
What Can Inflammation Tell Us about Therapeutic Strategies for Parkinson’s Disease?
Abstract
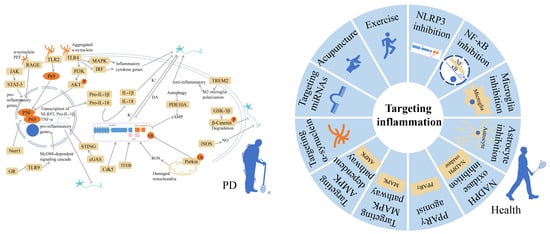
:
1. Introduction
2. Inflammatory Responses and Mechanisms
2.1. Genes Linked to PD and Inflammatory Reaction
2.1.1. LRRK2
2.1.2. SNCA
2.1.3. Pink1/Parkin
2.1.4. GBA1
2.1.5. DJ-1
2.2. Immune Signaling Pathways Associated with PD
2.2.1. Inflammasome
2.2.2. NF-κB Signaling
2.2.3. Toll-like Receptors (TLRs)
2.2.4. TREM2 Receptors
2.2.5. MAPK Signaling
2.2.6. JAK/STAT Pathway
2.2.7. RAGE
2.2.8. Others
3. Anti-Inflammatory Therapies for PD
3.1. NLRP3 Inflammasome Inhibition
3.2. NF-κB Inhibition
3.3. Microglia Inhibition
3.4. Astrocyte Inhibition
3.5. NADPH Oxidase Inhibition
3.6. PPARγ Agonist
3.7. Targeting MAPK Pathway
3.8. Targeting AMPK-Dependent Pathway
3.9. Targeting α-Synuclein
3.10. Targeting miRNAs
3.11. Acupuncture
3.12. Exercise
3.13. Others
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schüle, B.; et al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Mullin, S.; Schapira, A.H. Pathogenic mechanisms of neurodegeneration in Parkinson disease. Neurol. Clin. 2015, 33, 1–17. [Google Scholar] [CrossRef]
- Liang, Y.; Zhong, G.; Ren, M.; Sun, T.; Li, Y.; Ye, M.; Ma, C.; Guo, Y.; Liu, C. The Role of Ubiquitin-Proteasome System and Mitophagy in the Pathogenesis of Parkinson’s Disease. Neuromol. Med. 2023, 25, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Smeyne, R.J.; Noyce, A.J.; Byrne, M.; Savica, R.; Marras, C. Infection and Risk of Parkinson’s Disease. J. Park. Dis. 2021, 11, 31–43. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Lampe, J.B.; Gossrau, G.; Herting, B.; Kempe, A.; Sommer, U.; Füssel, M.; Weber, M.; Koch, R.; Reichmann, H. HLA typing and Parkinson’s disease. Eur. Neurol. 2003, 50, 64–68. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Wissemann, W.T.; Hill-Burns, E.M.; Zabetian, C.P.; Factor, S.A.; Patsopoulos, N.; Hoglund, B.; Holcomb, C.; Donahue, R.J.; Thomson, G.; Erlich, H.; et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am. J. Hum. Genet. 2013, 93, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Nemutlu Samur, D.; Akçay, G.; Yıldırım, S.; Özkan, A.; Çeker, T.; Derin, N.; Tanrıöver, G.; Aslan, M.; Ağar, A.; Özbey, G. Vortioxetine ameliorates motor and cognitive impairments in the rotenone-induced Parkinson’s disease via targeting TLR-2 mediated neuroinflammation. Neuropharmacology 2022, 208, 108977. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yu, S.Y.; Zuo, L.J.; Cao, C.J.; Hu, Y.; Chen, Z.J.; Piao, Y.S.; Wang, Y.J.; Wang, X.M.; Chen, S.D.; et al. Excessive Iron and α-Synuclein Oligomer in Brain are Relevant to Pure Apathy in Parkinson Disease. J. Geriatr. Psychiatry Neurol. 2016, 29, 187–194. [Google Scholar] [CrossRef]
- Yu, S.Y.; Sun, L.; Liu, Z.; Huang, X.Y.; Zuo, L.J.; Cao, C.J.; Zhang, W.; Wang, X.M. Sleep disorders in Parkinson’s disease: Clinical features, iron metabolism and related mechanism. PLoS ONE 2013, 8, e82924. [Google Scholar] [CrossRef]
- Hu, Y.; Yu, S.Y.; Zuo, L.J.; Piao, Y.S.; Cao, C.J.; Wang, F.; Chen, Z.J.; Du, Y.; Lian, T.H.; Liu, G.F.; et al. Investigation on Abnormal Iron Metabolism and Related Inflammation in Parkinson Disease Patients with Probable RBD. PLoS ONE 2015, 10, e0138997. [Google Scholar] [CrossRef]
- Nadig, A.P.R.; Huwaimel, B.; Alobaida, A.; Khafagy, E.S.; Alotaibi, H.F.; Moin, A.; Lila, A.S.A.; Suman, M.S.; Krishna, K.L. Manganese chloride (MnCl2) induced novel model of Parkinson’s disease in adult Zebrafish; Involvement of oxidative stress, neuroinflammation and apoptosis pathway. Biomed. Pharmacother. 2022, 155, 113697. [Google Scholar] [CrossRef]
- Liu, B.; Gao, H.M.; Hong, J.S. Parkinson’s disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: Role of neuroinflammation. Environ. Health Perspect. 2003, 111, 1065–1073. [Google Scholar] [CrossRef]
- Glushakova, O.Y.; Johnson, D.; Hayes, R.L. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J. Neurotrauma 2014, 31, 1180–1193. [Google Scholar] [CrossRef]
- Rosen, B.; Kurtishi, A.; Vazquez-Jimenez, G.R.; Møller, S.G. The Intersection of Parkinson’s Disease, Viral Infections, and COVID-19. Mol. Neurobiol. 2021, 58, 4477–4486. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Ferrero-Miliani, L.; Nielsen, O.H.; Andersen, P.S.; Girardin, S.E. Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1β generation. Clin. Exp. Immunol. 2007, 147, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.A. Inflammation and neurodegeneration: Chronicity matters. Aging 2018, 11, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Barbalace, M.C.; Malaguti, M.; Giusti, L.; Lucacchini, A.; Hrelia, S.; Angeloni, C. Anti-Inflammatory Activities of Marine Algae in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 61. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.W.; Stratton, M.S.; Ferguson, B.S. Dietary natural products as epigenetic modifiers in aging-associated inflammation and disease. Nat. Prod. Rep. 2020, 37, 653–676. [Google Scholar] [CrossRef]
- Lutters, B.; Foley, P.; Koehler, P.J. The centennial lesson of encephalitis lethargica. Neurology 2018, 90, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef]
- Krashia, P.; Cordella, A.; Nobili, A.; La Barbera, L.; Federici, M.; Leuti, A.; Campanelli, F.; Natale, G.; Marino, G.; Calabrese, V.; et al. Blunting neuroinflammation with resolvin D1 prevents early pathology in a rat model of Parkinson’s disease. Nat. Commun. 2019, 10, 3945. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef] [PubMed]
- Contaldi, E.; Magistrelli, L.; Milner, A.V.; Cosentino, M.; Marino, F.; Comi, C. Expression of Transcription Factors in CD4+T Cells as Potential Biomarkers of Motor Complications in Parkinson’s Disease. J. Park. Dis. 2021, 11, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Magistrelli, L.; Storelli, E.; Rasini, E.; Contaldi, E.; Comi, C.; Cosentino, M.; Marino, F. Relationship between circulating CD4+ T lymphocytes and cognitive impairment in patients with Parkinson’s disease. Brain Behav. Immun. 2020, 89, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Hall, S.; Surova, Y.; Nielsen, H.M.; Janelidze, S.; Brundin, L.; Hansson, O. Cerebrospinal fluid inflammatory markers in Parkinson’s disease—Associations with depression, fatigue, and cognitive impairment. Brain Behav. Immun. 2013, 33, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.F. Regionally-specific microglial activation in young mice over-expressing human wildtype α-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef]
- Brockmann, K.; Schulte, C.; Schneiderhan-Marra, N.; Apel, A.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.C.; Cormier, F.; et al. Inflammatory profile discriminates clinical subtypes in LRRK2-associated Parkinson’s disease. Eur. J. Neurol. 2017, 24, 427-e6. [Google Scholar] [CrossRef]
- Kozina, E.; Byrne, M.; Smeyne, R.J. Mutant LRRK2 in lymphocytes regulates neurodegeneration via IL-6 in an inflammatory model of Parkinson’s disease. NPJ Park. Dis. 2022, 8, 24. [Google Scholar] [CrossRef]
- Chen, Y.; Yin, Q.; Cheng, X.Y.; Zhang, J.R.; Jin, H.; Li, K.; Mao, C.J.; Wang, F.; Bei, H.Z.; Liu, C.F. G2019S LRRK2 Mutation Enhances MPP(+)-Induced Inflammation of Human Induced Pluripotent Stem Cells-Differentiated Dopaminergic Neurons. Front. Neurosci. 2022, 16, 947927. [Google Scholar] [CrossRef]
- Kim, K.S.; Marcogliese, P.C.; Yang, J.; Callaghan, S.M.; Resende, V.; Abdel-Messih, E.; Marras, C.; Visanji, N.P.; Huang, J.; Schlossmacher, M.G.; et al. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E5164–E5173. [Google Scholar] [CrossRef]
- Wang, S.; Chu, C.H.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.S.; Zhang, J. α-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.F.; Varanita, T.; Herrebout, M.A.C.; Plug, B.C.; Kole, J.; Musters, R.J.P.; Teunissen, C.E.; Hoozemans, J.J.M.; Bubacco, L.; Veerhuis, R. α-Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 2021, 69, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 receptor is critical in α-synuclein--mediated microglial NADPH oxidase activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. α-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; López-Royo, T.; Casarejos, M.; Lanciego, J.L.; Lastres-Becker, I. Cx3cr1-deficiency exacerbates α-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Glia 2018, 66, 1752–1762. [Google Scholar] [CrossRef]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambarè, D.; Bellini, E.; Ordazzo, G.; et al. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef]
- Lee, E.J.; Woo, M.S.; Moon, P.G.; Baek, M.C.; Choi, I.Y.; Kim, W.K.; Junn, E.; Kim, H.S. α-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.H.; Yan, W.F.; Zhang, Z.; Ma, K.L.; Peng, S.Y.; Cao, Y.L.; Yuan, Y.H.; Chen, N.H. Nurr1: A vital participant in the TLR4-NF-κB signal pathway stimulated by α-synuclein in BV-2 cells. Neuropharmacology 2019, 144, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Tran, T.; Ruhn, K.A.; Martinez, T.N.; Hong, J.; Marvin, M.; Hartley, M.; Treviño, I.; O’Brien, D.E.; Casey, B.; et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 10825–10834. [Google Scholar] [CrossRef] [PubMed]
- Dionísio, P.E.A.; Oliveira, S.R.; Amaral, J.; Rodrigues, C.M.P. Loss of Microglial Parkin Inhibits Necroptosis and Contributes to Neuroinflammation. Mol. Neurobiol. 2019, 56, 2990–3004. [Google Scholar] [CrossRef]
- Borsche, M.; König, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Brüggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain J. Neurol. 2020, 143, 3041–3051. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Wasner, K.; Smajic, S.; Ghelfi, J.; Delcambre, S.; Prada-Medina, C.A.; Knappe, E.; Arena, G.; Mulica, P.; Agyeah, G.; Rakovic, A.; et al. Parkin Deficiency Impairs Mitochondrial DNA Dynamics and Propagates Inflammation. Mov. Disord. Off. J. Mov. Disord. Soc. 2022, 37, 1405–1415. [Google Scholar] [CrossRef]
- Kim, J.; Byun, J.W.; Choi, I.; Kim, B.; Jeong, H.K.; Jou, I.; Joe, E. PINK1 Deficiency Enhances Inflammatory Cytokine Release from Acutely Prepared Brain Slices. Exp. Neurobiol. 2013, 22, 38–44. [Google Scholar] [CrossRef]
- Akundi, R.S.; Huang, Z.; Eason, J.; Pandya, J.D.; Zhi, L.; Cass, W.A.; Sullivan, P.G.; Büeler, H. Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS ONE 2011, 6, e16038. [Google Scholar] [CrossRef]
- Lee, H.J.; Jang, S.H.; Kim, H.; Yoon, J.H.; Chung, K.C. PINK1 stimulates interleukin-1β-mediated inflammatory signaling via the positive regulation of TRAF6 and TAK1. Cell. Mol. Life Sci. CMLS 2012, 69, 3301–3315. [Google Scholar] [CrossRef]
- Singh, K.; Han, K.; Tilve, S.; Wu, K.; Geller, H.M.; Sack, M.N. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia 2018, 66, 2427–2437. [Google Scholar] [CrossRef]
- Mullin, S.; Stokholm, M.G.; Hughes, D.; Mehta, A.; Parbo, P.; Hinz, R.; Pavese, N.; Brooks, D.J.; Schapira, A.H.V. Brain Microglial Activation Increased in Glucocerebrosidase (GBA) Mutation Carriers without Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Van Steenwinckel, J.; Schang, A.L.; Krishnan, M.L.; Degos, V.; Delahaye-Duriez, A.; Bokobza, C.; Csaba, Z.; Verdonk, F.; Montané, A.; Sigaut, S.; et al. Decreased microglial Wnt/β-catenin signalling drives microglial pro-inflammatory activation in the developing brain. Brain J. Neurol. 2019, 142, 3806–3833. [Google Scholar] [CrossRef] [PubMed]
- Awad, O.; Panicker, L.M.; Deranieh, R.M.; Srikanth, M.P.; Brown, R.A.; Voit, A.; Peesay, T.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Altered Differentiation Potential of Gaucher’s Disease iPSC Neuronal Progenitors due to Wnt/β-Catenin Downregulation. Stem Cell Rep. 2017, 9, 1853–1867. [Google Scholar] [CrossRef]
- Chahine, L.M.; Qiang, J.; Ashbridge, E.; Minger, J.; Yearout, D.; Horn, S.; Colcher, A.; Hurtig, H.I.; Lee, V.M.; Van Deerlin, V.M.; et al. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol. 2013, 70, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Ginns, E.I.; Mak, S.K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and α-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Graham, A.R.; Brown, E.; McLean, J.R.; Hayes, M.A.; Beagan, J.; Izen, S.C.; et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain α-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid. Redox Signal. 2015, 23, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Mus, L.; Siani, F.; Giuliano, C.; Ghezzi, C.; Cerri, S.; Blandini, F. Development and biochemical characterization of a mouse model of Parkinson’s disease bearing defective glucocerebrosidase activity. Neurobiol. Dis. 2019, 124, 289–296. [Google Scholar] [CrossRef]
- Waak, J.; Weber, S.S.; Waldenmaier, A.; Görner, K.; Alunni-Fabbroni, M.; Schell, H.; Vogt-Weisenhorn, D.; Pham, T.T.; Reumers, V.; Baekelandt, V.; et al. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 2478–2489. [Google Scholar] [CrossRef]
- Frøyset, A.K.; Edson, A.J.; Gharbi, N.; Khan, E.A.; Dondorp, D.; Bai, Q.; Tiraboschi, E.; Suster, M.L.; Connolly, J.B.; Burton, E.A.; et al. Astroglial DJ-1 over-expression up-regulates proteins involved in redox regulation and is neuroprotective in vivo. Redox Biol. 2018, 16, 237–247. [Google Scholar] [CrossRef]
- De Miranda, B.R.; Rocha, E.M.; Bai, Q.; El Ayadi, A.; Hinkle, D.; Burton, E.A.; Timothy Greenamyre, J. Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson’s disease. Neurobiol. Dis. 2018, 115, 101–114. [Google Scholar] [CrossRef]
- Trudler, D.; Weinreb, O.; Mandel, S.A.; Youdim, M.B.; Frenkel, D. DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J. Neurochem. 2014, 129, 434–447. [Google Scholar] [CrossRef]
- Ji, Y.J.; Wang, H.L.; Yin, B.L.; Ren, X.Y. Down-regulation of DJ-1 Augments Neuroinflammation via Nrf2/Trx1/NLRP3 Axis in MPTP-induced Parkinson’s Disease Mouse Model. Neuroscience 2020, 442, 253–263. [Google Scholar] [CrossRef]
- Choi, D.J.; An, J.; Jou, I.; Park, S.M.; Joe, E.H. A Parkinson’s disease gene, DJ-1, regulates anti-inflammatory roles of astrocytes through prostaglandin D2 synthase expression. Neurobiol. Dis. 2019, 127, 482–491. [Google Scholar] [CrossRef]
- Eguchi, T.; Kuwahara, T.; Sakurai, M.; Komori, T.; Fujimoto, T.; Ito, G.; Yoshimura, S.I.; Harada, A.; Fukuda, M.; Koike, M.; et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9115–E9124. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Reviews. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Chedid, J.; Labrador-Garrido, A.; Zhong, S.; Gao, J.; Zhao, Y.; Perera, G.; Kim, W.S.; Halliday, G.M.; Dzamko, N. A small molecule toll-like receptor antagonist rescues α-synuclein fibril pathology. J. Biol. Chem. 2022, 298, 102260. [Google Scholar] [CrossRef]
- Devine, M.J.; Gwinn, K.; Singleton, A.; Hardy, J. Parkinson’s disease and α-synuclein expression. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Westbroek, W.; Nguyen, M.; Siebert, M.; Lindstrom, T.; Burnett, R.A.; Aflaki, E.; Jung, O.; Tamargo, R.; Rodriguez-Gil, J.L.; Acosta, W.; et al. A new glucocerebrosidase-deficient neuronal cell model provides a tool to probe pathophysiology and therapeutics for Gaucher disease. Dis. Models Mech. 2016, 9, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Flavell, R.A. Molecular mechanism of NLRP3 inflammasome activation. J. Clin. Immunol. 2010, 30, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef]
- Mao, Z.; Liu, C.; Ji, S.; Yang, Q.; Ye, H.; Han, H.; Xue, Z. The NLRP3 Inflammasome is Involved in the Pathogenesis of Parkinson’s Disease in Rats. Neurochem. Res. 2017, 42, 1104–1115. [Google Scholar] [CrossRef]
- Gladkova, C.; Maslen, S.L.; Skehel, J.M.; Komander, D. Mechanism of parkin activation by PINK1. Nature 2018, 559, 410–414. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Kam, T.I.; Wang, H.; Neifert, S.; Chou, S.C.; Kumar, M.; Brahmachari, S.; Jhaldiyal, A.; Hinkle, J.T.; Akkentli, F.; et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron 2022, 110, 2422–2437.e9. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Liao, Y.; Dong, Y.; Hu, H.; Yang, N.; Kong, X.; Li, S.; Li, X.; Guo, J.; Qin, L.; et al. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 2020, 16, 2193–2205. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Qiu, J.; Wang, P.; Liu, J.; Zhao, Y.; Jiang, F.; Lou, H. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease. Brain Behav. Immun. 2021, 91, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mao, K.; Yu, H.; Wen, Y.; She, H.; Zhang, H.; Liu, L.; Li, M.; Li, W.; Zou, F. p38-TFEB pathways promote microglia activation through inhibiting CMA-mediated NLRP3 degradation in Parkinson’s disease. J. Neuroinflamm. 2021, 18, 295. [Google Scholar] [CrossRef] [PubMed]
- Su, C.J.; Feng, Y.; Liu, T.T.; Liu, X.; Bao, J.J.; Shi, A.M.; Hu, D.M.; Liu, T.; Yu, Y.L. Thioredoxin-interacting protein induced α-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 2017, 23, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Shao, X.Y.; Qi, G.J.; Chen, Q.; Bu, L.L.; Chen, L.J.; Shi, J.; Ming, J.; Tian, B. Cdk5-Dependent Activation of Neuronal Inflammasomes in Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.N.; Fang, J.N.; Hua, F.F.; Han, J.Y.; Yuan, Z.Q.; Xie, A.M. The mechanism behind activation of the Nod-like receptor family protein 3 inflammasome in Parkinson’s disease. Neural Regen. Res. 2022, 17, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Sarkar, S.; Harischandra, D.S.; Neal, M.; Kam, T.I.; Jin, H.; Saminathan, H.; Langley, M.; Charli, A.; Samidurai, M.; et al. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 2019, 216, 1411–1430. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.F.; Longhena, F.; Faustini, G.; van Eik, J.M.; Gombert, I.; Herrebout, M.A.C.; Fayed, M.; Sandre, M.; Varanita, T.; Teunissen, C.E.; et al. Dopamine signaling modulates microglial NLRP3 inflammasome activation: Implications for Parkinson’s disease. J. Neuroinflamm. 2022, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Zhang, Q.; Jiang, Q.; Zhang, T.; Chen, M.; Fan, Y.; Ding, J.; Lu, M.; Hu, G. Inhibition of the hepatic Nlrp3 protects dopaminergic neurons via attenuating systemic inflammation in a MPTP/p mouse model of Parkinson’s disease. J. Neuroinflamm. 2018, 15, 193. [Google Scholar] [CrossRef] [PubMed]
- Goes, A.T.R.; Jesse, C.R.; Antunes, M.S.; Lobo Ladd, F.V.; Lobo Ladd, A.A.B.; Luchese, C.; Paroul, N.; Boeira, S.P. Protective role of chrysin on 6-hydroxydopamine-induced neurodegeneration a mouse model of Parkinson’s disease: Involvement of neuroinflammation and neurotrophins. Chem.-Biol. Interact. 2018, 279, 111–120. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X.; Tuo, M.; Ma, J.; Xie, A. RAGE and its emerging role in the pathogenesis of Parkinson’s disease. Neurosci. Lett. 2018, 672, 65–69. [Google Scholar] [CrossRef]
- Hassanzadeh, K.; Rahimmi, A. Oxidative stress and neuroinflammation in the story of Parkinson’s disease: Could targeting these pathways write a good ending? J. Cell. Physiol. 2018, 234, 23–32. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef]
- Dutta, D.; Jana, M.; Majumder, M.; Mondal, S.; Roy, A.; Pahan, K. Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nat. Commun. 2021, 12, 5382. [Google Scholar] [CrossRef]
- Gao, H.; Wang, D.; Jiang, S.; Mao, J.; Yang, X. NF-κB is negatively associated with Nurr1 to reduce the inflammatory response in Parkinson’s disease. Mol. Med. Rep. 2021, 23, 396. [Google Scholar] [CrossRef]
- Yuan, Y.H.; Sun, J.D.; Wu, M.M.; Hu, J.F.; Peng, S.Y.; Chen, N.H. Rotenone could activate microglia through NFκB associated pathway. Neurochem. Res. 2013, 38, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, Y.; Lu, L.; Zhang, H.; Zhao, C.; Pu, Y.; Yin, L. Copper induces microglia-mediated neuroinflammation through ROS/NF-κB pathway and mitophagy disorder. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2022, 168, 113369. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, K.; Chapman, G.D.; Gunasekar, P.G. α-Synuclein overexpression enhances manganese-induced neurotoxicity through the NF-κB-mediated pathway. Toxicol. Mech. Methods 2011, 21, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.L.; Bantle, C.M.; Popichak, K.A.; Wright, K.A.; Thompson, D.; Forero, C.; Kirkley, K.S.; Damale, P.U.; Chong, E.K.P.; Tjalkens, R.B. NF-κB Signaling in Astrocytes Modulates Brain Inflammation and Neuronal Injury Following Sequential Exposure to Manganese and MPTP During Development and Aging. Toxicol. Sci. Off. J. Soc. Toxicol. 2020, 177, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Maatouk, L.; Compagnion, A.C.; Sauvage, M.C.; Bemelmans, A.P.; Leclere-Turbant, S.; Cirotteau, V.; Tohme, M.; Beke, A.; Trichet, M.; Bazin, V.; et al. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat. Commun. 2018, 9, 2450. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T.; Akira, S. Pleiotropic function of Toll-like receptors. Microbes Infect. 2004, 6, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef] [PubMed]
- Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83, 711–730. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.H.; Chen, Y.; Li, F.F.; Wang, S.; Zhang, X.L.; Yuan, Y.H.; Chen, N.H. TLR4 deficiency has a protective effect in the MPTP/probenecid mouse model of Parkinson’s disease. Acta Pharmacol. Sin. 2019, 40, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef]
- Santoro, A.; Spinelli, C.C.; Martucciello, S.; Nori, S.L.; Capunzo, M.; Puca, A.A.; Ciaglia, E. Innate immunity and cellular senescence: The good and the bad in the developmental and aged brain. J. Leukoc. Biol. 2018, 103, 509–524. [Google Scholar] [CrossRef]
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Reviews. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef]
- Guo, Y.; Wei, X.; Yan, H.; Qin, Y.; Yan, S.; Liu, J.; Zhao, Y.; Jiang, F.; Lou, H. TREM2 deficiency aggravates α-synuclein-induced neurodegeneration and neuroinflammation in Parkinson’s disease models. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 12164–12174. [Google Scholar] [CrossRef]
- Ren, M.; Guo, Y.; Wei, X.; Yan, S.; Qin, Y.; Zhang, X.; Jiang, F.; Lou, H. TREM2 overexpression attenuates neuroinflammation and protects dopaminergic neurons in experimental models of Parkinson’s disease. Exp. Neurol. 2018, 302, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, S.; Nie, K.; Li, Y.; Gao, Y.; Gan, R.; Wang, L.; Li, B.; Sun, X.; Wang, L.; et al. TREM2 modulates microglia phenotypes in the neuroinflammation of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2018, 499, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, D.; Hu, Q.; Wang, G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS ONE 2013, 8, e72046. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, B.; Lin, A. Novel strategies for inhibition of the p38 MAPK pathway. Trends Pharmacol. Sci. 2007, 28, 286–295. [Google Scholar] [CrossRef]
- Singh, R.K.; Diwan, M.; Dastidar, S.G.; Najmi, A.K. Differential effect of p38 and MK2 kinase inhibitors on the inflammatory and toxicity biomarkers in vitro. Hum. Exp. Toxicol. 2018, 37, 521–531. [Google Scholar] [CrossRef]
- Ray, A.; Sehgal, N.; Karunakaran, S.; Rangarajan, G.; Ravindranath, V. MPTP activates ASK1-p38 MAPK signaling pathway through TNF-dependent Trx1 oxidation in parkinsonism mouse model. Free Radic. Biol. Med. 2015, 87, 312–325. [Google Scholar] [CrossRef]
- Thomas, T.; Timmer, M.; Cesnulevicius, K.; Hitti, E.; Kotlyarov, A.; Gaestel, M. MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation--relevance in a mouse model of Parkinson’s disease. J. Neurochem. 2008, 105, 2039–2052. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H., 3rd; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y.; et al. Inhibition of the JAK/STAT Pathway Protects Against α-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 5144–5159. [Google Scholar] [CrossRef]
- Lashgari, N.A.; Roudsari, N.M.; Momtaz, S.; Sathyapalan, T.; Abdolghaffari, A.H.; Sahebkar, A. The involvement of JAK/STAT signaling pathway in the treatment of Parkinson’s disease. J. Neuroimmunol. 2021, 361, 577758. [Google Scholar] [CrossRef]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef]
- Kooshki, L.; Zarneshan, S.N.; Fakhri, S.; Moradi, S.Z.; Echeverria, J. The pivotal role of JAK/STAT and IRS/PI3K signaling pathways in neurodegenerative diseases: Mechanistic approaches to polyphenols and alkaloids. Phytomed. Int. J. Phytother. Phytopharm. 2023, 112, 154686. [Google Scholar] [CrossRef]
- Huang, M.; Guo, M.; Wang, K.; Wu, K.; Li, Y.; Tian, T.; Wang, Y.; Yan, W.; Zhou, Z.; Yang, H. HMGB1 Mediates Paraquat-Induced Neuroinflammatory Responses via Activating RAGE Signaling Pathway. Neurotox. Res. 2020, 37, 913–925. [Google Scholar] [CrossRef]
- Long, H.; Zhang, S.; Zeng, S.; Tong, Y.; Liu, J.; Liu, C.; Li, D. Interaction of RAGE with α-synuclein fibrils mediates inflammatory response of microglia. Cell Rep. 2022, 40, 111401. [Google Scholar] [CrossRef]
- Wang, X.; Sun, X.; Niu, M.; Zhang, X.; Wang, J.; Zhou, C.; Xie, A. RAGE Silencing Ameliorates Neuroinflammation by Inhibition of p38-NF-κB Signaling Pathway in Mouse Model of Parkinson’s Disease. Front. Neurosci. 2020, 14, 353. [Google Scholar] [CrossRef]
- Gasparotto, J.; Ribeiro, C.T.; Bortolin, R.C.; Somensi, N.; Rabelo, T.K.; Kunzler, A.; Souza, N.C.; Pasquali, M.A.B.; Moreira, J.C.F.; Gelain, D.P. Targeted inhibition of RAGE in substantia nigra of rats blocks 6-OHDA-induced dopaminergic denervation. Sci. Rep. 2017, 7, 8795. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 regulated Frizzled-1/β-Catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 2011, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Kumar Puli, L.; Färber, K.; Harkany, T.; Schulte, G. WNT signaling in activated microglia is proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Yu, J.; Gao, Y.; Kumar, G.; Guo, M.; Zhao, Y.; Fang, Q.; Zhang, H.; Yu, J.; Jiang, Y.; et al. Therapeutic potentials of the Rho kinase inhibitor Fasudil in experimental autoimmune encephalomyelitis and the related mechanisms. Metab. Brain Dis. 2019, 34, 377–384. [Google Scholar] [CrossRef]
- He, Q.; Li, Y.H.; Guo, S.S.; Wang, Y.; Lin, W.; Zhang, Q.; Wang, J.; Ma, C.G.; Xiao, B.G. Inhibition of Rho-kinase by Fasudil protects dopamine neurons and attenuates inflammatory response in an intranasal lipopolysaccharide-mediated Parkinson’s model. Eur. J. Neurosci. 2016, 43, 41–52. [Google Scholar] [CrossRef]
- Wang, H.M.; Zhang, T.; Li, Q.; Huang, J.K.; Chen, R.F.; Sun, X.J. Inhibition of glycogen synthase kinase-3β by lithium chloride suppresses 6-hydroxydopamine-induced inflammatory response in primary cultured astrocytes. Neurochem. Int. 2013, 63, 345–353. [Google Scholar] [CrossRef]
- Li, D.W.; Liu, Z.Q.; Chen, W.; Yao, M.; Li, G.R. Association of glycogen synthase kinase-3β with Parkinson’s disease (review). Mol. Med. Rep. 2014, 9, 2043–2050. [Google Scholar] [CrossRef]
- Ma, C.; Liu, Y.; Li, S.; Ma, C.; Huang, J.; Wen, S.; Yang, S.; Wang, B. Microglial cGAS drives neuroinflammation in the MPTP mouse models of Parkinson’s disease. CNS Neurosci. Ther. 2023, 29, 2018–2035. [Google Scholar] [CrossRef]
- Whitton, P.S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharmacol. 2007, 150, 963–976. [Google Scholar] [CrossRef]
- Broom, L.; Marinova-Mutafchieva, L.; Sadeghian, M.; Davis, J.B.; Medhurst, A.D.; Dexter, D.T. Neuroprotection by the selective iNOS inhibitor GW274150 in a model of Parkinson disease. Free Radic. Biol. Med. 2011, 50, 633–640. [Google Scholar] [CrossRef]
- Han, C.; Shen, H.; Yang, Y.; Sheng, Y.; Wang, J.; Li, W.; Zhou, X.; Guo, L.; Zhai, L.; Guan, Q. Antrodia camphorata polysaccharide resists 6-OHDA-induced dopaminergic neuronal damage by inhibiting ROS-NLRP3 activation. Brain Behav. 2020, 10, e01824. [Google Scholar] [CrossRef]
- Han, C.; Guo, L.; Yang, Y.; Li, W.; Sheng, Y.; Wang, J.; Guan, Q.; Zhang, X. Study on antrodia camphorata polysaccharide in alleviating the neuroethology of PD mice by decreasing the expression of NLRP3 inflammasome. Phytother. Res. PTR 2019, 33, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Sun, S.; Sun, Y.; Song, Q.; Zhu, J.; Song, N.; Chen, M.; Sun, T.; Xia, M.; Ding, J.; et al. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for Parkinson disease. Autophagy 2019, 15, 1860–1881. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Zhuang, W.; Lv, E.; Liu, Z.; Wang, Y.; Zhang, W.; Fu, W. Kaemperfol alleviates pyroptosis and microglia-mediated neuroinflammation in Parkinson’s disease via inhibiting p38MAPK/NF-κB signaling pathway. Neurochem. Int. 2022, 152, 105221. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xue, Z.; Zhu, L.; Zhou, J.; Zhuo, L.; Zhang, J.; Zhang, X.; Liu, W.; Han, L.; Liao, W. Rhynchophylline alleviates neuroinflammation and regulates metabolic disorders in a mouse model of Parkinson’s disease. Food Funct. 2023, 14, 3208–3219. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, P.; Cai, Y. Genkwanin suppresses MPP(+)-induced cytotoxicity by inhibiting TLR4/MyD88/NLRP3 inflammasome pathway in a cellular model of Parkinson’s disease. Neurotoxicology 2021, 87, 62–69. [Google Scholar] [CrossRef]
- Ahmed, S.; Kwatra, M.; Ranjan Panda, S.; Murty, U.S.N.; Naidu, V.G.M. Andrographolide suppresses NLRP3 inflammasome activation in microglia through induction of parkin-mediated mitophagy in in-vitro and in-vivo models of Parkinson disease. Brain Behav. Immun. 2021, 91, 142–158. [Google Scholar] [CrossRef]
- Yao, S.; Li, L.; Sun, X.; Hua, J.; Zhang, K.; Hao, L.; Liu, L.; Shi, D.; Zhou, H. FTY720 Inhibits MPP(+)-Induced Microglial Activation by Affecting NLRP3 Inflammasome Activation. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2019, 14, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, X.; Gao, Z.Y.; Lin, M.; Zhao, X.; Sun, Y.; Pu, X.P. Icaritin Provides Neuroprotection in Parkinson’s Disease by Attenuating Neuroinflammation, Oxidative Stress, and Energy Deficiency. Antioxidants 2021, 10, 529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, G.; He, J.; Yang, Q.; Li, D.; Li, J.; Zhang, F. Icariin attenuates neuroinflammation and exerts dopamine neuroprotection via an Nrf2-dependent manner. J. Neuroinflamm. 2019, 16, 92. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ojo-Amaize, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Desplats, P.; Wrasidlo, W.; Adame, A.; Nchekwube, E.; Oyemade, O.; et al. Hypoestoxide reduces neuroinflammation and α-synuclein accumulation in a mouse model of Parkinson’s disease. J. Neuroinflamm. 2015, 12, 236. [Google Scholar] [CrossRef]
- Sn, S.; Pandurangi, J.; Murumalla, R.; Dj, V.; Garimella, L.; Acharya, A.; Rai, S.; Paul, A.; Yarreiphang, H.; Pillai, M.S.; et al. Small molecule modulator of aggrephagy regulates neuroinflammation to curb pathogenesis of neurodegeneration. EBioMedicine 2019, 50, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Kumar, G.; Gedda, M.R.; Tiwari, N.; Patnaik, R.; Singh, R.K.; Singh, S.P. Effect of Chlorogenic Acid Supplementation in MPTP-Intoxicated Mouse. Front. Pharmacol. 2018, 9, 757. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Youdim, M.B.; Maor, G.; Mandel, S. Attenuation of 6-hydroxydopamine (6-OHDA)-induced nuclear factor-κB (NF-κB) activation and cell death by tea extracts in neuronal cultures. Biochem. Pharmacol. 2002, 63, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.T.; Cao, X.B.; Xiong, N.; Wang, H.C.; Huang, J.S.; Sun, S.G.; Wang, T. Morin exerts neuroprotective actions in Parkinson disease models in vitro and in vivo. Acta Pharmacol. Sin. 2010, 31, 900–906. [Google Scholar] [CrossRef]
- Lee, K.M.; Lee, Y.; Chun, H.J.; Kim, A.H.; Kim, J.Y.; Lee, J.Y.; Ishigami, A.; Lee, J. Neuroprotective and anti-inflammatory effects of morin in a murine model of Parkinson’s disease. J. Neurosci. Res. 2016, 94, 865–878. [Google Scholar] [CrossRef]
- Lee, E.; Park, H.R.; Ji, S.T.; Lee, Y.; Lee, J. Baicalein attenuates astroglial activation in the 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine-induced Parkinson’s disease model by downregulating the activations of nuclear factor-κB, ERK, and JNK. J. Neurosci. Res. 2014, 92, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jia, M.; Zhang, X.; Wang, P. Calycosin attenuates MPTP-induced Parkinson’s disease by suppressing the activation of TLR/NF-κB and MAPK pathways. Phytother. Res. PTR 2019, 33, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Koppula, S.; Kumar, H.; Park, J.Y.; Kim, I.W.; More, S.V.; Kim, I.S.; Han, S.D.; Kim, S.K.; Yoon, S.H.; et al. α-Asarone attenuates microglia-mediated neuroinflammation by inhibiting NFκB activation and mitigates MPTP-induced behavioral deficits in a mouse model of Parkinson’s disease. Neuropharmacology 2015, 97, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jing, H.; Yang, H.; Liu, Z.; Guo, H.; Chai, L.; Hu, L. Tanshinone I selectively suppresses pro-inflammatory genes expression in activated microglia and prevents nigrostriatal dopaminergic neurodegeneration in a mouse model of Parkinson’s disease. J. Ethnopharmacol. 2015, 164, 247–255. [Google Scholar] [CrossRef]
- Abdelsalam, R.M.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: Role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J. Neurochem. 2015, 133, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Mante, M.; Anderson, S.; Rockenstein, E.; Masliah, E. Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of Parkinson’s disease. J. Neuroinflamm. 2015, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Shin, W.H.; Baek, J.Y.; Cho, E.J.; Baik, H.H.; Kim, S.R.; Won, S.Y.; Jin, B.K. CB2 receptor activation prevents glial-derived neurotoxic mediator production, BBB leakage and peripheral immune cell infiltration and rescues dopamine neurons in the MPTP model of Parkinson’s disease. Exp. Mol. Med. 2016, 48, e205. [Google Scholar] [CrossRef]
- Kim, J.S.; Ryu, S.Y.; Yun, I.; Kim, W.J.; Lee, K.S.; Park, J.W.; Kim, Y.I. 1α,25-Dihydroxyvitamin D3 Protects Dopaminergic Neurons in Rodent Models of Parkinson’s Disease through Inhibition of Microglial Activation. J. Clin. Neurol. 2006, 2, 252–257. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, M.; Zhao, Y.; Zhang, Y.; Gao, Y.; Zhang, X.; Yang, G. α-lipoic acid improved motor function in MPTP-induced Parkinsonian mice by reducing neuroinflammation in the nigral and spinal cord. Neurosci. Lett. 2022, 781, 136669. [Google Scholar] [CrossRef]
- Jang, H.; Kim, S.; Lee, J.M.; Oh, Y.S.; Park, S.M.; Kim, S.R. Montelukast treatment protects nigral dopaminergic neurons against microglial activation in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neuroreport 2017, 28, 242–249. [Google Scholar] [CrossRef]
- Mansour, R.M.; Ahmed, M.A.E.; El-Sahar, A.E.; El Sayed, N.S. Montelukast attenuates rotenone-induced microglial activation/p38 MAPK expression in rats: Possible role of its antioxidant, anti-inflammatory and antiapoptotic effects. Toxicol. Appl. Pharmacol. 2018, 358, 76–85. [Google Scholar] [CrossRef]
- Kim, H.G.; Ju, M.S.; Ha, S.K.; Lee, H.; Lee, H.; Kim, S.Y.; Oh, M.S. Acacetin protects dopaminergic cells against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neuroinflammation in vitro and in vivo. Biol. Pharm. Bull. 2012, 35, 1287–1294. [Google Scholar] [CrossRef]
- Zhou, P.; Homberg, J.R.; Fang, Q.; Wang, J.; Li, W.; Meng, X.; Shen, J.; Luan, Y.; Liao, P.; Swaab, D.F.; et al. Histamine-4 receptor antagonist JNJ7777120 inhibits pro-inflammatory microglia and prevents the progression of Parkinson-like pathology and behaviour in a rat model. Brain Behav. Immun. 2019, 76, 61–73. [Google Scholar] [CrossRef]
- Kim, H.S.; Suh, Y.H. Minocycline and neurodegenerative diseases. Behav. Brain Res. 2009, 196, 168–179. [Google Scholar] [CrossRef]
- Tomás-Camardiel, M.; Rite, I.; Herrera, A.J.; de Pablos, R.M.; Cano, J.; Machado, A.; Venero, J.L. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood-brain barrier, and damage in the nigral dopaminergic system. Neurobiol. Dis. 2004, 16, 190–201. [Google Scholar] [CrossRef]
- Lv, J.; Zhu, J.; Wang, P.; Liu, T.; Yuan, J.; Yin, H.; Lan, Y.; Sun, Q.; Zhang, Z.; Ding, G.; et al. Artemisinin exerts a protective effect in the MPTP mouse model of Parkinson’s disease by inhibiting microglial activation via the TLR4/Myd88/NF-KB pathway. CNS Neurosci. Ther. 2023, 29, 1012–1023. [Google Scholar] [CrossRef]
- Huang, G.; Yuan, K.; Zhu, Q.; Zhang, S.; Lu, Q.; Zhu, M.; Sheng, H.; Yu, R.; Luo, G.; Xu, A. Triptolide inhibits the inflammatory activities of neutrophils to ameliorate chronic arthritis. Mol. Immunol. 2018, 101, 210–220. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Zhang, Q.; Zhang, J.N.; Zhang, Y.N.; Gu, L.; Yang, H.M.; Xia, N.; Wang, X.M.; Zhang, H. Triptolide up-regulates metabotropic glutamate receptor 5 to inhibit microglia activation in the lipopolysaccharide-induced model of Parkinson’s disease. Brain Behav. Immun. 2018, 71, 93–107. [Google Scholar] [CrossRef]
- Song, M.K.; Lee, J.H.; Kim, J.; Kim, J.H.; Hwang, S.; Kim, Y.S.; Kim, Y.J. Neuroprotective effect of NXP031 in the MPTP-induced Parkinson’s disease model. Neurosci. Lett. 2021, 740, 135425. [Google Scholar] [CrossRef]
- Kim, H.D.; Jeong, K.H.; Jung, U.J.; Kim, S.R. Naringin treatment induces neuroprotective effects in a mouse model of Parkinson’s disease in vivo, but not enough to restore the lesioned dopaminergic system. J. Nutr. Biochem. 2016, 28, 140–146. [Google Scholar] [CrossRef]
- Bok, E.; Chung, Y.C.; Kim, K.S.; Baik, H.H.; Shin, W.H.; Jin, B.K. Modulation of M1/M2 polarization by capsaicin contributes to the survival of dopaminergic neurons in the lipopolysaccharide-lesioned substantia nigra in vivo. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Zhou, T.T.; Zu, G.; Wang, X.; Zhang, X.G.; Li, S.; Liang, Z.H.; Zhao, J. Immunomodulatory and neuroprotective effects of ginsenoside Rg1 in the MPTP(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced mouse model of Parkinson’s disease. Int. Immunopharmacol. 2015, 29, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Chorny, A.; Gonzalez-Rey, E.; Ganea, D. Vasoactive intestinal peptide generates CD4+CD25+ regulatory T cells in vivo. J. Leukoc. Biol. 2005, 78, 1327–1338. [Google Scholar] [CrossRef]
- Fernandez-Martin, A.; Gonzalez-Rey, E.; Chorny, A.; Ganea, D.; Delgado, M. Vasoactive intestinal peptide induces regulatory T cells during experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2006, 36, 318–326. [Google Scholar] [CrossRef]
- Reynolds, A.D.; Stone, D.K.; Hutter, J.A.; Benner, E.J.; Mosley, R.L.; Gendelman, H.E. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J. Immunol. 2010, 184, 2261–2271. [Google Scholar] [CrossRef]
- Olson, K.E.; Kosloski-Bilek, L.M.; Anderson, K.M.; Diggs, B.J.; Clark, B.E.; Gledhill, J.M., Jr.; Shandler, S.J.; Mosley, R.L.; Gendelman, H.E. Selective VIP Receptor Agonists Facilitate Immune Transformation for Dopaminergic Neuroprotection in MPTP-Intoxicated Mice. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 16463–16478. [Google Scholar] [CrossRef]
- Olson, K.E.; Bade, A.N.; Schutt, C.R.; Dong, J.; Shandler, S.J.; Boska, M.D.; Mosley, R.L.; Gendelman, H.E.; Liu, Y. Manganese-Enhanced Magnetic Resonance Imaging for Detection of Vasoactive Intestinal Peptide Receptor 2 Agonist Therapy in a Model of Parkinson’s Disease. Neurother. J. Am. Soc. Exp. Neurother. 2016, 13, 635–646. [Google Scholar] [CrossRef]
- Mosley, R.L.; Lu, Y.; Olson, K.E.; Machhi, J.; Yan, W.; Namminga, K.L.; Smith, J.R.; Shandler, S.J.; Gendelman, H.E. A Synthetic Agonist to Vasoactive Intestinal Peptide Receptor-2 Induces Regulatory T Cell Neuroprotective Activities in Models of Parkinson’s Disease. Front. Cell. Neurosci. 2019, 13, 421. [Google Scholar] [CrossRef]
- Lee, Y.; Chun, H.J.; Lee, K.M.; Jung, Y.S.; Lee, J. Silibinin suppresses astroglial activation in a mouse model of acute Parkinson’s disease by modulating the ERK and JNK signaling pathways. Brain Res. 2015, 1627, 233–242. [Google Scholar] [CrossRef]
- Wang, L.; Bai, Y.; Tao, Y.; Shen, W.; Zhou, H.; He, Y.; Wu, H.; Huang, F.; Shi, H.; Wu, X. Bear bile powder alleviates Parkinson’s disease-like behavior in mice by inhibiting astrocyte-mediated neuroinflammation. Chin. J. Nat. Med. 2023, 21, 710–720. [Google Scholar] [CrossRef]
- Zhang, S.X.; Khalyfa, A.; Wang, Y.; Carreras, A.; Hakim, F.; Neel, B.A.; Brady, M.J.; Qiao, Z.; Hirotsu, C.; Gozal, D. Sleep fragmentation promotes NADPH oxidase 2-mediated adipose tissue inflammation leading to insulin resistance in mice. Int. J. Obes. 2014, 38, 619–624. [Google Scholar] [CrossRef]
- Wang, Q.; Chu, C.H.; Oyarzabal, E.; Jiang, L.; Chen, S.H.; Wilson, B.; Qian, L.; Hong, J.S. Subpicomolar diphenyleneiodonium inhibits microglial NADPH oxidase with high specificity and shows great potential as a therapeutic agent for neurodegenerative diseases. Glia 2014, 62, 2034–2043. [Google Scholar] [CrossRef]
- Che, Y.; Hou, L.; Sun, F.; Zhang, C.; Liu, X.; Piao, F.; Zhang, D.; Li, H.; Wang, Q. Taurine protects dopaminergic neurons in a mouse Parkinson’s disease model through inhibition of microglial M1 polarization. Cell Death Dis. 2018, 9, 435. [Google Scholar] [CrossRef]
- Lee, Y.; Cho, J.H.; Lee, S.; Lee, W.; Chang, S.C.; Chung, H.Y.; Moon, H.R.; Lee, J. Neuroprotective effects of MHY908, a PPAR α/γ dual agonist, in a MPTP-induced Parkinson’s disease model. Brain Res. 2019, 1704, 47–58. [Google Scholar] [CrossRef]
- Pisanu, A.; Lecca, D.; Mulas, G.; Wardas, J.; Simbula, G.; Spiga, S.; Carta, A.R. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis. 2014, 71, 280–291. [Google Scholar] [CrossRef]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-γ agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflamm. 2011, 8, 91. [Google Scholar] [CrossRef]
- Dehmer, T.; Heneka, M.T.; Sastre, M.; Dichgans, J.; Schulz, J.B. Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with IκBα induction and block of NFκB and iNOS activation. J. Neurochem. 2004, 88, 494–501. [Google Scholar] [CrossRef]
- Lazzarini, M.; Martin, S.; Mitkovski, M.; Vozari, R.R.; Stühmer, W.; Bel, E.D. Doxycycline restrains glia and confers neuroprotection in a 6-OHDA Parkinson model. Glia 2013, 61, 1084–1100. [Google Scholar] [CrossRef]
- Santa-Cecília, F.V.; Socias, B.; Ouidja, M.O.; Sepulveda-Diaz, J.E.; Acuña, L.; Silva, R.L.; Michel, P.P.; Del-Bel, E.; Cunha, T.M.; Raisman-Vozari, R. Doxycycline Suppresses Microglial Activation by Inhibiting the p38 MAPK and NF-kB Signaling Pathways. Neurotox. Res. 2016, 29, 447–459. [Google Scholar] [CrossRef]
- Yan, X.; Liu, D.F.; Zhang, X.Y.; Liu, D.; Xu, S.Y.; Chen, G.X.; Huang, B.X.; Ren, W.Z.; Wang, W.; Fu, S.P.; et al. Vanillin Protects Dopaminergic Neurons against Inflammation-Mediated Cell Death by Inhibiting ERK1/2, P38 and the NF-κB Signaling Pathway. Int. J. Mol. Sci. 2017, 18, 389. [Google Scholar] [CrossRef]
- Iba, M.; Kim, C.; Kwon, S.; Szabo, M.; Horan-Portelance, L.; Peer, C.J.; Figg, W.D.; Reed, X.; Ding, J.; Lee, S.J.; et al. Inhibition of p38α MAPK restores neuronal p38γ MAPK and ameliorates synaptic degeneration in a mouse model of DLB/PD. Sci. Transl. Med. 2023, 15, eabq6089. [Google Scholar] [CrossRef]
- Lee, J.A.; Kim, H.R.; Kim, J.; Park, K.D.; Kim, D.J.; Hwang, O. The Novel Neuroprotective Compound KMS99220 Has an Early Anti-neuroinflammatory Effect via AMPK and HO-1, Independent of Nrf2. Exp. Neurobiol. 2018, 27, 408–418. [Google Scholar] [CrossRef]
- Cao, B.; Zhang, Y.; Chen, J.; Wu, P.; Dong, Y.; Wang, Y. Neuroprotective effects of liraglutide against inflammation through the AMPK/NF-κB pathway in a mouse model of Parkinson’s disease. Metab. Brain Dis. 2022, 37, 451–462. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Li, C.; Zhu, D.; Cheng, O.; Cui, J. Dexmedetomidine alleviates pain in MPTP-treated mice by activating the AMPK/mTOR/NF-κB pathways in astrocytes. Neurosci. Lett. 2022, 791, 136933. [Google Scholar] [CrossRef]
- Mohamad, K.A.; El-Naga, R.N.; Wahdan, S.A. Neuroprotective effects of indole-3-carbinol on the rotenone rat model of Parkinson’s disease: Impact of the SIRT1-AMPK signaling pathway. Toxicol. Appl. Pharmacol. 2022, 435, 115853. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, M.; Luo, Y.; Jin, D.; Guo, X.; Yang, W.; Zheng, J.; Zhang, H.; Zhang, L.; Deng, C.; et al. Healthy Human Fecal Microbiota Transplantation into Mice Attenuates MPTP-Induced Neurotoxicity via AMPK/SOD2 Pathway. Aging Dis. 2023, 14, 2193. [Google Scholar] [CrossRef]
- Kim, C.; Spencer, B.; Rockenstein, E.; Yamakado, H.; Mante, M.; Adame, A.; Fields, J.A.; Masliah, D.; Iba, M.; Lee, H.J.; et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating α-synuclein transmission and neuroinflammation. Mol. Neurodegener. 2018, 13, 43. [Google Scholar] [CrossRef]
- Poewe, W.; Volc, D.; Seppi, K.; Medori, R.; Lührs, P.; Kutzelnigg, A.; Djamshidian, A.; Thun-Hohenstein, C.; Meissner, W.G.; Rascol, O.; et al. Safety and Tolerability of Active Immunotherapy Targeting α-Synuclein with PD03A in Patients with Early Parkinson’s Disease: A Randomized, Placebo-Controlled, Phase 1 Study. J. Park. Dis. 2021, 11, 1079–1089. [Google Scholar] [CrossRef]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef]
- Hung, K.C.; Huang, H.J.; Wang, Y.T.; Lin, A.M. Baicalein attenuates α-synuclein aggregation, inflammasome activation and autophagy in the MPP+-treated nigrostriatal dopaminergic system in vivo. J. Ethnopharmacol. 2016, 194, 522–529. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of α-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, M.; Yan, R.; Liu, J.; Maddila, S.; Junn, E.; Mouradian, M.M. MicroRNA-7 Protects Against Neurodegeneration Induced by α-Synuclein Preformed Fibrils in the Mouse Brain. Neurother. J. Am. Soc. Exp. Neurother. 2021, 18, 2529–2540. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Lv, R.; Du, L.; Zhou, F.; Yuan, X.; Liu, X.; Zhang, L. Rosmarinic Acid Alleviates Inflammation, Apoptosis, and Oxidative Stress through Regulating miR-155-5p in a Mice Model of Parkinson’s Disease. ACS Chem. Neurosci. 2020, 11, 3259–3266. [Google Scholar] [CrossRef]
- Lv, Q.; Zhong, Z.; Hu, B.; Yan, S.; Yan, Y.; Zhang, J.; Shi, T.; Jiang, L.; Li, W.; Huang, W. MicroRNA-3473b regulates the expression of TREM2/ULK1 and inhibits autophagy in inflammatory pathogenesis of Parkinson disease. J. Neurochem. 2021, 157, 599–610. [Google Scholar] [CrossRef]
- Kang, J.M.; Park, H.J.; Choi, Y.G.; Choe, I.H.; Park, J.H.; Kim, Y.S.; Lim, S. Acupuncture inhibits microglial activation and inflammatory events in the MPTP-induced mouse model. Brain Res. 2007, 1131, 211–219. [Google Scholar] [CrossRef]
- Jang, J.H.; Yeom, M.J.; Ahn, S.; Oh, J.Y.; Ji, S.; Kim, T.H.; Park, H.J. Acupuncture inhibits neuroinflammation and gut microbial dysbiosis in a mouse model of Parkinson’s disease. Brain Behav. Immun. 2020, 89, 641–655. [Google Scholar] [CrossRef]
- Spielman, L.J.; Little, J.P.; Klegeris, A. Physical activity and exercise attenuate neuroinflammation in neurological diseases. Brain Res. Bull. 2016, 125, 19–29. [Google Scholar] [CrossRef]
- Toy, W.A.; Petzinger, G.M.; Leyshon, B.J.; Akopian, G.K.; Walsh, J.P.; Hoffman, M.V.; Vučković, M.G.; Jakowec, M.W. Treadmill exercise reverses dendritic spine loss in direct and indirect striatal medium spiny neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. Neurobiol. Dis. 2014, 63, 201–209. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, Y.; Myers, K.G.; Heintz, R.; Holschneider, D.P. Recruitment of the prefrontal cortex and cerebellum in Parkinsonian rats following skilled aerobic exercise. Neurobiol. Dis. 2015, 77, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Koo, J.H.; Kwon, I.; Kang, E.B.; Um, H.S.; Soya, H.; Lee, Y.; Cho, J.Y. Neuroprotective effects of endurance exercise against neuroinflammation in MPTP-induced Parkinson’s disease mice. Brain Res. 2017, 1655, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.H.; Cho, J.Y. Treadmill Exercise Attenuates α-Synuclein Levels by Promoting Mitochondrial Function and Autophagy Possibly via SIRT1 in the Chronic MPTP/P-Induced Mouse Model of Parkinson’s Disease. Neurotox. Res. 2017, 32, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.H.; Kim, S.C.; Hong, H.P.; Park, C.Y.; Shin, M.S.; Kim, C.J.; Seo, J.H.; Kim, D.Y.; Kim, D.J.; Cho, H.J. Treadmill exercise ameliorates dopaminergic neuronal loss through suppressing microglial activation in Parkinson’s disease mice. Life Sci. 2012, 91, 1309–1316. [Google Scholar] [CrossRef]
- Al-Jarrah, M.; Obaidat, H.; Bataineh, Z.; Walton, L.; Al-Khateeb, A. Endurance exercise training protects against the upregulation of nitric oxide in the striatum of MPTP/probenecid mouse model of Parkinson’s disease. NeuroRehabilitation 2013, 32, 141–147. [Google Scholar] [CrossRef]
- Liu, B.; Arbogast, L.A. Progesterone decreases tyrosine hydroxylase phosphorylation state and increases protein phosphatase 2A activity in the stalk-median eminence on proestrous afternoon. J. Endocrinol. 2010, 204, 209–219. [Google Scholar] [CrossRef]
- Jang, Y.; Kwon, I.; Song, W.; Cosio-Lima, L.M.; Lee, Y. Endurance Exercise Mediates Neuroprotection Against MPTP-mediated Parkinson’s Disease via Enhanced Neurogenesis, Antioxidant Capacity, and Autophagy. Neuroscience 2018, 379, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients with Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef]
- During, M.J.; Cao, L.; Zuzga, D.S.; Francis, J.S.; Fitzsimons, H.L.; Jiao, X.; Bland, R.J.; Klugmann, M.; Banks, W.A.; Drucker, D.J.; et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 2003, 9, 1173–1179. [Google Scholar] [CrossRef]
- Kim, S.; Moon, M.; Park, S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J. Endocrinol. 2009, 202, 431–439. [Google Scholar] [CrossRef]
- Choi, S.H.; Aid, S.; Bosetti, F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: Implications for translational research. Trends Pharmacol. Sci. 2009, 30, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Gómez-Rodríguez, V.M.; González-Renovato, E.D.; Torres-Sánchez, E.D.; Ramírez-Anguiano, A.C. Fish oil, melatonin and vitamin E attenuates midbrain cyclooxygenase-2 activity and oxidative stress after the administration of 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine. Metab. Brain Dis. 2013, 28, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Tamburrino, A.; Churchill, M.J.; Wan, O.W.; Colino-Sanguino, Y.; Ippolito, R.; Bergstrand, S.; Wolf, D.A.; Herz, N.J.; Sconce, M.D.; Björklund, A.; et al. Cyclosporin promotes neurorestoration and cell replacement therapy in pre-clinical models of Parkinson’s disease. Acta Neuropathol. Commun. 2015, 3, 84. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Kliest, T.; Dodiya, H.B.; Broersen, L.M.; Garssen, J.; Keshavarzian, A.; Kraneveld, A.D. The gut-brain axis in Parkinson’s disease: Possibilities for food-based therapies. Eur. J. Pharmacol. 2017, 817, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Meng, L.B.; Chen, L.J.; Shi, X.; Tu, L.; Zhou, Q.; Yu, J.L.; Liao, X.; Zeng, Y.; Yuan, Q.Y. The role of the microbiota-gut-brain axis and intestinal microbiome dysregulation in Parkinson’s disease. Front. Neurol. 2023, 14, 1185375. [Google Scholar] [CrossRef] [PubMed]
- Leta, V.; Ray Chaudhuri, K.; Milner, O.; Chung-Faye, G.; Metta, V.; Pariante, C.M.; Borsini, A. Neurogenic and anti-inflammatory effects of probiotics in Parkinson’s disease: A systematic review of preclinical and clinical evidence. Brain Behav. Immun. 2021, 98, 59–73. [Google Scholar] [CrossRef]
- Chen, H.; Jacobs, E.; Schwarzschild, M.A.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol. 2005, 58, 963–967. [Google Scholar] [CrossRef]
- Swiątkiewicz, M.; Zaremba, M.; Joniec, I.; Członkowski, A.; Kurkowska-Jastrzębska, I. Potential neuroprotective effect of ibuprofen, insights from the mice model of Parkinson’s disease. Pharmacol. Rep. PR 2013, 65, 1227–1236. [Google Scholar] [CrossRef]
- Kurkowska-Jastrzebska, I.; Babiuch, M.; Joniec, I.; Przybyłkowski, A.; Członkowski, A.; Członkowska, A. Indomethacin protects against neurodegeneration caused by MPTP intoxication in mice. Int. Immunopharmacol. 2002, 2, 1213–1218. [Google Scholar] [CrossRef]
- Dong, X.L.; Wang, X.; Liu, F.; Liu, X.; Du, Z.R.; Li, R.W.; Xue, C.H.; Wong, K.H.; Wong, W.T.; Zhao, Q.; et al. Polymannuronic acid prevents dopaminergic neuronal loss via brain-gut-microbiota axis in Parkinson’s disease model. Int. J. Biol. Macromol. 2020, 164, 994–1005. [Google Scholar] [CrossRef]
- Gaballah, H.H.; Zakaria, S.S.; Elbatsh, M.M.; Tahoon, N.M. Modulatory effects of resveratrol on endoplasmic reticulum stress-associated apoptosis and oxido-inflammatory markers in a rat model of rotenone-induced Parkinson’s disease. Chem.-Biol. Interact. 2016, 251, 10–16. [Google Scholar] [CrossRef]
- Kujawska, M.; Jodynis-Liebert, J. Polyphenols in Parkinson’s Disease: A Systematic Review of In Vivo Studies. Nutrients 2018, 10, 642. [Google Scholar] [CrossRef]
- Ojha, R.P.; Rastogi, M.; Devi, B.P.; Agrawal, A.; Dubey, G.P. Neuroprotective effect of curcuminoids against inflammation-mediated dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2012, 7, 609–618. [Google Scholar] [CrossRef]
- Tseng, H.C.; Wang, M.H.; Chang, K.C.; Soung, H.S.; Fang, C.H.; Lin, Y.W.; Li, K.Y.; Yang, C.C.; Tsai, C.C. Protective Effect of (-)Epigallocatechin-3-gallate on Rotenone-Induced Parkinsonism-like Symptoms in Rats. Neurotox. Res. 2020, 37, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Tiefensee Ribeiro, C.; Gasparotto, J.; Petiz, L.L.; Brum, P.O.; Peixoto, D.O.; Kunzler, A.; da Rosa Silva, H.T.; Bortolin, R.C.; Almeida, R.F.; Quintans-Junior, L.J.; et al. Oral administration of carvacrol/β-cyclodextrin complex protects against 6-hydroxydopamine-induced dopaminergic denervation. Neurochem. Int. 2019, 126, 27–35. [Google Scholar] [CrossRef]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Fiske, B.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2023 Update. J. Park. Dis. 2023, 13, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Chen, H.; Schwarzschild, M.A.; Ascherio, A. Use of ibuprofen and risk of Parkinson disease. Neurology 2011, 76, 863–869. [Google Scholar] [CrossRef]
- Naren, P.; Cholkar, A.; Kamble, S.; Khan, S.S.; Srivastava, S.; Madan, J.; Mehra, N.; Tiwari, V.; Singh, S.B.; Khatri, D.K. Pathological and Therapeutic Advances in Parkinson’s Disease: Mitochondria in the Interplay. J. Alzheimer’s Dis. JAD 2023, 94, S399–S428. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Zhang, Y.; Santamaria, P.; Olson, K.E.; Schutt, C.R.; Bhatti, D.; Shetty, B.L.D.; Lu, Y.; Estes, K.A.; Standaert, D.G.; et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Park. Dis. 2017, 3, 10. [Google Scholar] [CrossRef]
- Mouhammad, Z.A.; Vohra, R.; Horwitz, A.; Thein, A.S.; Rovelt, J.; Cvenkel, B.; Williams, P.A.; Azuara-Blanco, A.; Kolko, M. Glucagon-Like Peptide 1 Receptor Agonists—Potential Game Changers in the Treatment of Glaucoma? Front. Neurosci. 2022, 16, 824054. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.T.; Chan, L.; Chen, K.Y.; Lee, H.H.; Huang, L.K.; Yang, Y.S.H.; Liu, Y.R.; Hu, C.J. Rifaximin Modifies Gut Microbiota and Attenuates Inflammation in Parkinson’s Disease: Preclinical and Clinical Studies. Cells 2022, 11, 3468. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, M.J.; Flores, A.J.; Ye, T.; Smidt, S.I.; Dollish, H.K.; Stancati, J.A.; Farrell, D.C.; Parent, K.L.; Doyle, K.P.; Besselsen, D.G.; et al. Preclinical evidence in support of repurposing sub-anesthetic ketamine as a treatment for L-DOPA-induced dyskinesia. Exp. Neurol. 2020, 333, 113413. [Google Scholar] [CrossRef] [PubMed]
- Huot, P.; Johnston, T.H.; Koprich, J.B.; Fox, S.H.; Brotchie, J.M. The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol. Rev. 2013, 65, 171–222. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain J. Neurol. 2013, 136 Pt 8, 2419–2431. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, Y.H.; Chen, N.H.; Wang, H.B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 2019, 67, 458–464. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Effects and Mechanisms | References |
|---|---|---|
| LRRK2 mutant, human | IL-8 ↑, MCP-1 ↑, MIP-1-β ↑, BDNF ↑ | [40] |
| C57BL/6J-Tg(LRRK2*R1441G)3IMjff/J mice, C57BL/6-Lrrk2 tm4.1 Arte mouse strain | DA neurons ↓, IL-6 ↑ | [41] |
| G2019S mutation in the LRRK2 gene, MPP+, induced pluripotent stem cells (iPSCs) | Neurites and neurite arborization ↓, survival rate ↓, apoptosis ↑, IL-1β ↑, TNF-α ↑, COX-2 ↑, IL-6 ↑, iNOS ↑ | [42] |
| Macrophages and microglia from PD patients and mice with LRRK2-G2019S mutation | DA neurons ↓, WAVE2 stabilization ↑, phagocytic response ↑ | [43] |
| AAV2 vectors harbor the cDNA encoding human α-synuclein, microglia, primary neurons | H2O2 ↑, microglial migration | [44] |
| α-Synuclein fibrils, primary microglia | IL-1β ↑, caspase-1 recruitment, NLRP3 activation ↑, | [45] |
| A53T mutant α-synuclein, microglia | oxidative stress, nicotinamide adenine dinucleotide phosphate oxidase activity ↑, P2X7 receptor ↑, PI3K/AKT ↑ | [46] |
| A53T mutant α-synuclein, A30P mutant α-synuclein, microglia | NF-κB/AP-1/Nrf2 pathway activation ↑, TNF-α ↑, CXCL10 ↑ | [47] |
| A53T mutant α-synuclein, Cx3cr1−/− mice | DA neurons ↓, RelA ↑, IL-1β ↑, TNF-α ↑, IL-6 ↑ | [48] |
| LV-FLEX-SNCAG420A, C57BL/6 mice | DA neurons ↓, phagocytic exhaustion, ROS ↑, NO ↑, inflammasome pathway, inflammatory cytokine, and inflammatory nuclear transcriptional pathway gene enrichment | [49] |
| α-Synuclein, microglia | MMPs ↑, NO ↑, ROS ↑, IL-1β ↑, TNF-α ↑, PAR-1 activity ↑, | [50] |
| α-Synuclein, microglia, C57BL/6 mice | TNF-α ↑, TLR4 activation, TLR4/PI3K/AKT/GSK3β pathway ↑, NF-κB ↑ | [51] |
| Exhaustive exercise, Parkin−/− mice, Pink1−/− mice | IL-6 ↑, IFNβ1 ↑, DA neurons ↓, circulating mtDNA ↑ | [52] |
| LPS, siRNA-mediated downregulation of Parkin, Parkin−/− mice, microglia | The vulnerability of DA neurons to inflammation ↑, survival of activated microglia ↑ | [53,54] |
| Pink1/Parkin mutant, human | IL-6 ↑, ccf-mtDNA ↑, mitophagy ↓, ccf-mtDNA-cGAS-STING-IL-6 pathway ↑ | [55] |
| Intestinal infection with Gram-negative bacteria, Pink1−/− mice, primary neuron | Mitochondrial antigen presentation, establishment of cytotoxic mitochondria-specific CD8+T cells | [56] |
| LPS, Parkin mutant, iPSC-derived neuron–microglia co-cultures | Mitochondrial biogenesis pathway ↓, mtDNA dyshomeostasis, sirtuin 1 ↓, immune response ↑ | [57] |
| LPS, Pink1−/− mice | IL-1β ↑, TNF-α ↑, IL-6 ↑, IL-1β ↑, TNF-α ↑, IL-6 ↑, Ca2+ storage capacity ↓, JNK activity ↑, DA level ↓ | [58,59] |
| Pink1−/− mouse embryonic fibroblasts | Autodimerization and autoubiquitination of TRAF6 ↑, polyubiquitination of TAK1 ↑, NF-κB activity ↑ | [60] |
| Parkin KO mice, astrocyte | NOD2 ↑, ER stress ↑, JNK activation ↑, cytokine release ↑, neurotropic factor ↓ | [61] |
| GBA mutant, human | Microglia activation | [62] |
| LPS, OF1 strain mice, zebrafish, GD iPSC-derived neuronal progenitor cells | Microglia activation, gene and protein expression of members of the Wnt/β-catenin signaling pathway ↓ | [63,64] |
| GBA mutant, human | Cognitive dysfunction ↑, IL-8 ↑, monocyte chemotactic protein 1 ↑, macrophage inflammatory protein 1α ↑ | [65] |
| GBA mutant, GCase inhibitor CBE, long-lived Gaucher mice | Glial activation, α-synuclein accumulation | [66] |
| GCase inhibitor CBE, BDF1 mice | α-synuclein aggregates, neuroinflammation ↑, complement C1q ↑, abnormalities in synaptic, axonal transport, and cytoskeletal proteins | [67] |
| GCase inhibitor CBE, C57BL/6N mice | Enhanced MPTP-induced neurodegeneration, α-synuclein ↑, microglia activation | [68] |
| Dj-1−/− astrocytes, primary neuron–astrocyte co-culture | NO ↑, iNOS ↑, p38 MAPK phosphorylation ↑, COX-2 ↑, IL-6 ↑, apoptosis ↑ | [69] |
| Increased astroglial DJ-1 expression, zebrafish, astrocyte | Protected from oxidative-stress-induced injuries as caused by MPP+ | [70] |
| Rotenone, overexpress human DJ-1 protein, Lewis rats | Astrocytes with DJ-1 overexpression showed a marked reduction in neuronal oxidative stress and microglial activation, α-synuclein accumulation and phosphorylation were decreased within DJ-1-transduced animals | [71] |
| DJ-1 depletion, microglia | IL-1β ↑, IL-6 ↑, monoamine oxidase activity ↑, ROS ↑, NO ↑, TREM2 ↓ | [72] |
| DJ-1 knockdown, C57BL/6 mice, BV2 murine microglial cells | Neuroinflammation ↑, apoptosis ↑, DA neurons ↓, Nrf2 ↓, NLRP3 ↑ | [73] |
| DJ-1 knockout, C57BL/6 mice, primary astrocytes | TNF-α ↑, HO-1 ↓, PTGDS levels increased in the injured brain of WT mice, but barely in that of knockdown mice | [74] |
| Strategies | Chemical Structure | Treatments | Effects and Mechanisms | References |
|---|---|---|---|---|
| NLRP3 inflammasome inhibition | Antrodia camphorata polysaccharide, 6-OHDA, MES23.5 | ROS-NLRP3 ↓, apoptosis of dopaminergic neurons ↓, dopamine ↑ | [145,146] | |
 | Kaempferol, LPS, C57 BL/6J mice, SD rats | NLRP3 inflammasome activation ↓, caspase-1 ↓, NLRP3-PYCARD-CASP1 complex assembly ↓, macroautophagy/autophagy ↑, the activation of microglia and astrocytes ↓, TH-positive neurons loss↓, IL-1β ↓, IL-18 ↓, iNOS ↓, COX-2 ↓, p38MAPK/NF-κB ↓ | [147,148] | |
 | MCC950, LPS, primary microglia | NLRP3 inflammasome activation ↓, α-synuclein-mediated inflammasome activation ↓ | [149] | |
 | Rhynchophylline, MPTP, C57 BL/6J mice | TLR4 ↓, NLRP3 ↓, COX2 ↓, | [150] | |
 | Genkwanin, MPP+, SH-SY5Y cells | Cell viability ↑, LDH release ↓, apoptosis ↓, ROS generation ↓, SOD activity ↑ | [151] | |
 | Andrographolide, microglia, MPP+, LPS | NLRP3 inflammasome activation ↓, ATP level ↑, dopaminergic neuron ↑, behavioral parameter improvement | [152] | |
 | Fingolimod, MPTP, C57 BL/6J mice | Behavioral deficits ↓, DA neurons loss ↓, dopamine levels ↑, microglial activation ↓, IL-6 ↓, IL-1β ↓, TNF-α ↓ | [153] | |
 | Icaritin and its glucoside, MPTP, 6-OH-DOPA, C57 BL/6 mice, Nrf2 knockout mice | NLRP3 inflammasome activation ↓, IL-1β ↓, VDAC and ATP5B stabilization, Nrf2 signaling activation ↑ | [154,155] | |
 | Selnoflast, human | NLRP3 inflammasome activation ↓, the trial is being tested | [246] | |
| NF-κB inhibition |  | Hypoestoxide, mThy1-α-syn transgenic mice | Decreased microgliosis, astrogliosis, and pro-inflammatory cytokine gene expression, DA neurons ↑, α-synuclein pathology ↓, phosphorylated NF-κB ↓ | [156] |
 | PD180970, MPTP, C57 BL/6J mice | IL-6 ↓, MCP-1 ↓, NF-κB ↓ | [157] | |
 | Chlorogenic acid, MPTP, Swiss albino male mice | Neuroinflammation ↓, TNF-α ↓, IL-1β ↓, IL-10 ↑, attenuation of astrocyte activation | [158] | |
| Tea extracts, 6-OHDA, PC12 cells, SH-SY5Y cells | NF-κB nuclear translocation and binding activity ↓ | [159] | ||
 | CU-CPT22, oligomeric α-synuclein, primary microglia | Nuclear translocation of NF-κB ↓, TNF-α ↓ | [118] | |
 | Morin, MPP+, PC12 cells, C57 BL/6 mice | Cell viability loss and apoptosis ↓, behavioral deficits ↓, DA neurons ↑, dopamine depletion ↓, astrocyte activation ↓, ROS ↓ | [160,161] | |
 | Baicalein, MPTP, C57 BL/6 mice | Motor ability ↑, DA neuron loss ↓, microglial and astrocyte activation ↓, nuclear translocation of NF-κB ↓, activation of JNK and ERK ↓ | [162] | |
 | Calycosin, MPTP, LPS, mice, BV2 cells | Behavioral dysfunctions ↓, inflammatory responses ↓, TLR/NF-κB and MAPK pathways ↓ | [163] | |
 | α-Asarone, MPTP, LPS, C57 BL/6 mice, BV-2 cells | Neuroinflammatory responses ↓, pro-inflammatory cytokine ↓, microglial activation ↓, behavioral impairments ↓ | [164] | |
 | Tanshinone I, LPS, MPTP, BV-2 cells, C57 BL/6 mice | NO ↓, TNF-α ↓, IL-1β ↓, IL-6 ↓, granulocyte colony-stimulating factor expression ↓, NF-κB activation ↓, improved motor functions, normalized striatal neurotransmitters | [165] | |
 | Vildagliptin, rotenone, Wistar rats | Neuronal demise ↓, cytochrome c ↓, caspase-3 ↓, RAGE/NFκB cascade ↓ | [166] | |
 | Lenalidomide, mThy1-α-syn transgenic mice | Behavioral deficits ↓, DA fiber loss ↓, microgliosis ↓, pro-inflammatory cytokines ↓, NF-κB activation ↓ | [167] | |
| Microglia inhibition |  | JWH133, MPTP, C57 BL/6 mice | BBB damage ↓, infiltration of peripheral immune cells ↓, iNOS ↓, pro-inflammatory cytokines ↓, microglial activation ↓ | [168] |
 | 1,25-dihydroxyvitamin D3, 6-OHDA, MPTP, SD rats, C57 BL/6 mice | Microglial activation ↓, pro-inflammatory cytokine expression ↓ | [169] | |
 | α-Lipoic acid, MPTP, C57 BL/6 mice | Step length and suspension time ↑, microglial activation ↓, NF-κB ↓, TNF-α ↓, iNOS ↓ | [170] | |
 | Montelukast, 6-OHDA, rotenone, C57 BL/6 mice, Wistar rats | Neuroinflammatory activities ↓, TNF-α ↓, IL-1β ↓, microglial activation ↓, p38 MAPK ↓, CysLT1 ↓ | [171,172] | |
 | Acacetin, MPTP, primary mesencephalic cells | DA neuron loss ↓, NO ↓, PGE2 ↓, TNF-α ↓, time of turning and locomotor activity ↓, dopamine level ↑, microglial activation ↓ | [173] | |
 | JNJ7777120, rotenone, SD rats | Microglial markers ↓, dopaminergic neuron degeneration ↓ | [174] | |
 | Minocycline, LPS, Wistar rats | IL-1α ↓, TNF-α ↓, OX-6 ↓, OX-42 ↓, 3-nitrotyrosine immunoreactivity ↓ | [176] | |
 | Artemisinin, MPTP, C57 mice | Improved behavioral symptoms, DA neurons ↑, microglial activation ↓ | [177] | |
 | Triptolide, LPS, BV2 cells, primary microglia | mGlu5 expression ↑, mGlu5 membrane localization ↑, NO ↓, iNOS ↓, TNF-α ↓, IL-1β ↓, IL-6 ↓, microglial activation ↓ | [179] | |
| NXP031, MPTP, C57 BL/6J mice | DA neuron loss ↓, motor impairment ↓, plasma ascorbic acid levels ↑, microglia activation-induced neuroinflammation ↓ | [180] | ||
 | Naringin, 6-OHDA, C57 BL/6 mice | mTORC1 activation ↑, microglial activation ↓, DA degeneration ↓ | [181] | |
 | Capsaicin, LPS, SD rats | DA neuron loss ↓, pro-inflammatory mediators ↓, shift the pro-inflammatory M1 microglia/macrophage population to an anti-inflammatory M2 state, iNOS ↓, arginase 1 ↑, peroxynitrate production ↓, ROS ↓, oxidative damage ↓ | [182] | |
 | Ginsenoside Rg1, MPTP, C57 BL/6J mice | TH-positive cells ↑, TNF-α ↓, IFN-γ ↓, IL-1β ↓, IL-6 ↓, microglial activation ↓, infiltration of CD3+ T cells into the SNpc region ↓ | [183] | |
| Vasoactive intestinal peptide, MPTP, 6-OHDA, α-synuclein, C57 BL/6J mice, SD rats | Microglial inflammatory responses ↓, robust nigrostriatal protection, increased neuronal sparing, astrogliosis ↓, inflammatory microglia ↓, DA neurons ↑, improved striatal densities | [186,187,188,189] | ||
| Astrocyte inhibition |  | Silibinin, MPTP, C57 BL/6 mice | Motor dysfunction ↓, DA neuron loss ↓, glial activation ↓, ERK and JNK phosphorylation ↓ | [190] |
| Bear bile powder, MPTP, C57 BL/6 mice | Dyskinesia ↓, tyrosine hydroxylase ↑, astrocyte hyperactivation ↓, glial fibrillary acidic protein ↓, COX2 ↓, iNOS ↓, TGR5 ↑, phosphorylation of protein kinase B ↓, NF-κB ↓ | [191] | ||
| NADPH oxidase inhibition |  | Diphenyleneiodonium, LPS, MPP+, rotenone, neuron–glia cultures | DA neurons ↑, NADPH oxidase activation ↓, superoxide production ↓ | [193] |
 | Taurine, paraquat and maneb, C57 BL/6J mice | Microglial M1 polarization ↓, gene expression levels of pro-inflammatory factors ↓, NADPH oxidase ↓, NF-κB ↓ | [194] | |
| PPARγ agonist |  | MHY908, MPTP, C57 BL/6 mice, SH-SY5Y neuroblastoma cells, primary astrocytes | Inflammatory response ↓, DA neuron loss ↓, motor deficit ↓, glial activation ↓, ROS ↓ | [195] |
 | Rosiglitazone, MPTP, C57 BL/6J mice | M2 anti-inflammatory microglia were stimulated and inflammatory microglia were inhibited | [196] | |
 | Pioglitazone, MPTP, rhesus monkeys, C57 BL/6 mice | DA neuron loss ↓, DA levels ↑, microglial activation ↓, NF-κB ↓ | [197,198] | |
| Targeting MAPK pathway |  | Doxycycline, 6-OHDA, LPS, C57BL/6 mice, primary microglial cell | DA neuron loss ↓, microglial activation marker IBA-1 ↓, ROS ↓, NO ↓, pro-inflammatory cytokines ↓, phosphorylation level of MAPK ↓ | [199,200] |
 | Vanillin, LPS, Wistar rats | Motor dysfunction ↓, DA neuron loss ↓, microglial activation ↓, iNOS ↓, COX-2 ↓, IL-1β ↓, IL-6 ↓, p-ERK1/2 ↓, p-p38 ↓ | [201] | |
 | SKF-86002, mice overexpressing α-synuclein | Neuroinflammation ↓, ameliorated synaptic, neurodegenerative, and motor behavioral deficits, α-synuclein accumulation ↓, p38α/β ↓ | [202] | |
| Targeting AMPK-dependent pathway |  | KMS-99220, LPS, BV2 cells | Phosphorylation of IκB ↓, nuclear translocation of NF-κB ↓, iNOS ↓, NO ↓ | [203] |
| Liraglutide, MPTP, C57 BL/6 mice | Iba1 and GFAP expression ↓, p-AMPK ↑, NF-κB ↓, neuroinflammation ↓ | [204] | ||
 | Dexmedetomidine, MPTP, C57BL/6J mice | Motor dysfunction ↓, mechanical allodynia ↓, thermal hyperalgesia ↓, astrocyte activation ↓, TNFα and IL-6 expression ↓, p-AMPK ↑, mTOR ↓, NF-κB ↑ | [205] | |
 | Indole-3-carbinol, rotenone, SD rats | Motor dysfunction ↓, striatal DA decrease ↓, weight loss ↓, TH expression reduction ↓, α-synuclein ↓, SIRT1-AMPK signaling pathway ↑ | [206] | |
| Healthy human fecal microbiota Transplantation, MPTP, C57BL/6J mice | Motor impairments ↓, DA neurodegeneration ↓, glial activation ↓, colonic inflammation ↓, AMPK/SOD2 signaling pathway ↑ | [207] | ||
| Targeting α-synuclein | Anti-TLR2, α-synuclein-overexpressing transgenic mice | α-Synuclein accumulation ↓, neuroinflammation ↓, neurodegeneration ↓, behavioral deficits ↓ | [208] | |
| PD03A, PD01A, patients with PD | A good safety and tolerability profile, humoral immune response against the α-synuclein target epitope | [209,210] | ||
 | Baicalein, MPP+, SD rats | Dopamine ↑, α-synuclein aggregates ↓, ED-1 ↓, caspase-1 ↓, IL-1β ↓, cathepsin B ↓ | [211] | |
| PRX002, human | Favorable safety, tolerability, and pharmacokinetic profiles at all doses tested, with no immunogenicity, serum α-synuclein ↓ | [247] | ||
| Targeting miRNAs | miR-7, MPTP, α-synuclein PFF or PBS injection, C57BL/6J mice | α-Synuclein ↓, oxidative stress ↓, less pronounced degeneration of the nigrostriatal pathway, better behavioral performance, neuroinflammatory reaction ↓, NLRP3 inflammasome activation ↓, DA neuron degeneration ↓, microglial activation ↓ | [212,213,214] | |
 | Rosmarinic acid, MPTP, C57BL/6 mice | Motor function improvement, inflammatory responses ↓ | [215] | |
| MicroRNA-3473b inhibitor, LPS, MPTP, BV2 cells, C57BL/6J mice | Secretion of inflammatory factors ↓, autophagy ↑, microglial activation ↓, TREM2/ULK1 ↑ | [216] | ||
| Acupuncture | Acupuncture, MPTP, C57BL/6 mice | MAC-1 ↓, COX-2 ↓, iNOS ↓, motor function improvement, comorbid anxiety ↓, DA fibers and neurons ↓, activation of microglia and astrocyte ↓, conversion of Bax and Bcl-2 expression, physiology function restoration, neuroinflammation ↓ | [217,218] | |
| Exercise | Exercise, human, MPTP, C57BL/6 mice | Dendritic spine density ↑, arborization ↑, synaptic protein PSD-95 ↑, synaptophysin ↑, cognition improvement, motor function restoration, pro-inflammatory cytokine ↓, α-synuclein ↓, TLR2 activation ↓, autophagy ↑, mitochondrial function improvement, Sirt1 pathway ↑, DA neuron loss ↓, apoptosis ↓, mitochondrial biogenesis ↑, oxidative stress ↓, PGC-1α ↑ | [219,220,221,222,223] | |
| Treadmill exercise, endurance exercise, ICR mice, albino mice | DA neuron loss ↓, motor balance ↑, coordination dysfunction ↓, iNOS ↓, MAPKs ↓, microglial activation ↓, nNOS ↓ | [224,225] | ||
| Exercise, MPTP, C57BL/6 mice | Motor deficits ↓, neurogenesis ↑, DA neuron loss ↓, antioxidant capacity ↑, autophagy ↑ | [227] | ||
| Others | Anti-TNF, human | Incidence of PD in patients with IBD ↓ | [228] | |
| Exendin, C57 BL/6 mice, MPTP | Microglial activation ↓, MMP3 ↓, pro-inflammatory molecules ↓ | [229,230] | ||
 | Melatonin, C57 BL/6 mice, MPTP | COX-2 activity ↓, lipid peroxides ↓, nitrite/nitrate ↓ | [232] | |
 | Cyclosporin, SD rats, AAV-α-synuclein | Motor performance ↑, NFATc3 ↓, mitochondrial stress ↓ | [233] | |
| Probiotics, patients with PD | Abdominal pain ↓, bloating ↓, bradykinesia ↓, rigidity ↓, tremor ↓, gait dysfunction ↓, inflammation ↓ | [235,236] | ||
| Nonsteroidal anti-inflammatory drugs, patients with PD | PD risk ↓ | [237] | ||
 | Ibuprofen, C57 BL mice, human, MPTP | Dopamine level ↑, ROS ↓, COX inhibition, PD risk ↓ | [238,248] | |
 | Indomethacin, C57 BL mice, MPTP | Microglial activation ↓, lymphocytic infiltration ↓ | [239] | |
 | Polymannuronic acid, C57 BL/6J mice, MPTP | Improved motor functions, DA neuronal loss ↓, HVA ↑, 5-HT ↑, 5-HIAA ↑, GABA ↑, pro-inflammatory cytokines ↓, MAPK ↓ | [240] | |
 | Resveratrol, Wistar albino rats, SD rats, 6-OHDA, rotenone | ER stress ↓, CHOP ↓, GRP78 ↓, caspase-3 activity ↓, glutathione peroxidase ↑, Nrf2 ↑, ROS ↓, COX-2 ↓, PLA2 ↓, TNF-α ↓, neuroinflammation ↓ | [241,242] | |
| Curcuminoids, C57 BL/6 mice, MPTP | Dopamine depletion ↓, iNOS ↓, GFAP ↓, pro-inflammatory cytokine ↓, total nitrite generation ↓, motor performance improvement, gross behavioral activity improvement | [243] | ||
 | EGCG, Wistar rats, C57 BL/6 mice, Lewis rats, rotenone, MPTP | Motor impairments ↓, NO level ↓, LPO ↓, SDH activity ↑, ATPase activity ↑, ETC enzyme activity ↑, catecholamines ↑, neuroinflammatory and apoptotic markers ↓, mitochondrial dysfunction ↓, neurochemical deficiency ↓, TH activity ↑ | [242,244] | |
 | Carvacrol, Wistar rats, 6-OHDA | TNF-α ↓, IL-1β ↓, DA neuron loss ↓ | [245] | |
| DNL151, human | Delay the worsening of pathology for patients with early-stage PD | [249] | ||
| Sargramostim, human | PD improvement | [250] | ||
| Exenatide, human | Exenatide suppress motor impairments and improves cognitive function of patients with PD | [251] | ||
 | Rifaximin, human, transgenic PD mice (MitoPark) | Relative abundance of Flavonifractor ↑, anti-inflammatory effect ↑ | [252] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, J.; Tao, K.; Wang, W.; Wang, X. What Can Inflammation Tell Us about Therapeutic Strategies for Parkinson’s Disease? Int. J. Mol. Sci. 2024, 25, 1641. https://doi.org/10.3390/ijms25031641
Xue J, Tao K, Wang W, Wang X. What Can Inflammation Tell Us about Therapeutic Strategies for Parkinson’s Disease? International Journal of Molecular Sciences. 2024; 25(3):1641. https://doi.org/10.3390/ijms25031641
Chicago/Turabian StyleXue, Jinsong, Keju Tao, Weijia Wang, and Xiaofei Wang. 2024. "What Can Inflammation Tell Us about Therapeutic Strategies for Parkinson’s Disease?" International Journal of Molecular Sciences 25, no. 3: 1641. https://doi.org/10.3390/ijms25031641