transporters Regulate the Cellular Pharmacology and Sensitivity to Pt Drugs. Crit. Rev. Oncol. Hematol. 2005, 53, 13–23. [Google Scholar] [CrossRef]
- Safaei, R. Role of Copper Transporters in the Uptake and Efflux of Platinum Containing Drugs. Cancer Lett. 2006, 234, 34–39. [Google Scholar] [CrossRef]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic Cation Transporters Are Determinants of Oxaliplatin Cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific Organic Cation Transporters: Structure, Function, Physiological Roles, and Biopharmaceutical Implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef]
- Kido, Y.; Matsson, P.; Giacomini, K.M. Profiling of a Prescription Drug Library for Potential Renal Drug-Drug Interactions Mediated by the Organic Cation Transporter 2. J. Med. Chem. 2011, 54, 4548–4558. [Google Scholar] [CrossRef]
- Jehn, C.F.; Boulikas, T.; Kourvetaris, A.; Possinger, K.; Lüftner, D. Pharmacokinetics of Liposomal Cisplatin (Lipoplatin) in Combination with 5-FU in Patients with Advanced Head and Neck Cancer: First Results of a Phase III Study. Anticancer. Res. 2007, 27, 471–475. [Google Scholar]
- Rajkumar, P. Cisplatin Concentrations in Long and Short Duration Infusion: Implications for the Optimal Time of Radiation Delivery. J. Clin. Diagn. Res. 2016, 10, XC01–XC04. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Inui, K.-I. Organic Cation Transporter OCT/SLC22A and H(+)/Organic Cation Antiporter MATE/SLC47A Are Key Molecules for Nephrotoxicity of Platinum Agents. Biochem. Pharmacol. 2011, 81, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K.-I. Cisplatin and Oxaliplatin, but Not Carboplatin and Nedaplatin, Are Substrates for Human Organic Cation Transporters (SLC22A1-3 and Multidrug and Toxin Extrusion Family). J. Pharmacol. Exp. Ther. 2006, 319, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Elia, N.; Lippincott-Schwartz, J. Culturing MDCK Cells in Three Dimensions for Analyzing Intracellular Dynamics. Curr. Protoc. Cell Biol. 2009, 43, 4.22.1–4.22.18. [Google Scholar] [CrossRef]
- Muth, T.R.; Caplan, M.J. Transport Protein Trafficking in Polarized Cells. Annu. Rev. Cell Dev. Biol. 2003, 19, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Kantauskaitė, M.; Hucke, A.; Reike, M.; Ahmed Eltayeb, S.; Xiao, C.; Barz, V.; Ciarimboli, G. Rapid Regulation of Human Multidrug and Extrusion Transporters HMATE1 and HMATE2K. Int. J. Mol. Sci. 2020, 21, 5157. [Google Scholar] [CrossRef] [PubMed]
- Harashima, N.; Tanaka, K.; Sasatomi, T.; Shimizu, K.; Miyagi, Y.; Yamada, A.; Tamura, M.; Yamana, H.; Itoh, K.; Shichijo, S. Recognition of the Lck Tyrosine Kinase as a Tumor Antigen by Cytotoxic T Lymphocytes of Cancer Patients with Distant Metastases. Eur. J. Immunol. 2001, 31, 323–332. [Google Scholar] [CrossRef]
- Rudd, C.E. How the Discovery of the CD4/CD8-P56lck Complexes Changed Immunology and Immunotherapy. Front. Cell Dev. Biol. 2021, 9, 626095. [Google Scholar] [CrossRef]
- Mahabeleshwar, G.H.; Das, R.; Kundu, G.C. Tyrosine Kinase, P56 -Induced Cell Motility, and Urokinase-Type Plasminogen Activator Secretion Involve Activation of Epidermal Growth Factor Receptor/Extracellular Signal Regulated Kinase Pathways. J. Biol. Chem. 2004, 279, 9733–9742. [Google Scholar] [CrossRef]
- Weiße, J.; Rosemann, J.; Müller, L.; Kappler, M.; Eckert, A.W.; Glaß, M.; Misiak, D.; Hüttelmaier, S.; Ballhausen, W.G.; Hatzfeld, M.; et al. Identification of Lymphocyte Cell-Specific Protein-Tyrosine Kinase (LCK) as a Driver for Invasion and Migration of Oral Cancer by Tumor Heterogeneity Exploitation. Mol. Cancer 2021, 20, 88. [Google Scholar] [CrossRef]
- Wischnjow, A.; Sarko, D.; Janzer, M.; Kaufman, C.; Beijer, B.; Brings, S.; Haberkorn, U.; Larbig, G.; Kübelbeck, A.; Mier, W. Renal Targeting: Peptide-Based Drug Delivery to Proximal Tubule Cells. Bioconjug. Chem. 2016, 27, 1050–1057. [Google Scholar] [CrossRef]
- Lee, W.-K.; Reichold, M.; Edemir, B.; Ciarimboli, G.; Warth, R.; Koepsell, H.; Thévenod, F. Organic Cation Transporters OCT1, 2, and 3 Mediate High-Affinity Transport of the Mutagenic Vital Dye Ethidium in the Kidney Proximal Tubule. Am. J. Physiol. Renal Physiol. 2009, 296, F1504–F1513. [Google Scholar] [CrossRef] [PubMed]
- Schlatter, E.; Klassen, P.; Massmann, V.; Holle, S.K.; Guckel, D.; Edemir, B.; Pavenstädt, H.; Ciarimboli, G. Mouse Organic Cation Transporter 1 Determines Properties and Regulation of Basolateral Organic Cation Transport in Renal Proximal Tubules. Pflugers Arch. 2014, 466, 1581–1589. [Google Scholar] [CrossRef]
- Wilde, S.; Schlatter, E.; Koepsell, H.; Edemir, B.; Reuter, S.; Pavenstädt, H.; Neugebauer, U.; Schröter, R.; Brast, S.; Ciarimboli, G. Calmodulin-Associated Post-Translational Regulation of Rat Organic Cation Transporter 2 in the Kidney Is Gender Dependent. Cell Mol. Life Sci. 2009, 66, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Salomon, J.J.; Endter, S.; Tachon, G.; Falson, F.; Buckley, S.T.; Ehrhardt, C. Transport of the Fluorescent Organic Cation 4-(4-(Dimethylamino)Styryl)-N-Methylpyridinium Iodide (ASP+) in Human Respiratory Epithelial Cells. Eur. J. Pharm. Biopharm. 2012, 81, 351–359. [Google Scholar] [CrossRef]
- Wittwer, M.B.; Zur, A.A.; Khuri, N.; Kido, Y.; Kosaka, A.; Zhang, X.; Morrissey, K.M.; Sali, A.; Huang, Y.; Giacomini, K.M. Discovery of Potent, Selective Multidrug and Toxin Extrusion Transporter 1 (MATE1, SLC47A1) Inhibitors Through Prescription Drug Profiling and Computational Modeling. J. Med. Chem. 2013, 56, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, M.V.; Saadatmand, A.R.; Bokelmann, K.; Meineke, I.; Kaiser, R.; Brockmöller, J. Effects of OCT1 Polymorphisms on the Cellular Uptake, Plasma Concentrations and Efficacy of the 5-HT(3) Antagonists Tropisetron and Ondansetron. Pharmacogenomics J. 2012, 12, 22–29. [Google Scholar] [CrossRef]
- Mayer, F.P.; Schmid, D.; Holy, M.; Yang, J.-W.; Salzer, I.; Boehm, S.; Chiba, P.; Sitte, H.H. Real-Time Uptake of Fluorescent ASP+ via the Organic Cation Transporter 3. BMC Pharmacol. Toxicol. 2012, 13, A80. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Wehe, C.A.; Beyer, G.; Sperling, M.; Ciarimboli, G.; Karst, U. Assessing the Intracellular Concentration of Platinum in Medulloblastoma Cell Lines after Cisplatin Incubation. J. Trace Elem. Med. Biol. 2014, 28, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (logIC50 ± SEM) (µM) | |||
|---|---|---|---|
| Cell Line/Substance | Disopyramide | Imipramine | Orphenadrine |
| hOCT2-HEK | 1.5 * (−5.82 ± 0.06) n = 15–44 | 0.9 (−6.05 ± 0.09) n = 8–38 | 2.0 (−5.72 ± 0.08) n = 16–70 |
| mOCT1-HEK | 0.2 * (−6.79 ± 0.12) n = 8–31 | 1.0 (−5.99 ± 0.30) n = 8–48 | 0.8 (−6.08 ± 0.13) n = 8–47 |
| mOCT2-HEK | 11.8 * (−4.93 ± 0.09) n = 8–32 | 0.4 (−6.39 ± 0.06) n = 8–28 | 1.0 (−6.00 ± 0.07) n = 16–56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hucke, A.; Kantauskaite, M.; Köpp, T.N.; Wehe, C.A.; Karst, U.; Nedvetsky, P.I.; Ciarimboli, G. Modulating the Activity of the Human Organic Cation Transporter 2 Emerges as a Potential Strategy to Mitigate Unwanted Toxicities Associated with Cisplatin Chemotherapy. Int. J. Mol. Sci. 2024, 25, 2922. https://doi.org/10.3390/ijms25052922
Hucke A, Kantauskaite M, Köpp TN, Wehe CA, Karst U, Nedvetsky PI, Ciarimboli G. Modulating the Activity of the Human Organic Cation Transporter 2 Emerges as a Potential Strategy to Mitigate Unwanted Toxicities Associated with Cisplatin Chemotherapy. International Journal of Molecular Sciences. 2024; 25(5):2922. https://doi.org/10.3390/ijms25052922
Chicago/Turabian StyleHucke, Anna, Marta Kantauskaite, Tim N. Köpp, Christoph A. Wehe, Uwe Karst, Pavel I. Nedvetsky, and Giuliano Ciarimboli. 2024. "Modulating the Activity of the Human Organic Cation Transporter 2 Emerges as a Potential Strategy to Mitigate Unwanted Toxicities Associated with Cisplatin Chemotherapy" International Journal of Molecular Sciences 25, no. 5: 2922. https://doi.org/10.3390/ijms25052922