

Mechanisms of Oxidative Stress in Metabolic Syndrome



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Metabolic Syndrome Components
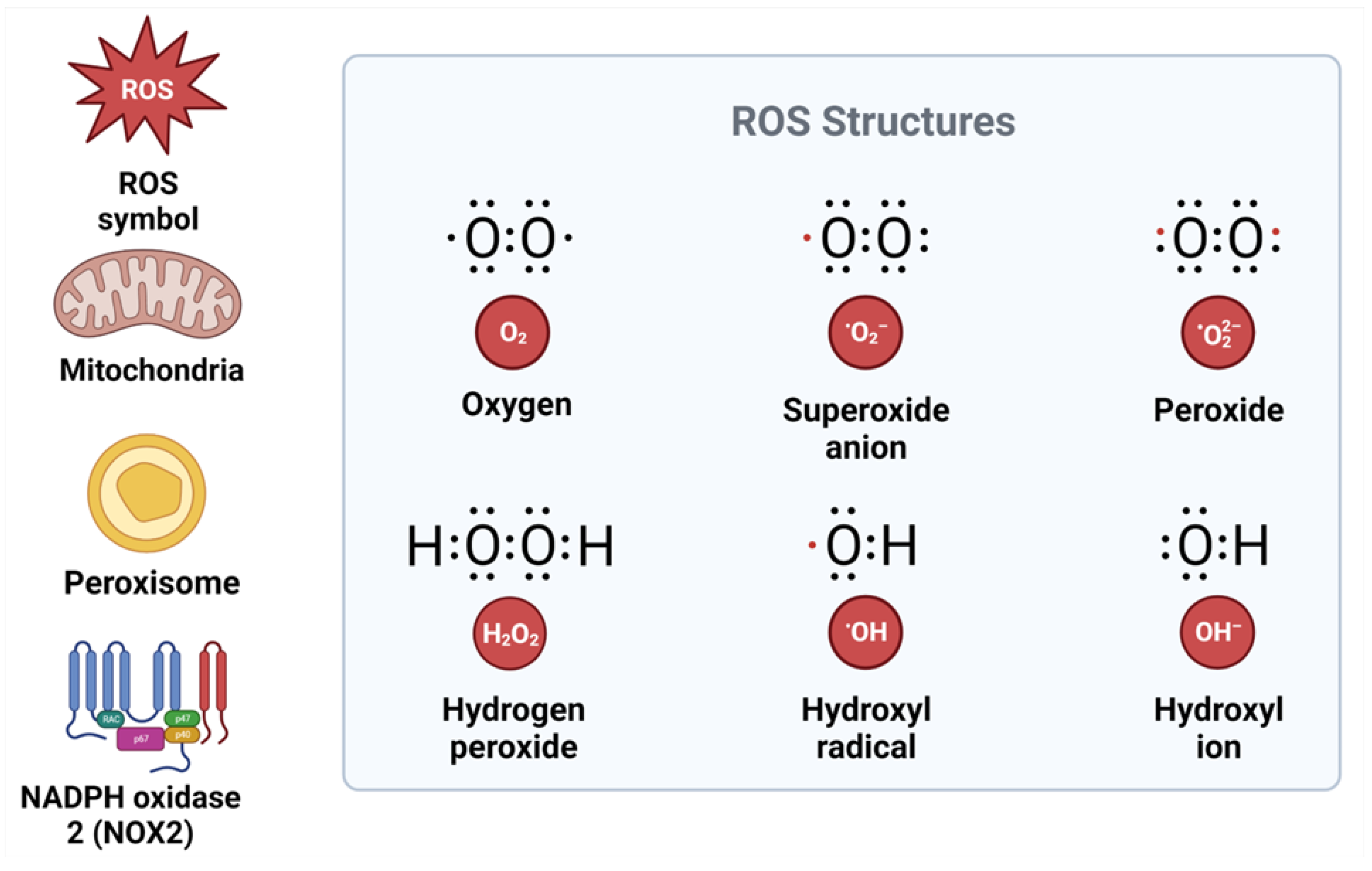
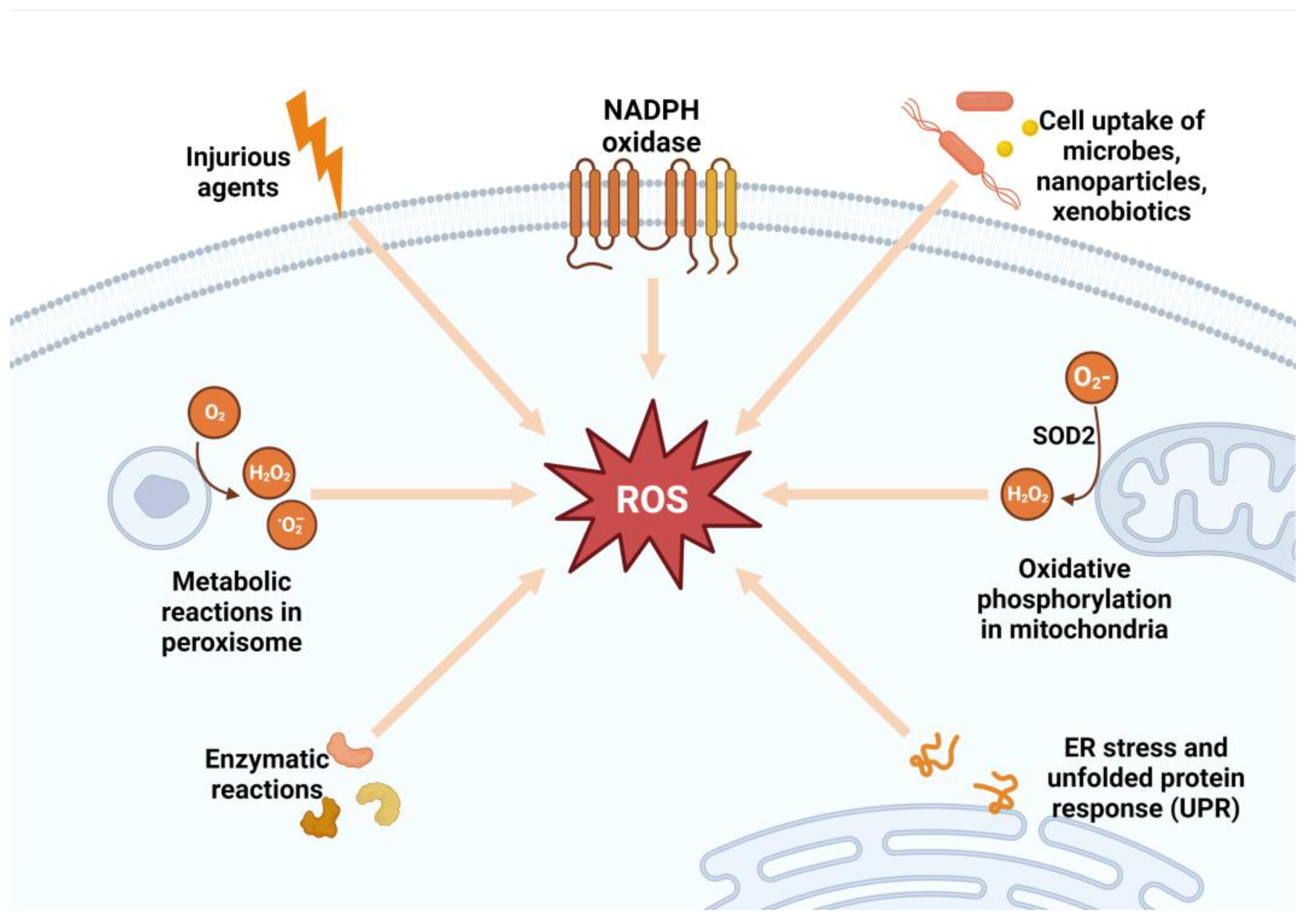
3. Mechanisms of Reactive Oxygen Species and Their Role in the Development and Progression of Metabolic Syndrome
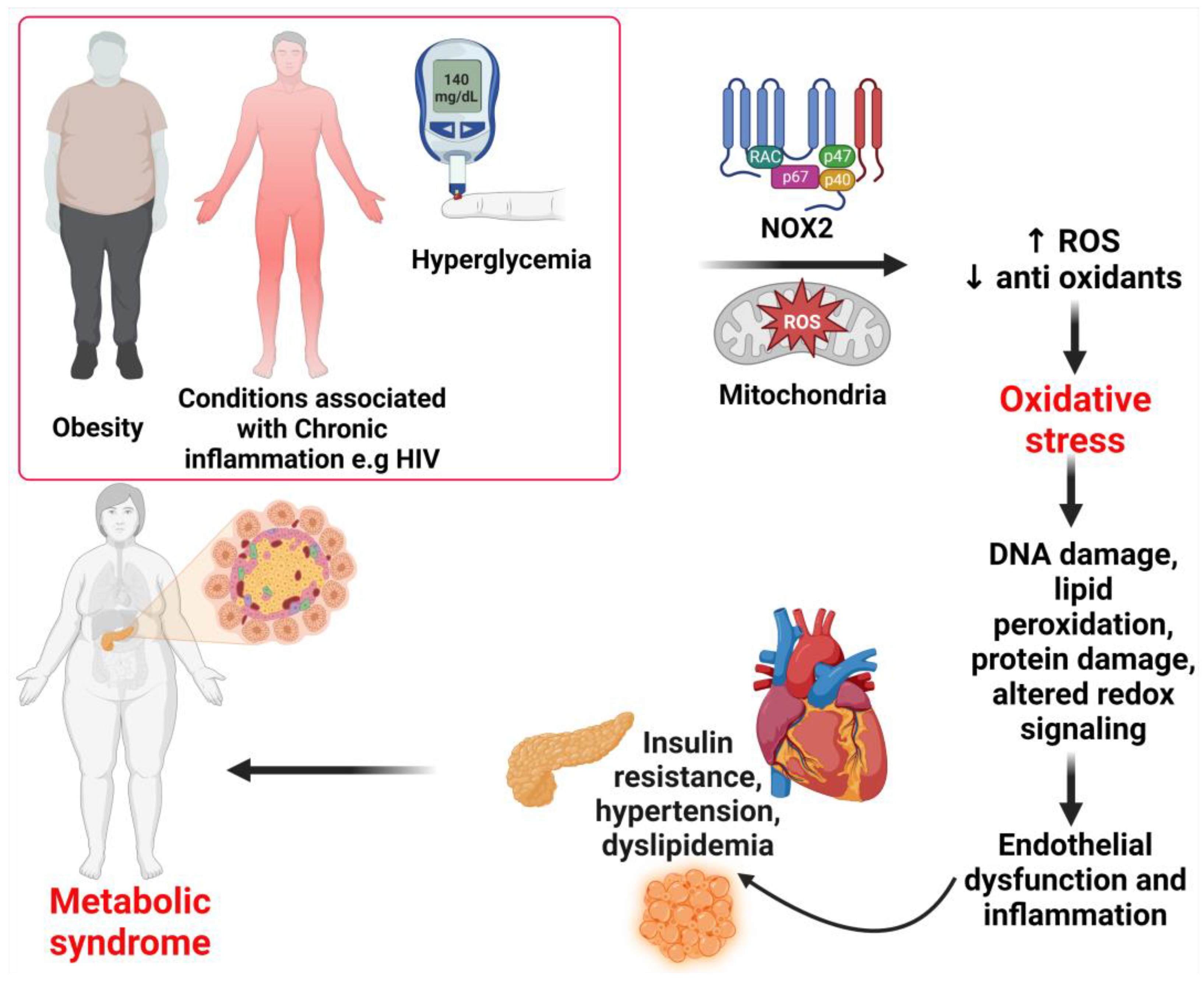
4. Mechanisms of Oxidative Stress Associated with Abdominal Obesity
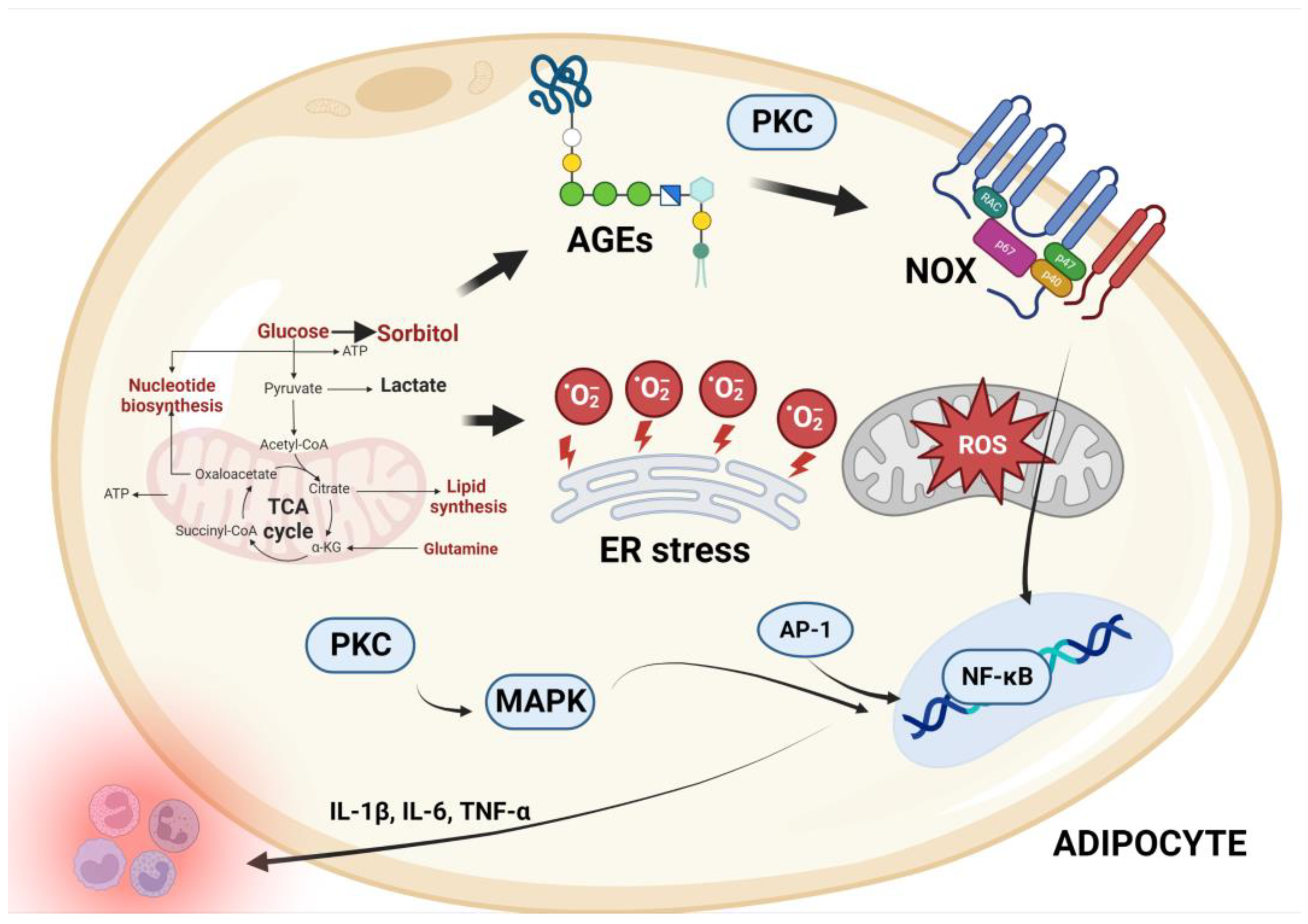
4.1. Inflammation and Free Radical Production via Several Pathways
4.2. Adipokines
4.3. Food Intake
5. Mechanisms of Oxidative Stress Associated with Abnormal Lipogram Levels
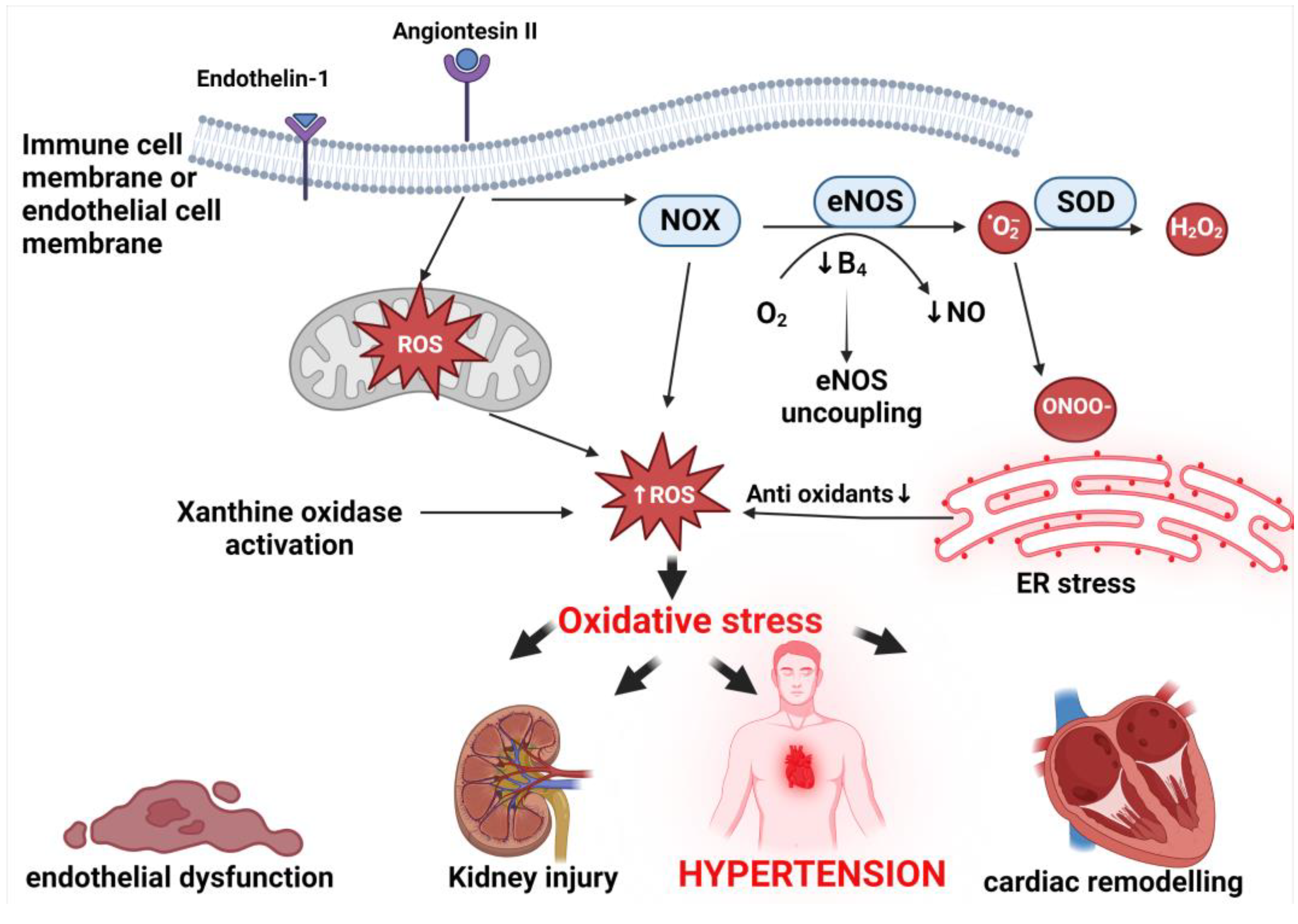
6. Mechanisms of Oxidative Stress Associated with Hypertension
6.1. Endoplasmic Reticulum
6.2. Mitochondrial Oxidative Stress
6.3. Nitric Oxide Synthase Uncoupling
7. Mechanisms of Oxidative Stress Associated with Impaired Fasting Glucose and Insulin Resistance
7.1. Lipid-Induced Insulin Resistance
7.2. Mitochondrial Dysfunction
7.3. Low-Grade Inflammation
7.4. Glucose Transporters
8. Immune Activation Mechanisms of Oxidative Stress in Metabolic Syndrome
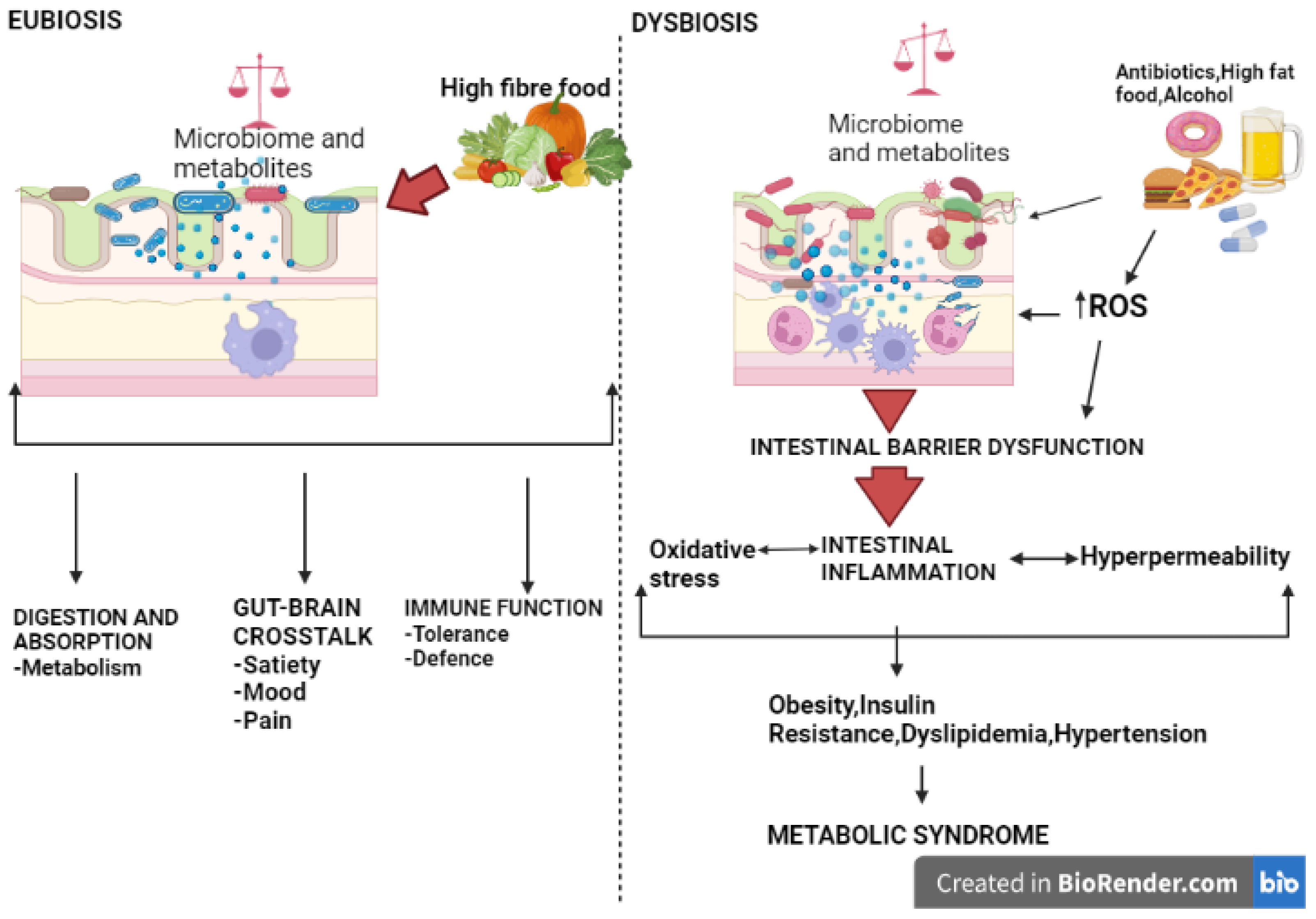
9. Gut Microbiota, Oxidative Stress, and Metabolic Syndrome
10. Comorbidities Associated with Risk for Metabolic Syndrome
Metabolic Syndrome and Cardiovascular Disease Risk
11. Biomarkers of Oxidative Stress in Metabolic Syndrome
12. Targeted Therapeutic Strategies for Metabolic Disease
13. Conclusions
14. What Is Known
- Oxidative stress plays a role in metabolic derangements in obesity, diabetes, and cardiovascular pathogenesis;
- Biomarkers and molecular targets may help us develop innovative methods for preventing, diagnosing, and treating inflammatory and metabolic disorders;
- Antioxidants can be used as a preventative or therapeutic treatment for metabolic diseases.
15. What Is New
- Mitochondrial oxidative stress and dysfunction may be the primary causes of oxidative damage and metabolic abnormalities in metabolic syndrome;
- Several signaling pathways involving NF-kB, PKC, MAPK, polyol, JNK, ERK, and NOX are activated to induce metabolic syndrome and multiple organ damage;
- Adiposity plays a vital role in inducing oxidative stress that results in endothelial dysfunction, cardiovascular remodeling, and hypertension;
- Components of the Mediterranean diet, such as polyphenols found in olives, can lower oxidative stress and reduce the risk of the development of metabolic syndrome.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic Syndrome: Pathophysiology, Management, and Modulation by Natural Compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225. [Google Scholar] [CrossRef]
- Gallardo-Alfaro, L.; Bibiloni, M.d.M.; Mascaró, C.M.; Montemayor, S.; Ruiz-Canela, M.; Salas-Salvadó, J.; Corella, D.; Fitó, M.; Romaguera, D.; Vioque, J.; et al. Leisure-Time Physical Activity, Sedentary Behaviour and Diet Quality Are Associated with Metabolic Syndrome Severity: The PREDIMED-Plus Study. Nutrients 2020, 12, 1013. [Google Scholar] [CrossRef]
- Wilkinson, M.J.; Manoogian, E.N.C.; Zadourian, A.; Lo, H.; Fakhouri, S.; Shoghi, A.; Wang, X.; Fleischer, J.G.; Navlakha, S.; Panda, S.; et al. Ten-Hour Time-Restricted Eating Reduces Weight, Blood Pressure, and Atherogenic Lipids in Patients with Metabolic Syndrome. Cell Metab. 2020, 31, 92–104.e5. [Google Scholar] [CrossRef] [PubMed]
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Capó, X.; Bouzas, C.; Mateos, D.; Pons, A.; Tur, J.A.; Sureda, A. Metabolic Syndrome Is Associated with Oxidative Stress and Proinflammatory State. Antioxidants 2020, 9, 236. [Google Scholar] [CrossRef]
- Dieli-Conwright, C.M.; Courneya, K.S.; Demark-Wahnefried, W.; Sami, N.; Lee, K.; Buchanan, T.A.; Spicer, D.V.; Tripathy, D.; Bernstein, L.; Mortimer, J.E. Effects of Aerobic and Resistance Exercise on Metabolic Syndrome, Sarcopenic Obesity, and Circulating Biomarkers in Overweight or Obese Survivors of Breast Cancer: A Randomized Controlled Trial. J. Clin. Oncol. 2018, 36, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 8267234. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.; Sciatti, E.; Favero, G.; Bonomini, F.; Vizzardi, E.; Rezzani, R. Essential Hypertension and Oxidative Stress: Novel Future Perspectives. Int. J. Mol. Sci. 2022, 23, 14489. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Touyz, R.M.; Rios, F.J.; Alves-Lopes, R.; Neves, K.B.; Camargo, L.L.; Montezano, A.C. Oxidative Stress: A Unifying Paradigm in Hypertension. Can. J. Cardiol. 2020, 36, 659. [Google Scholar] [CrossRef]
- Han, T.S.; Lean, M.E. A Clinical Perspective of Obesity, Metabolic Syndrome and Cardiovascular Disease. JRSM Cardiovasc. Dis. 2016, 5, 2048004016633371. [Google Scholar] [CrossRef] [PubMed]
- Todowede, O.O.; Sartorius, B. Prevalence of Metabolic Syndrome, Discrete or Comorbid Diabetes and Hypertension in Sub-Saharan Africa among People Living with HIV versus HIV-Negative Populations: A Systematic Review and Meta-Analysis Protocol. BMJ Open 2017, 7, e016602. [Google Scholar] [CrossRef]
- Kunduraci, Y.E.; Ozbek, H. Does the Energy Restriction Intermittent Fasting Diet Alleviate Metabolic Syndrome Biomarkers? A Randomized Controlled Trial. Nutrients 2020, 12, 3213. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Suh, Y.J. Association between Serum Uric Acid and Metabolic Syndrome in Koreans. J. Korean Med. Sci. 2019, 34, e307. [Google Scholar] [CrossRef]
- Thomas, M.S.; Huang, L.; Garcia, C.; Sakaki, J.R.; Blesso, C.N.; Chun, O.K.; Fernandez, M.L. The Effects of Eggs in a Plant-Based Diet on Oxidative Stress and Inflammation in Metabolic Syndrome. Nutrients 2022, 14, 2548. [Google Scholar] [CrossRef]
- Bhupathiraju, S.N.; Hu, F.B. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ. Res. 2016, 118, 1723–1735. [Google Scholar] [CrossRef]
- Grant, B.; Sandelson, M.; Agyemang-Prempeh, B.; Zalin, A. Managing Obesity in People with Type 2 Diabetes. Clin. Med. 2021, 21, e327–e331. [Google Scholar] [CrossRef]
- Schroder, J.D.; Falqueto, H.; Mânica, A.; Zanini, D.; de Oliveira, T.; de Sá, C.A.; Cardoso, A.M.; Manfredi, L.H. Effects of Time-Restricted Feeding in Weight Loss, Metabolic Syndrome and Cardiovascular Risk in Obese Women. J. Transl. Med. 2021, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Pedro-Botet, J.; Ascaso, J.F.; Barrios, V.; De la Sierra, A.; Escalada, J.; Millán, J.; Mostaza, J.M.; Pérez-Martínez, P.; Pintó, X.; Salas-Salvadó, J.; et al. COSMIC Project: Consensus on the Objectives of the Metabolic Syndrome in Clinic. Diabetes Metab. Syndr. Obes. Targets Ther. 2018, 11, 683–697. [Google Scholar] [CrossRef]
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Wolosowicz, M.; Prokopiuk, S.; Kaminski, T.W. Recent Advances in the Treatment of Insulin Resistance Targeting Molecular and Metabolic Pathways: Fighting a Losing Battle? Medicina 2022, 58, 472. [Google Scholar] [CrossRef]
- Kaur, J. A Comprehensive Review on Metabolic Syndrome. Cardiol. Res. Pract. 2014, 2014, 943162. [Google Scholar] [CrossRef]
- Marušić, M.; Paić, M.; Knobloch, M.; Liberati Pršo, A.-M. NAFLD, Insulin Resistance, and Diabetes Mellitus Type 2. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6613827. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, Y.K.; Kytikova, O.Y.; Novgorodtseva, T.P.; Antonyuk, M.V.; Gvozdenko, T.A.; Kantur, T.A. Lipid-Induced Mechanisms of Metabolic Syndrome. J. Obes. 2020, 2020, 5762395. [Google Scholar] [CrossRef] [PubMed]
- Otamas, A.; Grant, P.J.; Ajjan, R.A. Diabetes and Atherothrombosis: The Circadian Rhythm and Role of Melatonin in Vascular Protection. Diab. Vasc. Dis. Res. 2020, 17, 1479164120920582. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Samson, S.L.; Garber, A.J. Metabolic Syndrome. Endocrinol. Metab. Clin. N. Am. 2014, 43, 1–23. [Google Scholar] [CrossRef]
- Jakubczyk, K.; Dec, K.; Kałduńska, J.; Kawczuga, D.; Kochman, J.; Janda, K. Reactive Oxygen Species—Sources, Functions, Oxidative Damage. Pol. Merkur. Lek. Organ Pol. Tow. Lek. 2020, 48, 124–127. [Google Scholar]
- DeVallance, E.; Li, Y.; Jurczak, M.J.; Cifuentes-Pagano, E.; Pagano, P.J. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid. Redox Signal. 2019, 31, 687–709. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive Oxygen Species (ROS) Homeostasis and Redox Regulation in Cellular Signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, F.; Yue, S.; Chen, C.; Song, S.; Wang, H.; Zhao, M. Functions of Reactive Oxygen Species in Apoptosis and Ganoderic Acid Biosynthesis in Ganoderma Lucidum. FEMS Microbiol. Lett. 2019, 366, fnaa015. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of Cytochrome P450s in the Generation and Metabolism of Reactive Oxygen Species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10, 23–48. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Wang, S.-B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine Oxidative Post-Translational Modifications: Emerging Regulation in the Cardiovascular System. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.P.; Bhatnagar, A. Role of thiols in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Tomin, T.; Schittmayer, M.; Honeder, S.; Heininger, C.; Birner-Gruenberger, R. Irreversible Oxidative Post-Translational Modifications in Heart Disease. Expert Rev. Proteom. 2019, 16, 681–693. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, R.; Wang, Z. Contribution of Oxidative Stress to HIF-1-Mediated Profibrotic Changes during the Kidney Damage. Oxid. Med. Cell Longev. 2021, 2021, 6114132. [Google Scholar] [CrossRef] [PubMed]
- Padgett, L.E.; Broniowska, K.A.; Hansen, P.A.; Corbett, J.A.; Tse, H.M. The Role of Reactive Oxygen Species and Proinflammatory Cytokines in Type 1 Diabetes Pathogenesis. Ann. N. Y. Acad. Sci. 2013, 1281, 16–35. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Čolak, E.; Pap, D. The Role of Oxidative Stress in the Development of Obesity and Obesity-Related Metabolic Disorders. J. Med. Biochem. 2021, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid Peroxidation in Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Bekkouche, L.; Bouchenak, M.; Malaisse, W.J.; Yahia, D.A. The Mediterranean Diet Adoption Improves Metabolic, Oxidative, and Inflammatory Abnormalities in Algerian Metabolic Syndrome Patients. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Metab. 2014, 46, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Céspedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef]
- Mayoral, L.P.-C.; Andrade, G.M.; Mayoral, E.P.-C.; Huerta, T.H.; Canseco, S.P.; Rodal Canales, F.J.; Cabrera-Fuentes, H.A.; Cruz, M.M.; Pérez Santiago, A.D.; Alpuche, J.J.; et al. Obesity Subtypes, Related Biomarkers & Heterogeneity. Indian J. Med. Res. 2020, 151, 11–21. [Google Scholar] [CrossRef]
- Aras, M.; Tchang, B.G.; Pape, J. Obesity and Diabetes. Nurs. Clin. N. Am. 2021, 56, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Catani, M.V.; Evangelista, D.; Gasperi, V.; Avigliano, L. Obesity-Associated Oxidative Stress: Strategies Finalized to Improve Redox State. Int. J. Mol. Sci. 2013, 14, 10497–10538. [Google Scholar] [CrossRef] [PubMed]
- Nono Nankam, P.A.; Nguelefack, T.B.; Goedecke, J.H.; Blüher, M. Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry. Antioxidants 2021, 10, 622. [Google Scholar] [CrossRef]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, Á.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, Oxidative Stress, and Obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Huang, C.-J.; McAllister, M.J.; Slusher, A.L.; Webb, H.E.; Mock, J.T.; Acevedo, E.O. Obesity-Related Oxidative Stress: The Impact of Physical Activity and Diet Manipulation. Sports Med. Open 2015, 1, 32. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative Stress in Obesity: A Critical Component in Human Diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, S. Oxidative Stress, Insulin Resistance, Dyslipidemia and Type 2 Diabetes Mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef]
- Arnold, P.K.; Finley, L.W.S. Regulation and Function of the Mammalian Tricarboxylic Acid Cycle. J. Biol. Chem. 2022, 299, 102838. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Delehanty, J.B.; Das, S.; Goldberg, E.; Sangtani, A.; Knight, D.A. Synthesis of a Reactive Oxygen Species-Responsive Doxorubicin Derivative. Molecules 2018, 23, 1809. [Google Scholar] [CrossRef]
- Yan, L. Redox Imbalance Stress in Diabetes Mellitus: Role of the Polyol Pathway. Anim. Models Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Physiological and Pathological Roles of Aldose Reductase. Metabolites 2021, 11, 655. [Google Scholar] [CrossRef]
- Srikanth, K.K.; Orrick, J.A. Biochemistry, Polyol or Sorbitol Pathways. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Lai, S.W.T.; Lopez Gonzalez, E.D.J.; Zoukari, T.; Ki, P.; Shuck, S.C. Methylglyoxal and Its Adducts: Induction, Repair, and Association with Disease. Chem. Res. Toxicol. 2022, 35, 1720–1746. [Google Scholar] [CrossRef] [PubMed]
- Jubaidi, F.F.; Zainalabidin, S.; Taib, I.S.; Abdul Hamid, Z.; Mohamad Anuar, N.N.; Jalil, J.; Mohd Nor, N.A.; Budin, S.B. The Role of PKC-MAPK Signalling Pathways in the Development of Hyperglycemia-Induced Cardiovascular Complications. Int. J. Mol. Sci. 2022, 23, 8582. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.C. Plasma Free Fatty Acid Concentration as a Modifiable Risk Factor for Metabolic Disease. Nutrients 2021, 13, 2590. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Miwa, A.; Ohsaka, F.; Karatsu, Y.; Tsuruta, T.; Hino, S.; Morita, T.; Sonoyama, K. Local Free Fatty Acids Trigger the Expression of Lipopolysaccharide-Binding Protein in Murine White Adipose Tissue. Biosci. Microbiota Food Health 2022, 41, 54–65. [Google Scholar] [CrossRef]
- Shi, C.; Zhu, L.; Chen, X.; Gu, N.; Chen, L.; Zhu, L.; Yang, L.; Pang, L.; Guo, X.; Ji, C.; et al. IL-6 and TNF-α Induced Obesity-Related Inflammatory Response Through Transcriptional Regulation of MiR-146b. J. Interferon Cytokine Res. 2014, 34, 342–348. [Google Scholar] [CrossRef]
- Glass, C.K.; Olefsky, J.M. Inflammation and Lipid Signaling in the Etiology of Insulin Resistance. Cell Metab. 2012, 15, 635–645. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and Inflammation: The Linking Mechanism and the Complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Gutiérrez-Cuevas, J.; Galicia-Moreno, M.; Monroy-Ramírez, H.C.; Sandoval-Rodriguez, A.; García-Bañuelos, J.; Santos, A.; Armendariz-Borunda, J. The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants 2022, 11, 235. [Google Scholar] [CrossRef]
- Mohammadi, M.; Gozashti, M.H.; Aghadavood, M.; Mehdizadeh, M.R.; Hayatbakhsh, M.M. Clinical Significance of Serum IL-6 and TNF-α Levels in Patients with Metabolic Syndrome. Rep. Biochem. Mol. Biol. 2017, 6, 74. [Google Scholar]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Lukic, L.; Lalic, N.M.; Rajkovic, N.; Jotic, A.; Lalic, K.; Milicic, T.; Seferovic, J.P.; Macesic, M.; Stanarcic Gajovic, J. Hypertension in Obese Type 2 Diabetes Patients Is Associated with Increases in Insulin Resistance and IL-6 Cytokine Levels: Potential Targets for an Efficient Preventive Intervention. Int. J. Environ. Res. Public Health 2014, 11, 3586–3598. [Google Scholar] [CrossRef] [PubMed]
- Estienne, A.; Bongrani, A.; Reverchon, M.; Ramé, C.; Ducluzeau, P.-H.; Froment, P.; Dupont, J. Involvement of Novel Adipokines, Chemerin, Visfatin, Resistin and Apelin in Reproductive Functions in Normal and Pathological Conditions in Humans and Animal Models. Int. J. Mol. Sci. 2019, 20, 4431. [Google Scholar] [CrossRef] [PubMed]
- Recinella, L.; Orlando, G.; Ferrante, C.; Chiavaroli, A.; Brunetti, L.; Leone, S. Adipokines: New Potential Therapeutic Target for Obesity and Metabolic, Rheumatic, and Cardiovascular Diseases. Front. Physiol. 2020, 11, 578966. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.S. Direct and Indirect Effects of Leptin on Adipocyte Metabolism. Biochim. Biophys. Acta 2014, 1842, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Bełtowski, J.; Wójcicka, G.; Jamroz, A. Leptin Decreases Plasma Paraoxonase 1 (PON1) Activity and Induces Oxidative Stress: The Possible Novel Mechanism for Proatherogenic Effect of Chronic Hyperleptinemia. Atherosclerosis 2003, 170, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Praticó, D.; Savino, K.; Rokach, J.; Stahl, W.; Mecocci, P. Increased F2 Isoprostane Plasma Levels in Patients with Congestive Heart Failure Are Correlated with Antioxidant Status and Disease Severity. J. Card. Fail. 2004, 10, 334–338. [Google Scholar] [CrossRef]
- Esfahani, M.; Movahedian, A.; Baranchi, M.; Goodarzi, M.T. Adiponectin: An Adipokine with Protective Features against Metabolic Syndrome. Iran. J. Basic Med. Sci. 2015, 18, 430–442. [Google Scholar]
- AL-Suhaimi, E.A.; Shehzad, A. Leptin, Resistin and Visfatin: The Missing Link between Endocrine Metabolic Disorders and Immunity. Eur. J. Med. Res. 2013, 18, 12. [Google Scholar] [CrossRef]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef] [PubMed]
- Parvaresh Rizi, E.; Baig, S.; Shabeer, M.; Teo, Y.; Mok, S.F.; Loh, T.P.; Magkos, F.; Virtue, S.; Vidal-Puig, A.; Tai, E.S.; et al. Meal Rich in Carbohydrate, but Not Protein or Fat, Reveals Adverse Immunometabolic Responses Associated with Obesity. Nutr. J. 2016, 15, 100. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Castrejón-Téllez, V.; Soto, M.E.; Rubio-Ruiz, M.E.; Manzano-Pech, L.; Guarner-Lans, V. Oxidative Stress, Plant Natural Antioxidants, and Obesity. Int. J. Mol. Sci. 2021, 22, 1786. [Google Scholar] [CrossRef]
- Basu, D.; Goldberg, I.J. Regulation of Lipoprotein Lipase-Mediated Lipolysis of Triglycerides. Curr. Opin. Lipidol. 2020, 31, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Svecla, M.; Catapano, A.L.; Holleboom, A.G.; Norata, G.D. Impact of Protein Glycosylation on Lipoprotein Metabolism and Atherosclerosis. Cardiovasc. Res. 2021, 117, 1033–1045. [Google Scholar] [CrossRef]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive Oxygen Species (ROS) in Macrophage Activation and Function in Diabetes. Immunobiology 2019, 224, 242–253. [Google Scholar] [CrossRef]
- Masenga, S.K.; Elijovich, F.; Koethe, J.R.; Hamooya, B.M.; Heimburger, D.C.; Munsaka, S.M.; Laffer, C.L.; Kirabo, A. Hypertension and Metabolic Syndrome in Persons with HIV. Curr. Hypertens. Rep. 2020, 22, 78. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E.D.; Dustan, H.P.; Bumpus, F.M. Irvine, H. Page: 1901–1991—The Celebration of a Leader. Hypertension 1991, 18, 443–445. [Google Scholar] [CrossRef]
- Harrison, D.G. The Mosaic Theory Revisited: Common Molecular Mechanisms Coordinating Diverse Organ and Cellular Events in Hypertension. J. Am. Soc. Hypertens. 2013, 7, 68–74. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. The Discovery of Nitric Oxide and Its Role in Vascular Biology. Br. J. Pharmacol. 2006, 147, S193–S201. [Google Scholar] [CrossRef] [PubMed]
- Renna, N.F. Oxidative Stress, Vascular Remodeling, and Vascular Inflammation in Hypertension. Int. J. Hypertens. 2013, 2013, 710136. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Camargo, L.L.; Rios, F.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993. [Google Scholar] [CrossRef]
- Tain, Y.-L.; Hsu, C.-N. Oxidative Stress-Induced Hypertension of Developmental Origins: Preventive Aspects of Antioxidant Therapy. Antioxidants 2022, 11, 511. [Google Scholar] [CrossRef]
- Baradaran, A.; Nasri, H.; Rafieian-Kopaei, M. Oxidative Stress and Hypertension: Possibility of Hypertension Therapy with Antioxidants. J. Res. Med. Sci. 2014, 19, 358–367. [Google Scholar]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Münzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (ENOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- González, J.; Valls, N.; Brito, R.; Rodrigo, R. Essential Hypertension and Oxidative Stress: New Insights. World J. Cardiol. 2014, 6, 353–366. [Google Scholar] [CrossRef]
- Bellucci, M.; De Marchis, F.; Pompa, A. The Endoplasmic Reticulum Is a Hub to Sort Proteins toward Unconventional Traffic Pathways and Endosymbiotic Organelles. J. Exp. Bot. 2018, 69, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Marahatta, A.; Kim, H.-R.; Chae, H.-J. An Involvement of Oxidative Stress in Endoplasmic Reticulum Stress and Its Associated Diseases. Int. J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef]
- Young, C.N. Endoplasmic Reticulum Stress in the Pathogenesis of Hypertension. Exp. Physiol. 2017, 102, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Lenna, S.; Han, R.; Trojanowska, M. ER Stress and Endothelial Dysfunction. IUBMB Life 2014, 66, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-K.; Lim, M.; Byeon, S.-H.; Lee, Y.-H. Inhibition of Endoplasmic Reticulum Stress Improves Coronary Artery Function in the Spontaneously Hypertensive Rats. Sci. Rep. 2016, 6, 31925. [Google Scholar] [CrossRef] [PubMed]
- Camargo, L.L.; Harvey, A.P.; Rios, F.J.; Tsiropoulou, S.; de Oliveira Silva, R.D.N.; Cao, Z.; Graham, D.; McMaster, C.; Burchmore, R.J.; Hartley, R.C.; et al. Vascular Nox Compartmentalization, Protein Hyperoxidation and ER Stress Response in Hypertension. Hypertension 2018, 72, 235–246. [Google Scholar] [CrossRef]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic Reticulum Stress Sensing in the Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef]
- Loperena, R.; Harrison, D.G. Oxidative Stress and Hypertensive Diseases. Med. Clin. N. Am. 2017, 101, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of Superoxide and Hydrogen Peroxide from Specific Mitochondrial Sites under Different Bioenergetic Conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-Induced Production of Mitochondrial Reactive Oxygen Species: Potential Mechanisms and Relevance for Cardiovascular Disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Kröller-Schön, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinßius, E.; et al. Molecular Mechanisms of the Crosstalk Between Mitochondria and NADPH Oxidase Through Reactive Oxygen Species—Studies in White Blood Cells and in Animal Models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Feng, Y.; Feng, Y.; Gu, L.; Liu, P.; Cao, J.; Zhang, S. The Critical Role of Tetrahydrobiopterin (BH4) Metabolism in Modulating Radiosensitivity: BH4/NOS Axis as an Angel or a Devil. Front. Oncol. 2021, 11, 720632. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH Oxidases and Oxidase Crosstalk in Cardiovascular Diseases: Novel Therapeutic Targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, J.; Song, P.; Wu, Y.; Zhang, J.; Choi, H.C.; Zou, M.-H. Acute Inhibition of GTP Cyclohydrolase 1 Uncouples Endothelial Nitric Oxide Synthase and Elevates Blood Pressure. Hypertension 2008, 52, 484–490. [Google Scholar] [CrossRef]
- Gao, L.; Siu, K.L.; Chalupsky, K.; Nguyen, A.; Chen, P.; Weintraub, N.L.; Galis, Z.; Cai, H. Role of Uncoupled ENOS in Abdominal Aortic Aneurysm Formation: Treatment with Folic Acid. Hypertension 2012, 59, 158. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E. Does Increased Oxidative Stress Cause Hypertension? Diabetes Care 2008, 31 (Suppl. 2), S185–S189. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-Alcoholic Fatty Liver Disease (NAFLD) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a Link between Obesity, Metabolic Syndrome and Type 2 Diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose Tissue and Insulin Resistance in Obese. Biomed. Pharmacother. 2021, 137, 111315. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and Cellular Properties of Insulin Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin Signaling in Health and Disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Hurrle, S.; Hsu, W.H. The Etiology of Oxidative Stress in Insulin Resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef]
- Antoniolli, L.P.; Nedel, B.L.; Pazinato, T.C.; de Andrade Mesquita, L.; Gerchman, F. Accuracy of Insulin Resistance Indices for Metabolic Syndrome: A Cross-Sectional Study in Adults. Diabetol. Metab. Syndr. 2018, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative Stress and the Etiology of Insulin Resistance and Type 2 Diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef]
- Shum, M.; Ngo, J.; Shirihai, O.S.; Liesa, M. Mitochondrial Oxidative Function in NAFLD: Friend or Foe? Mol. Metab. 2020, 50, 101134. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Estall, J.L. An Intimate Relationship between ROS and Insulin Signalling: Implications for Antioxidant Treatment of Fatty Liver Disease. Int. J. Cell Biol. 2014, 2014, 519153. [Google Scholar] [CrossRef] [PubMed]
- Kolka, C.M.; Bergman, R.N. The Endothelium in Diabetes: Its Role in Insulin Access and Diabetic Complications. Rev. Endocr. Metab. Disord. 2013, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte Dysfunctions Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef]
- Sears, B.; Perry, M. The Role of Fatty Acids in Insulin Resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef]
- Gilbert, M. Role of Skeletal Muscle Lipids in the Pathogenesis of Insulin Resistance of Obesity and Type 2 Diabetes. J. Diabetes Investig. 2021, 12, 1934–1941. [Google Scholar] [CrossRef]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac Pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Sangwung, P.; Petersen, K.F.; Shulman, G.I.; Knowles, J.W. Mitochondrial Dysfunction, Insulin Resistance, and Potential Genetic Implications. Endocrinology 2020, 161, bqaa017. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.-G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial Dysfunction and Insulin Resistance: An Update. Endocr. Connect. 2014, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed]
- Weksler-Zangen, S. Is Type 2 Diabetes a Primary Mitochondrial Disorder? Cells 2022, 11, 1617. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H. Oxidative Stress in Pancreatic Beta Cell Regeneration. Oxid. Med. Cell Longev. 2017, 2017, 1930261. [Google Scholar] [CrossRef]
- Park, S.Y.; Gautier, J.-F.; Chon, S. Assessment of Insulin Secretion and Insulin Resistance in Human. Diabetes Metab. J. 2021, 45, 641–654. [Google Scholar] [CrossRef]
- Jesinkey, S.R.; Madiraju, A.K.; Alves, T.C.; Yarborough, O.H.; Cardone, R.L.; Zhao, X.; Parsaei, Y.; Nasiri, A.R.; Butrico, G.; Liu, X.; et al. Mitochondrial GTP Links Nutrient Sensing to β Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Rep. 2019, 28, 759–772.e10. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.-Y.; Jun, H.-S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Prasun, P. Role of Mitochondria in Pathogenesis of Type 2 Diabetes Mellitus. J. Diabetes Metab. Disord. 2020, 19, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Dinić, S.; Arambašić Jovanović, J.; Uskoković, A.; Mihailović, M.; Grdović, N.; Tolić, A.; Rajić, J.; Đorđević, M.; Vidaković, M. Oxidative Stress-Mediated Beta Cell Death and Dysfunction as a Target for Diabetes Management. Front. Endocrinol. 2022, 13, 1006376. [Google Scholar] [CrossRef]
- Eguchi, N.; Vaziri, N.D.; Dafoe, D.C.; Ichii, H. The Role of Oxidative Stress in Pancreatic β Cell Dysfunction in Diabetes. Int. J. Mol. Sci. 2021, 22, 1509. [Google Scholar] [CrossRef]
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Rains, J.L.; Jain, S.K. Oxidative Stress, Insulin Signaling and Diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Scheidereit, C. The IκB Kinase Complex in NF-ΚB Regulation and Beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Ijomone, O.M.; Iroegbu, J.D.; Aschner, M.; Bornhorst, J. Impact of Environmental Toxicants on P38- and ERK-MAPK Signaling Pathways in the Central Nervous System. Neurotoxicology 2021, 86, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of Insulin Sensitivity by Serine/Threonine Phosphorylation of Insulin Receptor Substrate Proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Hançer, N.J.; Qiu, W.; Cherella, C.; Li, Y.; Copps, K.D.; White, M.F. Insulin and Metabolic Stress Stimulate Multisite Serine/Threonine Phosphorylation of Insulin Receptor Substrate 1 and Inhibit Tyrosine Phosphorylation. J. Biol. Chem. 2014, 289, 12467–12484. [Google Scholar] [CrossRef]
- Blanco, C.L.; McGill-Vargas, L.L.; Gastaldelli, A.; Seidner, S.R.; McCurnin, D.C.; Leland, M.M.; Anzueto, D.G.; Johnson, M.C.; Liang, H.; DeFronzo, R.A.; et al. Peripheral Insulin Resistance and Impaired Insulin Signaling Contribute to Abnormal Glucose Metabolism in Preterm Baboons. Endocrinology 2015, 156, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Nakagawa, Y.; Kojima, I.; Shibata, H. Prolonged Insulin Stimulation Down-Regulates GLUT4 through Oxidative Stress-Mediated Retromer Inhibition by a Protein Kinase CK2-Dependent Mechanism in 3T3-L1 Adipocytes. J. Biol. Chem. 2014, 289, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Ismail, H.M.; Scapozza, L.; Ruegg, U.T.; Dorchies, O.M. Diapocynin, a Dimer of the NADPH Oxidase Inhibitor Apocynin, Reduces ROS Production and Prevents Force Loss in Eccentrically Contracting Dystrophic Muscle. PLoS ONE 2014, 9, e110708. [Google Scholar] [CrossRef]
- Hachiya, R.; Tanaka, M.; Itoh, M.; Suganami, T. Molecular Mechanism of Crosstalk between Immune and Metabolic Systems in Metabolic Syndrome. Inflamm. Regen. 2022, 42, 13. [Google Scholar] [CrossRef]
- Saitoh, S.; Van Wijk, K.; Nakajima, O. Crosstalk between Metabolic Disorders and Immune Cells. Int. J. Mol. Sci. 2021, 22, 10017. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, C. Pro-Inflammatory Cytokines: The Link between Obesity and Osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Al-Mansoori, L.; Al-Jaber, H.; Prince, M.S.; Elrayess, M.A. Role of Inflammatory Cytokines, Growth Factors and Adipokines in Adipogenesis and Insulin Resistance. Inflammation 2022, 45, 31–44. [Google Scholar] [CrossRef]
- McDonnell, W.J.; Koethe, J.R.; Mallal, S.A.; Pilkinton, M.A.; Kirabo, A.; Ameka, M.K.; Cottam, M.A.; Hasty, A.H.; Kennedy, A.J. High CD8 T-Cell Receptor Clonality and Altered CDR3 Properties Are Associated with Elevated Isolevuglandins in Adipose Tissue During Diet-Induced Obesity. Diabetes 2018, 67, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 Inflammasome Activation and Cell Death. Cell Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Yang, D.; Oppenheim, J.J. Interleukin-8: An Evolving Chemokine. Cytokine 2022, 153, 155828. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkova, N.N.; Starkov, A.A. Metabolic ROS Signaling: To Immunity and Beyond. Biochem. Biokhimiia 2020, 85, 1650–1667. [Google Scholar] [CrossRef]
- Wang, P.-X.; Deng, X.-R.; Zhang, C.-H.; Yuan, H.-J. Gut Microbiota and Metabolic Syndrome. Chin. Med. J. 2020, 133, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, F.S.; Luaces-Regueira, M.; Ton, A.M.; Campagnaro, B.P.; Campos-Toimil, M.; Pereira, T.M.; Vasquez, E.C. Mechanisms of Action of Kefir in Chronic Cardiovascular and Metabolic Diseases. Cell. Physiol. Biochem. 2018, 48, 1901–1914. [Google Scholar] [CrossRef]
- Festi, D.; Schiumerini, R.; Eusebi, L.H.; Marasco, G.; Taddia, M.; Colecchia, A. Gut Microbiota and Metabolic Syndrome. World J. Gastroenterol. 2014, 20, 16079–16094. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef] [PubMed]
- Dudek-Wicher, R.K.; Junka, A.; Bartoszewicz, M. The Influence of Antibiotics and Dietary Components on Gut Microbiota. Przegla̜d Gastroenterol. 2018, 13, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Ventura, M.; Buttó, L.F.; Duranti, S.; O’Toole, P.W.; Motherway, M.O.; van Sinderen, D. Molecular Dialogue between the Human Gut Microbiota and the Host: A Lactobacillus and Bifidobacterium Perspective. Cell. Mol. Life Sci. 2014, 71, 183–203. [Google Scholar] [CrossRef]
- Croci, S.; D’Apolito, L.I.; Gasperi, V.; Catani, M.V.; Savini, I. Dietary Strategies for Management of Metabolic Syndrome: Role of Gut Microbiota Metabolites. Nutrients 2021, 13, 1389. [Google Scholar] [CrossRef]
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-Grade Inflammation, Diet Composition and Health: Current Research Evidence and Its Translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef]
- Rastelli, M.; Knauf, C.; Cani, P.D. Gut Microbes and Health: A Focus on the Mechanisms Linking Microbes, Obesity, and Related Disorders. Obesity 2018, 26, 792–800. [Google Scholar] [CrossRef]
- Malard, F.; Dore, J.; Gaugler, B.; Mohty, M. Introduction to Host Microbiome Symbiosis in Health and Disease. Mucosal Immunol. 2021, 14, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Veiga, P. Rethinking Diet to Aid Human–Microbe Symbiosis. Trends Microbiol. 2017, 25, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Gu, S.; Liu, D.; Zhao, L.; Xia, S.; He, X.; Chen, H.; Ge, J. Lactobacillus Brevis 23017 Relieves Mercury Toxicity in the Colon by Modulation of Oxidative Stress and Inflammation Through the Interplay of MAPK and NF-ΚB Signaling Cascades. Front. Microbiol. 2018, 9, 2425. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, B.; Xu, H.; Tang, L.; Li, Y.; Gong, L.; Wang, Y.; Li, W. Probiotic Bacillus Attenuates Oxidative Stress- Induced Intestinal Injury via P38-Mediated Autophagy. Front. Microbiol. 2019, 10, 2185. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, Y.; Cheng, Y.; Yan, Q.; Zhou, C.; He, Z.; Zeng, J.; He, J.; Tan, Z. Supplementation of Lactobacillus Plantarum or Macleaya Cordata Extract Alleviates Oxidative Damage Induced by Weaning in the Lower Gut of Young Goats. Animals 2020, 10, 548. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Wang, Y.; Xu, H.; Mei, X.; Yu, D.; Wang, Y.; Li, W. Antioxidant Properties of Probiotic Bacteria. Nutrients 2017, 9, 521. [Google Scholar] [CrossRef]
- Jones, R.M.; Luo, L.; Ardita, C.S.; Richardson, A.N.; Kwon, Y.M.; Mercante, J.W.; Alam, A.; Gates, C.L.; Wu, H.; Swanson, P.A.; et al. Symbiotic Lactobacilli Stimulate Gut Epithelial Proliferation via Nox-Mediated Generation of Reactive Oxygen Species. EMBO J. 2013, 32, 3017–3028. [Google Scholar] [CrossRef]
- Jones, R.M.; Mercante, J.W.; Neish, A.S. Reactive Oxygen Production Induced by the Gut Microbiota: Pharmacotherapeutic Implications. Curr. Med. Chem. 2012, 19, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Neish, A.S. Redox Signaling Mediated by the Gut Microbiota. Free Radic. Res. 2013, 47, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Carrizales-Sánchez, A.K.; García-Cayuela, T.; Hernández-Brenes, C.; Senés-Guerrero, C. Gut Microbiota Associations with Metabolic Syndrome and Relevance of Its Study in Pediatric Subjects. Gut Microbes 2021, 13, 1960135. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, E.C.; Pereira, T.M.C.; Campos-Toimil, M.; Baldo, M.P.; Peotta, V.A. Gut Microbiota, Diet, and Chronic Diseases: The Role Played by Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019, 7092032. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of Gut Microbiota in Type 2 Diabetes Pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef]
- Davis, C.D. The Gut Microbiome and Its Role in Obesity. Nutr. Today 2016, 51, 167–174. [Google Scholar] [CrossRef]
- Tai, N.; Wong, F.S.; Wen, L. The Role of Gut Microbiota in the Development of Type 1, Obesity and Type 2 Diabetes Mellitus. Rev. Endocr. Metab. Disord. 2015, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Guirro, M.; Costa, A.; Gual-Grau, A.; Herrero, P.; Torrell, H.; Canela, N.; Arola, L. Effects from Diet-Induced Gut Microbiota Dysbiosis and Obesity Can Be Ameliorated by Fecal Microbiota Transplantation: A Multiomics Approach. PLoS ONE 2019, 14, e0218143. [Google Scholar] [CrossRef]
- Tomas, J.; Mulet, C.; Saffarian, A.; Cavin, J.-B.; Ducroc, R.; Regnault, B.; Kun Tan, C.; Duszka, K.; Burcelin, R.; Wahli, W.; et al. High-Fat Diet Modifies the PPAR-γ Pathway Leading to Disruption of Microbial and Physiological Ecosystem in Murine Small Intestine. Proc. Natl. Acad. Sci. USA 2016, 113, E5934–E5943. [Google Scholar] [CrossRef]
- Thingholm, L.B.; Rühlemann, M.C.; Koch, M.; Fuqua, B.; Laucke, G.; Boehm, R.; Bang, C.; Franzosa, E.A.; Hübenthal, M.; Rahnavard, A.; et al. Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition. Cell Host Microbe 2019, 26, 252–264.e10. [Google Scholar] [CrossRef]
- Aoun, A.; Darwish, F.; Hamod, N. The Influence of the Gut Microbiome on Obesity in Adults and the Role of Probiotics, Prebiotics, and Synbiotics for Weight Loss. Prev. Nutr. Food Sci. 2020, 25, 113–123. [Google Scholar] [CrossRef]
- Noubiap, J.J.; Nansseu, J.R.; Lontchi-Yimagou, E.; Nkeck, J.R.; Nyaga, U.F.; Ngouo, A.T.; Tounouga, D.N.; Tianyi, F.L.; Foka, A.J.; Ndoadoumgue, A.L.; et al. Global, Regional, and Country Estimates of Metabolic Syndrome Burden in Children and Adolescents in 2020: A Systematic Review and Modelling Analysis. Lancet Child Adolesc. Health 2022, 6, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Shariq, O.A.; McKenzie, T.J. Obesity-Related Hypertension: A Review of Pathophysiology, Management, and the Role of Metabolic Surgery. Gland Surg. 2020, 9, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Swarup, S.; Goyal, A.; Grigorova, Y.; Zeltser, R. Metabolic Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Tune, J.D.; Goodwill, A.G.; Sassoon, D.J.; Mather, K.J. Cardiovascular Consequences of Metabolic Syndrome. Transl. Res. J. Lab. Clin. Med. 2017, 183, 57–70. [Google Scholar] [CrossRef]
- Saeed, A.; Kampangkaew, J.; Nambi, V. Prevention of Cardiovascular Disease in Women. Methodist DeBakey Cardiovasc. J. 2017, 13, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Saheera, S.; Krishnamurthy, P. Cardiovascular Changes Associated with Hypertensive Heart Disease and Aging. Cell Transplant. 2020, 29, 0963689720920830. [Google Scholar] [CrossRef]
- Dhawan, D.; Sharma, S. Abdominal Obesity, Adipokines and Non-Communicable Diseases. J. Steroid Biochem. Mol. Biol. 2020, 203, 105737. [Google Scholar] [CrossRef]
- Tao, L.-X.; Yang, K.; Liu, X.-T.; Cao, K.; Zhu, H.-P.; Luo, Y.-X.; Guo, J.; Wu, L.-J.; Li, X.; Guo, X.-H. Longitudinal Associations between Triglycerides and Metabolic Syndrome Components in a Beijing Adult Population, 2007–2012. Int. J. Med. Sci. 2016, 13, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.; Kokkinos, P.; Nyelin, E. Physical Activity, Cardiorespiratory Fitness, and the Metabolic Syndrome. Nutrients 2019, 11, 1652. [Google Scholar] [CrossRef]
- Ho, E.; Karimi Galougahi, K.; Liu, C.-C.; Bhindi, R.; Figtree, G.A. Biological Markers of Oxidative Stress: Applications to Cardiovascular Research and Practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Mocan, M.; Vesa, Ş.; Suciu, Ş.; Blaga, S.N. Systemic Markers of Oxidative Stress in Relation to Metabolic Syndrome Components. Clujul Med. 2013, 86, 227–234. [Google Scholar] [PubMed]
- Espinosa-Moncada, J.; Marín-Echeverri, C.; Galvis-Pérez, Y.; Ciro-Gómez, G.; Aristizábal, J.C.; Blesso, C.N.; Fernandez, M.L.; Barona-Acevedo, J. Evaluation of Agraz Consumption on Adipocytokines, Inflammation, and Oxidative Stress Markers in Women with Metabolic Syndrome. Nutrients 2018, 10, 1639. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Namkung, J. Effects of Exercise Intervention on Mitochondrial Stress Biomarkers in Metabolic Syndrome Patients: A Randomized Controlled Trial. Int. J. Environ. Res. Public Health 2021, 18, 2242. [Google Scholar] [CrossRef]
- Cojic, M.; Kocic, R.; Klisic, A.; Kocic, G. The Effects of Vitamin D Supplementation on Metabolic and Oxidative Stress Markers in Patients with Type 2 Diabetes: A 6-Month Follow Up Randomized Controlled Study. Front. Endocrinol. 2021, 12, 610893. [Google Scholar] [CrossRef] [PubMed]
- Schönknecht, Y.B.; Crommen, S.; Stoffel-Wagner, B.; Coenen, M.; Fimmers, R.; Stehle, P.; Ramirez, A.; Egert, S. APOE Ɛ4 Is Associated with Postprandial Inflammation in Older Adults with Metabolic Syndrome Traits. Nutrients 2021, 13, 3924. [Google Scholar] [CrossRef]
- Sangaleti, C.T.; Katayama, K.Y.; De Angelis, K.; Lemos de Moraes, T.; Araújo, A.A.; Lopes, H.F.; Camacho, C.; Bortolotto, L.A.; Michelini, L.C.; Irigoyen, M.C.; et al. The Cholinergic Drug Galantamine Alleviates Oxidative Stress Alongside Anti-Inflammatory and Cardio-Metabolic Effects in Subjects With the Metabolic Syndrome in a Randomized Trial. Front. Immunol. 2021, 12, 613979. [Google Scholar] [CrossRef]
- Grabež, M.; Škrbić, R.; Stojiljković, M.P.; Vučić, V.; Rudić Grujić, V.; Jakovljević, V.; Djuric, D.M.; Suručić, R.; Šavikin, K.; Bigović, D.; et al. A Prospective, Randomized, Double-Blind, Placebo-Controlled Trial of Polyphenols on the Outcomes of Inflammatory Factors and Oxidative Stress in Patients with Type 2 Diabetes Mellitus. Rev. Cardiovasc. Med. 2022, 23, 57. [Google Scholar] [CrossRef]
- Jeong, J.H.; Jung, S.; Kim, K.-N. Considering Serum Alanine Aminotransferase and Gamma-Glutamyltransferase Levels Together Strengthen the Prediction of Impaired Fasting Glucose Risk: A Cross-Sectional and Longitudinal Study. Sci. Rep. 2021, 11, 3333. [Google Scholar] [CrossRef]
- Jeong, H.; Baek, S.-Y.; Kim, S.W.; Park, E.-J.; Lee, J.; Kim, H.; Jeon, C.H. C Reactive Protein Level as a Marker for Dyslipidaemia, Diabetes and Metabolic Syndrome: Results from the Korea National Health and Nutrition Examination Survey. BMJ Open 2019, 9, e029861. [Google Scholar] [CrossRef] [PubMed]
- Felipe, A.; Guadalupe, E.; Druso, P.; Carlos, M.; Pablo, S.; Oscar, C.; Luis, V.; Diego, M.; Jaime, R.; Inés, U.; et al. Serum Ferritin Is Associated with Metabolic Syndrome and Red Meat Consumption. Oxid. Med. Cell. Longev. 2015, 2015, 769739. [Google Scholar] [CrossRef]
- Ryo, M.; Nakamura, T.; Kihara, S.; Kumada, M.; Shibazaki, S.; Takahashi, M.; Nagai, M.; Matsuzawa, Y.; Funahashi, T. Adiponectin as a Biomarker of the Metabolic Syndrome. Circ. J. 2004, 68, 975–981. [Google Scholar] [CrossRef]
- Ahonen, T.M.; Saltevo, J.T.; Kautiainen, H.J.; Kumpusalo, E.A.; Vanhala, M.J. The Association of Adiponectin and Low-Grade Inflammation with the Course of Metabolic Syndrome. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, S.; Jensen, J.S.; Bjerre, M.; Frystyk, J.; Flyvbjerg, A.; Jeppesen, J.; Mogelvang, R. Low Adiponectin Levels at Baseline and Decreasing Adiponectin Levels over 10 Years of Follow-up Predict Risk of the Metabolic Syndrome. Diabetes Metab. 2017, 43, 134–139. [Google Scholar] [CrossRef]
- Madeira, I.; Bordallo, M.A.; Rodrigues, N.C.; Carvalho, C.; Gazolla, F.; Collett-Solberg, P.; Medeiros, C.; Bordallo, A.P.; Borges, M.; Monteiro, C.; et al. Leptin as a Predictor of Metabolic Syndrome in Prepubertal Children. Arch. Endocrinol. Metab. 2017, 61, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Shin, D. Prospective Associations of Serum Adiponectin, Leptin, and Leptin-Adiponectin Ratio with Incidence of Metabolic Syndrome: The Korean Genome and Epidemiology Study. Int. J. Environ. Res. Public Health 2020, 17, 3287. [Google Scholar] [CrossRef] [PubMed]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.V.S.; Dias, M.M.G.; Brilhante, A.J.V.C.; Terra, M.F.; García-Arévalo, M.; Figueira, A.C.M. Multifactorial Basis and Therapeutic Strategies in Metabolism-Related Diseases. Nutrients 2021, 13, 2830. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.; Adams, H.; Kubena, K.; Guo, S. Etiology of Metabolic Syndrome and Dietary Intervention. Int. J. Mol. Sci. 2019, 20, 128. [Google Scholar] [CrossRef]
- Perrone, S.; Laschi, E.; Buonocore, G. Biomarkers of Oxidative Stress in the Fetus and in the Newborn. Free Radic. Biol. Med. 2019, 142, 23–31. [Google Scholar] [CrossRef]
- Li, Y.; Hruby, A.; Bernstein, A.M.; Ley, S.H.; Wang, D.D.; Chiuve, S.E.; Sampson, L.; Rexrode, K.M.; Rimm, E.B.; Willett, W.C.; et al. Saturated Fat as Compared with Unsaturated Fats and Sources of Carbohydrates in Relation to Risk of Coronary Heart Disease: A Prospective Cohort Study. J. Am. Coll. Cardiol. 2015, 66, 1538–1548. [Google Scholar] [CrossRef]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated Fatty Acid-Enriched High-Fat Diets Impede Adipose NLRP3 Inflammasome-Mediated IL-1β Secretion and Insulin Resistance despite Obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef] [PubMed]
- Di Daniele, N.; Noce, A.; Vidiri, M.F.; Moriconi, E.; Marrone, G.; Annicchiarico-Petruzzelli, M.; D’Urso, G.; Tesauro, M.; Rovella, V.; De Lorenzo, A. Impact of Mediterranean Diet on Metabolic Syndrome, Cancer and Longevity. Oncotarget 2016, 8, 8947–8979. [Google Scholar] [CrossRef] [PubMed]
- Finicelli, M.; Squillaro, T.; Di Cristo, F.; Di Salle, A.; Melone, M.A.B.; Galderisi, U.; Peluso, G. Metabolic Syndrome, Mediterranean Diet, and Polyphenols: Evidence and Perspectives. J. Cell. Physiol. 2019, 234, 5807–5826. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; González-Manzano, S.; González-Paramás, A.M. Wine, Polyphenols, and Mediterranean Diets. What Else Is There to Say? Molecules 2021, 26, 5537. [Google Scholar] [CrossRef]
- Saibandith, B.; Spencer, J.P.E.; Rowland, I.R.; Commane, D.M. Olive Polyphenols and the Metabolic Syndrome. Molecules 2017, 22, 1082. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, S.; Damasceno, N.R.T.; Braga, R.A.M.; Martinez, R.; Kris-Etherton, P.; Sala-Vila, A. Effect of Nuts on Markers of Inflammation and Oxidative Stress: A Narrative Review. Nutrients 2023, 15, 1099. [Google Scholar] [CrossRef]
- Fatahi, S.; Daneshzad, E.; Lotfi, K.; Azadbakht, L. The Effects of Almond Consumption on Inflammatory Biomarkers in Adults: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Adv. Nutr. 2022, 13, 1462–1475. [Google Scholar] [CrossRef]
- Mateș, L.; Popa, D.-S.; Rusu, M.E.; Fizeșan, I.; Leucuța, D. Walnut Intake Interventions Targeting Biomarkers of Metabolic Syndrome and Inflammation in Middle-Aged and Older Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Antioxidants 2022, 11, 1412. [Google Scholar] [CrossRef]
- Mateș, L.; Rusu, M.E.; Popa, D.-S. Phytochemicals and Biological Activities of Walnut Septum: A Systematic Review. Antioxidants 2023, 12, 604. [Google Scholar] [CrossRef]
- Vassalle, C.; Maltinti, M.; Sabatino, L. Targeting Oxidative Stress for Disease Prevention and Therapy: Where Do We Stand, and Where Do We Go from Here. Molecules 2020, 25, 2653. [Google Scholar] [CrossRef]
- Asgharzadeh, F.; Hashemzehi, M.; Moradi-Marjaneh, R.; Hassanian, S.M.; Ferns, G.A.; Khazaei, M.; Avan, A. Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers as Therapeutic Options in the Treatment of Renal Cancer: A Meta-Analysis. Life Sci. 2020, 242, 117181. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Demirci-Çekiç, S.; Özkan, G.; Avan, A.N.; Uzunboy, S.; Çapanoğlu, E.; Apak, R. Biomarkers of Oxidative Stress and Antioxidant Defense. J. Pharm. Biomed. Anal. 2022, 209, 114477. [Google Scholar] [CrossRef] [PubMed]
- Carrizzo, A.; Forte, M.; Lembo, M.; Formisano, L.; Puca, A.A.; Vecchione, C. Rac-1 as a New Therapeutic Target in Cerebro- and Cardio-Vascular Diseases. Curr. Drug Targets 2014, 15, 1231–1246. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. https://doi.org/10.3390/ijms24097898
Masenga SK, Kabwe LS, Chakulya M, Kirabo A. Mechanisms of Oxidative Stress in Metabolic Syndrome. International Journal of Molecular Sciences. 2023; 24(9):7898. https://doi.org/10.3390/ijms24097898
Chicago/Turabian StyleMasenga, Sepiso K., Lombe S. Kabwe, Martin Chakulya, and Annet Kirabo. 2023. "Mechanisms of Oxidative Stress in Metabolic Syndrome" International Journal of Molecular Sciences 24, no. 9: 7898. https://doi.org/10.3390/ijms24097898