
Deletion of Smooth Muscle O-GlcNAc Transferase Prevents Development of Atherosclerosis in Western Diet-Fed Hyperglycemic ApoE-/- Mice In Vivo
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Validation of Inducible SMC-Specific OGT Knockout Mice
2.2. Metabolic Phenotype of SMC-Specific OGT Knockout Mice under Basal and STZ-Induced Hyperglycemia
2.3. Reduced O-GlcNAc Protein Expression Increases SM Contractile Marker Expression in the Aortic Vasculature of STZ-Induced Hyperglycemic Mice with SMC-Specific OGT Deficiency
2.4. Effect of SMC-Specific OGT Deletion on Glycemic Index and Plasma Lipid Levels in Western Diet-Fed ApoE-/- Mice In Vivo
2.5. SMC-Specific OGT Deficiency Impedes Lesion Burden in Western Diet-Fed Hyperglycemic ApoE-/- Mice
2.6. SMC-Specific OGT Deficiency Abrogates Inflammatory and Smooth Muscle Cell Lesion Abundance in Western Diet-Fed Hyperglycemic ApoE-/- Mice
2.7. Lack of SMC-Specific OGT Increases SM Contractile Marker Expression While Reducing Inflammatory and Proliferative Marker Expression in Western Diet-Fed ApoE-/- Aortic Vasculature
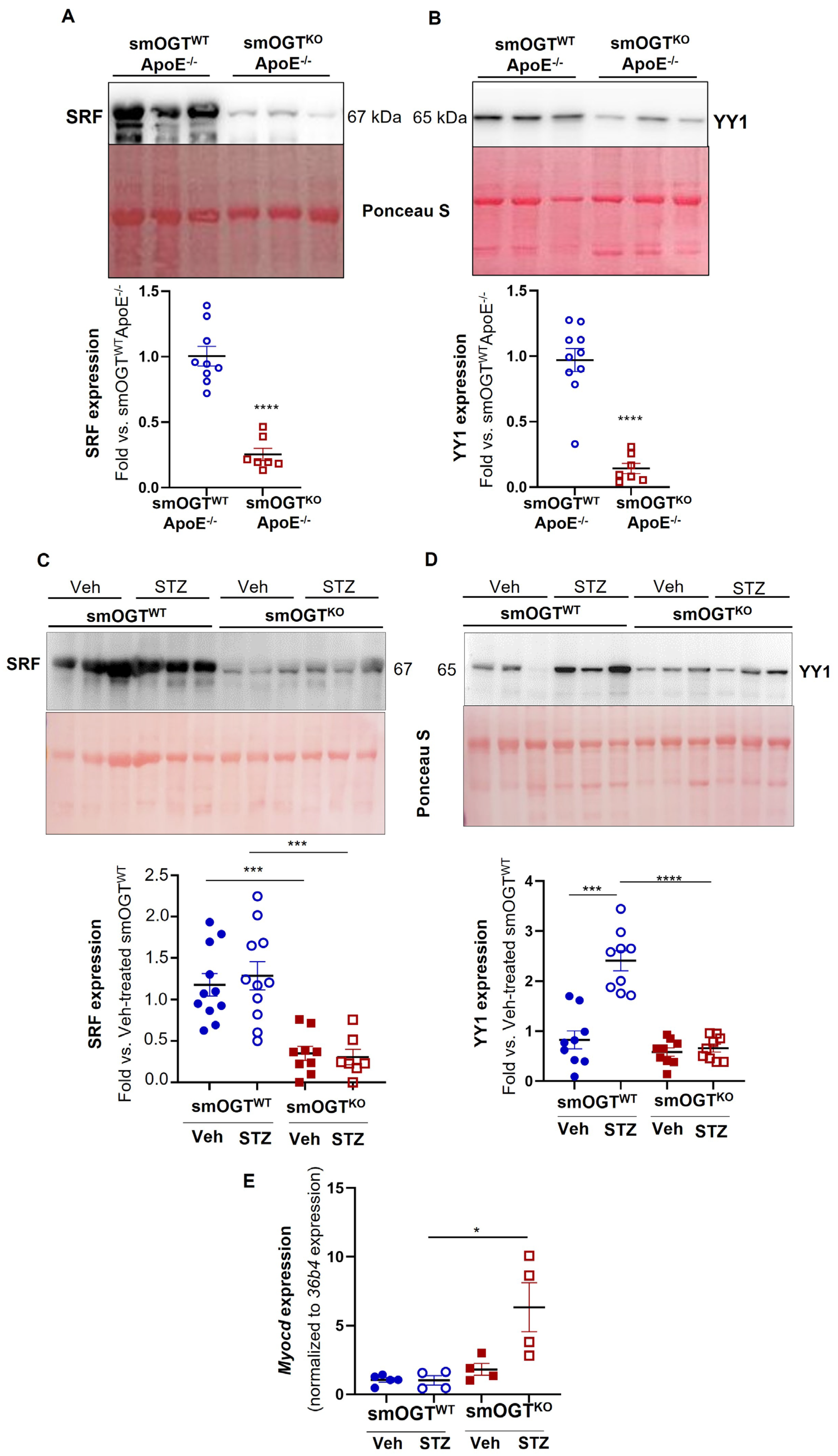
2.8. SMC-Specific OGT Deletion Attenuates YY1 and SRF Expression in Aortic Vessels of Hyperglycemic Mice In Vivo
3. Discussion
4. Materials and Methods
4.1. Mouse Models
4.2. Study Design
4.3. Plasma Lipid Analyses
4.4. Glucose Tolerance Test
4.5. Echocardiographic Analysis of Cardiac Function
4.6. Metabolic Phenotyping
4.7. Aortic Root Morphometry
4.8. Immunohistochemistry
4.9. Immunoblotting and Quantitative Real Time PCR
4.10. Image Quantification
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Alla, V.M.; Agrawal, V.; DeNazareth, A.; Mohiuddin, S.; Ravilla, S.; Rendell, M. A Reappraisal of the Risks and Benefits of Treating to Target with Cholesterol Lowering Drugs. Drugs 2013, 73, 1025–1054. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Tett, S.; Peeters, G.M.E.E.; Mishra, G.D.; Dobson, A. New-Onset Diabetes After Statin Exposure in Elderly Women: The Australian Longitudinal Study on Women’s Health. Drugs Aging 2017, 34, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H. Cardiovascular events and atherosclerosis in patients with type 2 diabetes and impaired glucose tolerance: What are the medical treatments to prevent cardiovascular events in such patients? J. Diabetes Investig. 2022, 13, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; Bornfeldt, K.E. Diabetes and atherosclerosis: Is there a role for hyperglycemia? J. Lipid Res. 2009, 50, S335–S339. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.-S.; et al. Hyperglycemia Promotes Myelopoiesis and Impairs the Resolution of Atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef]
- Kanter, J.E.; Johansson, F.; LeBoeuf, R.C.; Bornfeldt, K.E. Do glucose and lipids exert independent effects on atherosclerotic lesion initiation or progression to advanced plaques? Circ. Res. 2007, 100, 769–781. [Google Scholar] [CrossRef]
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 8, 2545–2559. [Google Scholar] [CrossRef]
- Nathan, D.M.; Lachin, J.; Cleary, P.; Orchard, T.; Brillon, D.J.; Backlund, J.-Y.; O’Leary, D.H.; Genuth, S.; Trial, D.C.A.C.; Epidemiology of Diabetes Interventions and Complications Research Group. Intensive Diabetes Therapy and Carotid Intima–Media Thickness in Type 1 Diabetes Mellitus. New Engl. J. Med. 2003, 348, 2294–2303. [Google Scholar] [CrossRef]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.-Y.C.; Genuth, S.M.; Lachin, J.; Orchard, T.; Raskin, P.; Zinman, B.; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive Diabetes Treatment and Cardiovascular Disease in Patients with Type 1 Diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [CrossRef]
- Faries, P.L.; Rohan, D.I.; Takahara, H.; Wyers, M.C.; Contreras, M.A.; Quist, W.C.; King, G.L.; LoGerfo, F.W. Human vascular smooth muscle cells of diabetic origin exhibit increased proliferation, adhesion, and migration. J. Vasc. Surg. 2001, 33, 601–607. [Google Scholar] [CrossRef]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef]
- Miano, J.M.; Fisher, E.A.; Majesky, M.W. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021, 143, 2110–2116. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.-L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef]
- Zachara, N.E.; Hart, G.W. Cell signaling, the essential role of O-GlcNAc! Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2006, 1761, 599–617. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Gonzalez-Rellan, M.J.; Fondevila, M.F.; Dieguez, C.; Nogueiras, R. O-GlcNAcylation: A Sweet Hub in the Regulation of Glucose Metabolism in Health and Disease. Front. Endocrinol. 2022, 13. [Google Scholar] [CrossRef]
- Hardivillé, S.; Hart, G.W. Nutrient Regulation of Signaling, Transcription, and Cell Physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef]
- Harwood, K.R.; Hanover, J.A. Nutrient-driven O-GlcNAc cycling—Think globally but act locally. J. Cell Sci. 2014, 127, 1857–1867. [Google Scholar] [CrossRef]
- Masaki, N.; Feng, B.; Bretón-Romero, R.; Inagaki, E.; Weisbrod, R.M.; Fetterman, J.L.; Hamburg, N.M. O-GlcNAcylation Mediates Glucose-Induced Alterations in Endothelial Cell Phenotype in Human Diabetes Mellitus. J. Am. Heart Assoc. 2020, 9, e014046. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, R.; Sahu, S.; Chavez, R.J.; Raman, P. Trivalent chromium inhibits TSP-1 expression, proliferation, and O-GlcNAc signaling in vascular smooth muscle cells in response to high glucose in vitro. Am. J. Physiol. Physiol. 2015, 308, C111–C122. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.B.; Hart, G.W. New insights: A role for O-GlcNAcylation in diabetic complications. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Prakoso, D.; Lim, S.Y.; Erickson, J.R.; Wallace, R.S.; Lees, J.G.; Tate, M.; Kiriazis, H.; Donner, D.G.; Henstridge, D.C.; Davey, J.R.; et al. Fine-tuning the cardiac O-GlcNAcylation regulatory enzymes governs the functional and structural phenotype of the diabetic heart. Cardiovasc. Res. 2021, 118, 212–225. [Google Scholar] [CrossRef]
- Heath, J.M.; Sun, Y.; Yuan, K.; Bradley, W.E.; Litovsky, S.; Dell’italia, L.J.; Chatham, J.C.; Wu, H.; Chen, Y. Activation of AKT by O-Linked N-Acetylglucosamine Induces Vascular Calcification in Diabetes Mellitus. Circ. Res. 2014, 114, 1094–1102. [Google Scholar] [CrossRef]
- Fricovsky, E.S.; Suarez, J.; Ihm, S.H.; Scott, B.; Suarez-Ramirez, J.A.; Banerjee, I.; Torres-Gonzalez, M.; Wang, H.; Ellrott, I.; Maya-Ramos, L.; et al. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am. J. Physiol. Integr. Comp. Physiol. 2012, 303, R689–R699. [Google Scholar] [CrossRef]
- Weigert, C.; Friess, U.; Brodbeck, K.; Schleicher, E.D. Glutamine:fructose-6-phosphate aminotransferase enzyme activity is necessary for the induction of TGF-?1 and fibronectin expression in mesangial cells. Diabetologia 2003, 46, 852–855. [Google Scholar] [CrossRef]
- James, L.R.; Fantus, I.G.; Goldberg, H.; Ly, H.; Scholey, J.W. Overexpression of GFAT activates PAI-1 promoter in mesangial cells. Am. J. Physiol. Physiol. 2000, 279, F718–F727. [Google Scholar] [CrossRef]
- James, L.R.; Tang, D.; Ingram, A.; Ly, H.; Thai, K.; Cai, L.; Scholey, J.W. Flux through the hexosamine pathway is a determinant of nuclear factor kappaB- dependent promoter activation. Diabetes 2002, 51, 1146–1156. [Google Scholar] [CrossRef]
- Raman, P.; Krukovets, I.; Marinic, T.E.; Bornstein, P.; Stenina, O.I. Glycosylation Mediates Up-regulation of a Potent Antiangiogenic and Proatherogenic Protein, Thrombospondin-1, by Glucose in Vascular Smooth Muscle Cells. J. Biol. Chem. 2007, 282, 5704–5714. [Google Scholar] [CrossRef]
- Federici, M.; Menghini, R.; Mauriello, A.; Hribal, M.L.; Ferrelli, F.; Lauro, D.; Sbraccia, P.; Spagnoli, L.G.; Sesti, G.; Lauro, R. Insulin-Dependent Activation of Endothelial Nitric Oxide Synthase Is Impaired by O-Linked Glycosylation Modification of Signaling Proteins in Human Coronary Endothelial Cells. Circulation 2002, 106, 466–472. [Google Scholar] [CrossRef]
- Carr, J.J.; Register, T.C.; Hsu, F.-C.; Lohman, K.; Lenchik, L.; Bowden, D.W.; Langefeld, C.D.; Xu, J.; Rich, S.S.; Wagenknecht, L.E.; et al. Calcified atherosclerotic plaque and bone mineral density in type 2 diabetes: The diabetes heart study. Bone 2008, 42, 43–52. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wu, H. Metabolic Stress and Cardiovascular Disease in Diabetes Mellitus. Arter. Thromb. Vasc. Biol. 2019, 39, 1911–1924. [Google Scholar] [CrossRef]
- Ganguly, R.; Sahu, S.; Ohanyan, V.; Haney, R.; Chavez, R.J.; Shah, S.; Yalamanchili, S.; Raman, P. Oral chromium picolinate impedes hyperglycemia-induced atherosclerosis and inhibits proatherogenic protein TSP-1 expression in STZ-induced type 1 diabetic ApoE−/− mice. Sci. Rep. 2017, 7, srep45279. [Google Scholar] [CrossRef]
- Runager, K.; Bektas, M.; Berkowitz, P.; Rubenstein, D.S. Targeting O-Glycosyltransferase (OGT) to Promote Healing of Diabetic Skin Wounds. J. Biol. Chem. 2014, 289, 5462–5466. [Google Scholar] [CrossRef]
- Zhang, B.; Li, M.-D.; Yin, R.; Liu, Y.; Yang, Y.; Mitchell-Richards, K.A.; Nam, J.H.; Li, R.; Wang, L.; Iwakiri, Y.; et al. O-GlcNAc transferase suppresses necroptosis and liver fibrosis. J. Clin. Investig. 2019, 4, e127709. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, R.-Z.; Lian, S.; Liu, P.; Hu, Y.-J.; Shi, H.-Z.; Lv, H.-M.; Yang, Y.-Y.; Xu, B.; Li, S.-Z. O-GlcNAcylation: The “stress and nutrition receptor” in cell stress response. Cell Stress Chaperones 2020, 26, 297–309. [Google Scholar] [CrossRef]
- Ng, Y.H.; Okolo, C.A.; Erickson, J.R.; Baldi, J.C.; Jones, P.P. Protein O-GlcNAcylation in the heart. Acta Physiol. 2021, 233, e13696. [Google Scholar] [CrossRef]
- Ramirez-Correa, G.A.; Ma, J.; Slawson, C.; Zeidan, Q.; Lugo-Fagundo, N.S.; Xu, M.; Shen, X.; Gao, W.D.; Caceres, V.; Chakir, K.; et al. Removal of Abnormal Myofilament O-GlcNAcylation Restores Ca2+ Sensitivity in Diabetic Cardiac Muscle. Diabetes 2015, 64, 3573–3587. [Google Scholar] [CrossRef]
- Wright, J.N.; Collins, H.E.; Wende, A.; Chatham, J.C. O-GlcNAcylation and cardiovascular disease. Biochem. Soc. Trans. 2017, 45, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.V.; Andreadou, I.; Hausenloy, D.J.; Bøtker, H.E. The Role of O-GlcNAcylation for Protection against Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2019, 20, 404. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.J.; Long, B.W.; DeMartino, A.M.; Brittian, K.R.; Readnower, R.D.; Brainard, R.E.; Cummins, T.D.; Annamalai, L.; Hill, B.G.; Jones, S.P. Cardiomyocyte Ogt is essential for postnatal viability. Am. J. Physiol. Circ. Physiol. 2014, 306, H142–H153. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Saudek, C.D.; Hart, G.W. Increased Expression of β-N-Acetylglucosaminidase in Erythrocytes From Individuals With Pre-diabetes and Diabetes. Diabetes 2010, 59, 1845–1850. [Google Scholar] [CrossRef]
- Lehman, D.M.; Fu, D.J.; Freeman, A.B.; Hunt, K.J.; Leach, R.J.; Johnson-Pais, T.; Hamlington, J.; Dyer, T.D.; Arya, R.; Abboud, H.; et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes 2005, 54, 1214–1221. [Google Scholar] [CrossRef]
- Phillips, J.W.; Barringhaus, K.G.; Sanders, J.M.; Yang, Z.; Chen, M.; Hesselbacher, S.; Czarnik, A.C.; Ley, K.; Nadler, J.; Sarembock, I.J. Rosiglitazone Reduces the Accelerated Neointima Formation After Arterial Injury in a Mouse Injury Model of Type 2 Diabetes. Circulation 2003, 108, 1994–1999. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.; Chai, W.; Chen, M.-H.; Liu, Z.; Shi, W. Hyperglycemia in apolipoprotein E-deficient mouse strains with different atherosclerosis susceptibility. Cardiovasc. Diabetol. 2011, 10, 117. [Google Scholar] [CrossRef]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef]
- Leslie, K.; Xie, Y.; Ostriker, A.; Martin, K. Abstract 176: O-GlcNAc Transferase Promotes Vascular Smooth Muscle Cell De-differentiation. Arter. Thromb. Vasc. Biol. 2016, 36. [Google Scholar] [CrossRef]
- Bauer, A.J.; Leslie, K.L.; Ostriker, A.C.; A Martin, K. Abstract 459: O-Linked N-Acetylglucosamine Promotes Vascular Smooth Muscle Cell Dedifferentiation and Intimal Hyperplasia. Arter. Thromb. Vasc. Biol. 2017, 37. [Google Scholar] [CrossRef]
- Xu, D.; Wang, W.; Bian, T.; Yang, W.; Shao, M.; Yang, H. Increased expression of O-GlcNAc transferase (OGT) is a biomarker for poor prognosis and allows tumorigenesis and invasion in colon cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 1305–1314. [Google Scholar]
- Barnes, J.W.; Tian, L.; Heresi, G.A.; Farver, C.F.; Asosingh, K.; Comhair, S.A.A.; Aulak, K.S.; Dweik, R.A. O -Linked β- N -Acetylglucosamine Transferase Directs Cell Proliferation in Idiopathic Pulmonary Arterial Hypertension. Circulation 2015, 131, 1260–1268. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, L.; Qi, J.; Shan, H.; Yu, W.; Gu, Y. MAPK/ERK signaling pathway-induced hyper-O-GlcNAcylation enhances cancer malignancy. Mol. Cell. Biochem. 2015, 410, 101–110. [Google Scholar] [CrossRef]
- Fülöp, N.; Zhang, Z.; Marchase, R.B.; Chatham, J.C. Glucosamine cardioprotection in perfused rat hearts associated with increased O-linked N-acetylglucosamine protein modification and altered p38 activation. Am. J. Physiol. Circ. Physiol. 2007, 292, H2227–H2236. [Google Scholar] [CrossRef]
- Mack, C.P. Signaling Mechanisms That Regulate Smooth Muscle Cell Differentiation. Arter. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef]
- Muto, A.; Fitzgerald, T.N.; Pimiento, J.M.; Maloney, S.P.; Teso, D.; Paszkowiak, J.J.; Westvik, T.S.; Kudo, F.A.; Nishibe, T.; Dardik, A. Smooth muscle cell signal transduction: Implications of vascular biology for vascular surgeons. J. Vasc. Surg. 2007, 45, A15–A24. [Google Scholar] [CrossRef]
- Zhao, M.; Xiong, X.; Ren, K.; Xu, B.; Cheng, M.; Sahu, C.; Wu, K.; Nie, Y.; Huang, Z.; Blumberg, R.S.; et al. Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. EMBO Mol. Med. 2018, 10, e8736. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.-B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Miano, J.M. Myocardin in biology and disease. J. Biomed. Res. 2015, 29, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-P.; He, X.; Liu, F.; Yin, S.; Wu, S.; Yang, M.; Zhao, J.; Dai, X.; Jiang, H.; Yu, L.; et al. YY1 directly interacts with myocardin to repress the triad myocardin/SRF/CArG box-mediated smooth muscle gene transcription during smooth muscle phenotypic modulation. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.-Z.; Hockemeyer, D.; McAnally, J.; Nordheim, A.; Olson, E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004, 428, 185–189. [Google Scholar] [CrossRef]
- Liu, Y.; Sinha, S.; McDonald, O.G.; Shang, Y.; Hoofnagle, M.H.; Owens, G.K. Kruppel-like Factor 4 Abrogates Myocardin-induced Activation of Smooth Muscle Gene Expression. J. Biol. Chem. 2005, 280, 9719–9727. [Google Scholar] [CrossRef]
- Onuh, J.O.; Qiu, H. Serum response factor-cofactor interactions and their implications in disease. FEBS J. 2020, 288, 3120–3134. [Google Scholar] [CrossRef]
- Miano, J.M. Serum response factor: Toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 2003, 35, 577–593. [Google Scholar] [CrossRef]
- Werth, D.; Grassi, G.; Konjer, N.; Dapas, B.; Farra, R.; Giansante, C.; Kandolf, R.; Guarnieri, G.; Nordheim, A.; Heidenreich, O. Proliferation of human primary vascular smooth muscle cells depends on serum response factor. Eur. J. Cell Biol. 2010, 89, 216–224. [Google Scholar] [CrossRef]
- Kim, S.; Ip, H.S.; Lu, M.M.; Clendenin, C.; Parmacek, M.S. A Serum Response Factor-Dependent Transcriptional Regulatory Program Identifies Distinct Smooth Muscle Cell Sublineages. Mol. Cell. Biol. 1997, 17, 2266–2278. [Google Scholar] [CrossRef]
- Itoh, S.; Katoh, Y.; Konishi, H.; Takaya, N.; Kimura, T.; Periasamy, M.; Yamaguchi, H. Nitric Oxide Regulates Smooth-muscle-specific Myosin Heavy Chain Gene Expression at the Transcriptional Level—Possible Role of SRF and YY1 Through CArG Element. J. Mol. Cell. Cardiol. 2001, 33, 95–107. [Google Scholar] [CrossRef]
- Ellis, P.D.; Martin, K.M.; Rickman, C.; Metcalfe, J.C.; Kemp, P. Increased actin polymerization reduces the inhibition of serum response factor activity by Yin Yang 1. Biochem. J. 2002, 364, 547–554. [Google Scholar] [CrossRef]
- Santiago, F.S.; Lowe, H.C.; Bobryshev, Y.V.; Khachigian, L.M. Induction of the Transcriptional Repressor Yin Yang-1 by Vascular Cell Injury. J. Biol. Chem. 2001, 276, 41143–41149. [Google Scholar] [CrossRef]
- Yue, Y.; Lv, W.; Zhang, L.; Kang, W. MiR-147b influences vascular smooth muscle cell proliferation and migration via targeting YY1 and modulating Wnt/β-catenin activities. Acta Biochim. Biophys. Sin. 2018, 50, 905–913. [Google Scholar] [CrossRef]
- Warthi, G.; Faulkner, J.L.; Doja, J.; Ghanam, A.R.; Gao, P.; Yang, A.C.; Slivano, O.J.; Barris, C.T.; Kress, T.C.; Zawieja, S.D.; et al. Generation and comparative analysis of an Itga8-CreERT2 mouse with preferential activity in vascular smooth muscle cells. Nat. Cardiovasc. Res. 2022, 1, 1084–1100. [Google Scholar] [CrossRef]
- Ohanyan, V.; Yin, L.; Bardakjian, R.; Kolz, C.; Enrick, M.; Hakobyan, T.; Kmetz, J.; Bratz, I.; Luli, J.; Nagane, M.; et al. Requisite Role of Kv1.5 Channels in Coronary Metabolic Dilation. Circ. Res. 2015, 117, 612–621. [Google Scholar] [CrossRef]
- Ganguly, R.; Khanal, S.; Mathias, A.; Gupta, S.; Lallo, J.; Sahu, S.; Ohanyan, V.; Patel, A.; Storm, K.; Datta, S.; et al. TSP-1 (Thrombospondin-1) Deficiency Protects ApoE −/− Mice Against Leptin-Induced Atherosclerosis. Arter. Thromb. Vasc. Biol. 2021, 41, e112–e127. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khanal, S.; Bhavnani, N.; Mathias, A.; Lallo, J.; Gupta, S.; Ohanyan, V.; Ferrell, J.M.; Raman, P. Deletion of Smooth Muscle O-GlcNAc Transferase Prevents Development of Atherosclerosis in Western Diet-Fed Hyperglycemic ApoE-/- Mice In Vivo. Int. J. Mol. Sci. 2023, 24, 7899. https://doi.org/10.3390/ijms24097899
Khanal S, Bhavnani N, Mathias A, Lallo J, Gupta S, Ohanyan V, Ferrell JM, Raman P. Deletion of Smooth Muscle O-GlcNAc Transferase Prevents Development of Atherosclerosis in Western Diet-Fed Hyperglycemic ApoE-/- Mice In Vivo. International Journal of Molecular Sciences. 2023; 24(9):7899. https://doi.org/10.3390/ijms24097899
Chicago/Turabian StyleKhanal, Saugat, Neha Bhavnani, Amy Mathias, Jason Lallo, Shreya Gupta, Vahagn Ohanyan, Jessica M. Ferrell, and Priya Raman. 2023. "Deletion of Smooth Muscle O-GlcNAc Transferase Prevents Development of Atherosclerosis in Western Diet-Fed Hyperglycemic ApoE-/- Mice In Vivo" International Journal of Molecular Sciences 24, no. 9: 7899. https://doi.org/10.3390/ijms24097899