
SIGMAR1 Confers Innate Resilience against Neurodegeneration


Abstract
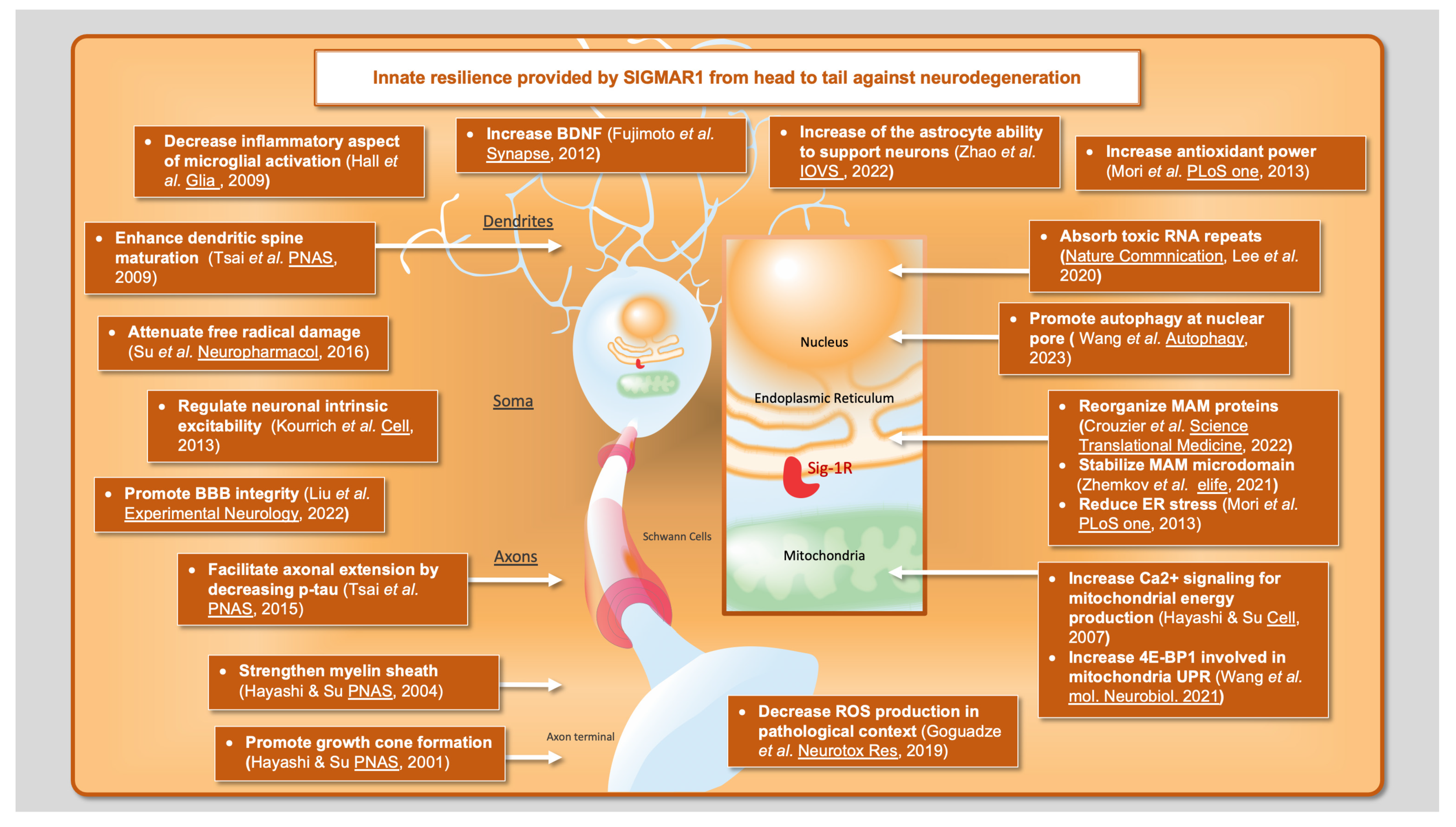
:1. Introduction
2. The Relevance of the Unfolded Protein Response (UPR) in Neurodegenerative Contexts
3. Mitochondria-Associated ER Membrane (MAM): Critical Interface between Two Fundamental Organelles
4. SIGMAR1 and the Control of Oxidative Stress
5. Beyond the MAM: Other Entry Points for SIGMAR1 to Counteract Neurodegenerative Disorders
5.1. BDNF/TrkB
5.2. Voltage-Dependent Potassium Channel
5.3. Neuron’s Morphology
5.4. Tau Phosphorylation
5.5. Nuclear Pore Complex
6. SIGMAR1 and Glial Cells
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Kitaichi, K.; Chabot, J.-G.; Moebius, F.F.; Flandorfer, A.; Glossmann, H.; Quirion, R. Expression of the purported sigma1 (σ1) receptor in the mammalian brain and its possible relevance in deficits induced by antagonism of the NMDA receptor complex as revealed using an antisense strategy. J. Chem. Neuroanat. 2000, 20, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Novakova, M.; Ela, C.; Barg, J.; Vogel, Z.; Hasin, Y.; Eilam, Y. Inotropic action of σ receptor ligands in isolated cardiac myocytes from adult rats. Eur. J. Pharmacol. 1995, 286, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca(2+) Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Epstein, M.; Guo, L.-W. Subcellular Localization of the Sigma-1 Receptor in Retinal Neurons—An Electron Microscopy Study. Sci. Rep. 2015, 5, 10689. [Google Scholar] [CrossRef]
- Seth, P.; Leibach, F.H.; Ganapathy, V. Cloning and Structural Analysis of the cDNA and the Gene Encoding the Murine Type 1 Sigma Receptor. Biochem. Biophys. Res. Commun. 1997, 241, 535–540. [Google Scholar] [CrossRef]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef]
- Kekuda, R.; Prasad, P.D.; Fei, Y.-J.; Leibach, F.H.; Ganapathy, V. Cloning and Functional Expression of the Human Type 1 Sigma Receptor (hSigmaR1). Biochem. Biophys. Res. Commun. 1996, 229, 553–558. [Google Scholar] [CrossRef]
- Su, T.P. Evidence for sigma opioid receptor: Binding of [3H]SKF-10047 to etorphine-inaccessible sites in guinea-pig brain. Experiment 1982, 223, 284–290. [Google Scholar]
- Ossa, F.; Schnell, J.R.; Ortega-Roldan, J.L. A Review of the Human Sigma-1 Receptor Structure. Adv. Exp. Med. Biol. 2017, 964, 15–29. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human σ1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.-H.; Ye, Y.; Wan, B.-B.; Yu, Y.-D.; Liu, C.; Chen, Q.-J. Emerging Benefits: Pathophysiological Functions and Target Drugs of the Sigma-1 Receptor in Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 5649–5666. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.A.; Cavendish, J.Z.; Robson, M.J.; Scandinaro, A.L.; Matsumoto, R.R. Role of sigma-1 receptors in neurodegenerative diseases. J. Pharmacol. Sci. 2015, 127, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Weng, T.-Y.; Tsai, S.-Y.A.; Su, T.-P. Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases. J. Biomed. Sci. 2017, 24, 74. [Google Scholar] [CrossRef]
- Penke, B.; Fulop, L.; Szucs, M.; Frecska, E. The Role of Sigma-1 Receptor, an Intracellular Chaperone in Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 97–116. [Google Scholar] [CrossRef]
- Resende, R.; Fernandes, T.; Pereira, A.C.; Marques, A.P.; Pereira, C.F. Endoplasmic Reticulum-Mitochondria Contacts Modulate Reactive Oxygen Species-Mediated Signaling and Oxidative Stress in Brain Disorders: The Key Role of Sigma-1 Receptor. Antioxid. Redox Signal. 2022, 37, 758–780. [Google Scholar] [CrossRef]
- Smith, S.B.; Wang, J.; Cui, X.; Mysona, B.A.; Zhao, J.; Bollinger, K.E. Sigma 1 receptor: A novel therapeutic target in retinal disease. Prog. Retin. Eye Res. 2018, 67, 130–149. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Futschik, M.E.; Kalathur, R.K.R.; Ayasolla, K. The Unfolded Protein Response and its potential role in Huntington’s disease. Nat. Précéd. 2012. [Google Scholar] [CrossRef]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.-P. Sigma-1 Receptor Chaperone at the ER-Mitochondrion Interface Mediates the Mitochondrion-ER-Nucleus Signaling for Cellular Survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [PubMed]
- Dreser, A.; Vollrath, J.T.; Sechi, A.; Johann, S.; Roos, A.; Yamoah, A.; Katona, I.; Bohlega, S.; Wiemuth, D.; Tian, Y.; et al. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017, 24, 1655–1671. [Google Scholar] [CrossRef]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217 Pt 1, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, S.G.; Pham, M.T.; Mitchell, M.B.; Yeom, S.G.; Jordan, S.; Chang, A.; Sopher, B.L.; La Spada, A.R. 4E-BP1 Protects Neurons from Misfolded Protein Stress and Parkinson’s Disease Toxicity by Inducing the Mitochondrial Unfolded Protein Response. J. Neurosci. 2020, 40, 8734–8745. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Goguadze, N.; Kimura, Y.; Yasui, Y.; Pan, B.; Wang, T.-Y.; Nakamura, Y.; Lin, Y.-T.; Hogan, Q.H.; Wilson, K.L.; et al. Genomic Action of Sigma-1 Receptor Chaperone Relates to Neuropathic Pain. Mol. Neurobiol. 2021, 58, 2523–2541. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-Associated ER Membranes—The Origin Site of Autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.; Mueller, S.; Miller, T.; Miller, C.C. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Delprat, B.; Crouzier, L.; Su, T.P.; Maurice, T. At the Crossing of ER Stress and MAMs: A Key Role of Sigma-1 Receptor? Adv. Exp. Med. Biol. 2020, 1131, 699–718. [Google Scholar]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef]
- Zaman, M.; Shutt, T.E. The Role of Impaired Mitochondrial Dynamics in MFN2-Mediated Pathology. Front. Cell Dev. Biol. 2022, 10, 858286. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Suaga, P.; Pérez-Nievas, B.G.; Glennon, E.B.; Lau, D.H.W.; Paillusson, S.; Mórotz, G.M.; Calì, T.; Pizzo, P.; Noble, W.; Miller, C.C.J. The VAPB-PTPIP51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol. Commun. 2019, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2011, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Zhemkov, V.; Ditlev, J.A.; Lee, W.R.; Wilson, M.; Liou, J.; Rosen, M.K.; Bezprozvanny, I. The role of sigma 1 receptor in organization of endoplasmic reticulum signaling microdomains. eLife 2021, 10, e65192. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; Médard, J.-J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138 Pt 4, 875–890. [Google Scholar] [CrossRef]
- Crouzier, L.; Danese, A.; Yasui, Y.; Richard, E.M.; Liévens, J.-C.; Patergnani, S.; Couly, S.; Diez, C.; Denus, M.; Cubedo, N.; et al. Activation of the sigma-1 receptor chaperone alleviates symptoms of Wolfram syndrome in preclinical models. Sci. Transl. Med. 2022, 14, eabh3763. [Google Scholar] [CrossRef] [PubMed]
- Tagashira, H.; Bhuiyan, S.; Shinoda, Y.; Kawahata, I.; Numata, T.; Fukunaga, K. Sigma-1 receptor is involved in modification of ER-mitochondria proximity and Ca(2+) homeostasis in cardiomyocytes. J. Pharmacol. Sci. 2023, 151, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. α-Synuclein Controls Mitochondrial Calcium Homeostasis by Enhancing Endoplasmic Reticulum-Mitochondria Interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef]
- Lau, D.H.; Paillusson, S.; Hartopp, N.; Rupawala, H.; Mórotz, G.M.; Gomez-Suaga, P.; Greig, J.; Troakes, C.; Noble, W.; Miller, C.C. Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 2020, 143, 105020. [Google Scholar] [CrossRef]
- Zhemkov, V.; Geva, M.; Hayden, M.R.; Bezprozvanny, I. Sigma-1 Receptor (S1R) Interaction with Cholesterol: Mechanisms of S1R Activation and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4082. [Google Scholar] [CrossRef] [PubMed]
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.; Maurice, T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox. Res. 2019, 35, 1–18. [Google Scholar] [CrossRef]
- Su, T.C.; Lin, S.H.; Lee, P.T.; Yeh, S.H.; Hsieh, T.H.; Chou, S.Y.; Su, T.P.; Hung, J.J.; Chang, W.C.; Lee, Y.C.; et al. The sigma-1 receptor-zinc finger protein 179 pathway protects against hydrogen peroxide-induced cell injury. Neuropharmacology 2016, 105, 1–9. [Google Scholar] [CrossRef]
- Colucci-D’amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Kikuchi-Utsumi, K.; Nakaki, T. Chronic treatment with a selective ligand for the sigma-1 receptor chaperone, SA4503, up-regulates BDNF protein levels in the rat hippocampus. Neurosci. Lett. 2008, 440, 19–22. [Google Scholar] [CrossRef]
- Fujimoto, M.; Hayashi, T.; Urfer, R.; Mita, S.; Su, T.-P. Sigma-1 receptor chaperones regulate the secretion of brain-derived neurotrophic factor. Synapse 2012, 66, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Mysona, B.A.; Zhao, J.; Smith, S.; Bollinger, K.E. Relationship between Sigma-1 receptor and BDNF in the visual system. Exp. Eye Res. 2018, 167, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, A.; Gradus, T.; Altman, T.; Maimon, R.; Avraham, N.S.; Geva, M.; Hayden, M.; Perlson, E. Targeting the Sigma-1 Receptor via Pridopidine Ameliorates Central Features of ALS Pathology in a SOD1(G93A) Model. Cell Death Dis. 2019, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, S.; Lahaye, R.A.; Vitet, H.; Scaramuzzino, C.; Virlogeux, A.; Capellano, L.; Genoux, A.; Gershoni-Emek, N.; Geva, M.; Hayden, M.R.; et al. Pridopidine rescues BDNF/TrkB trafficking dynamics and synapse homeostasis in a Huntington disease brain-on-a-chip model. Neurobiol. Dis. 2022, 173, 105857. [Google Scholar] [CrossRef] [PubMed]
- Francardo, V.; Bez, F.; Wieloch, T.; Nissbrandt, H.; Ruscher, K.; Cenci, M.A. Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 2014, 137 Pt 7, 1998–2014. [Google Scholar] [CrossRef]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.K. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. 2020, 257, 118020. [Google Scholar] [CrossRef]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef]
- Shruthi, S.; Sumitha, R.; Varghese, A.M.; Ashok, S.; Chandrasekhar Sagar, B.K.; Sathyaprabha, T.N.; Nalini, A.; Kramer, B.W.; Raju, T.R.; Vijayalakshmi, K.; et al. Brain-Derived Neurotrophic Factor Facilitates Functional Recovery from ALS-Cerebral Spinal Fluid-Induced Neurodegenerative Changes in the NSC-34 Motor Neuron Cell Line. Neuro Degener. Dis. 2017, 17, 44–58. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Kourrich, S.; Hayashi, T.; Chuang, J.-Y.; Tsai, S.-Y.; Su, T.-P.; Bonci, A. Dynamic Interaction between Sigma-1 Receptor and Kv1.2 Shapes Neuronal and Behavioral Responses to Cocaine. Cell 2013, 152, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Hayashi, T.; Harvey, B.K.; Wang, Y.; Wu, W.W.; Shen, R.F.; Zhang, Y.; Becker, K.G.; Hoffer, B.J.; Su, T.P. Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1xGTP pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 22468–22473. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, M.; Hayashi, T.; Su, T. Nerve growth factor-induced neurite sprouting in PC12 cells involves sigma-1 receptors: Implications for antidepressants. J. Pharmacol. Exp. Ther. 2002, 303, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.-P. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Mysona, B.A.; Zhao, J.; De Greef, O.; Beisel, A.; Patel, P.A.; Berman, L.; Smith, S.B.; Bollinger, K. Sigma-1 receptor agonist, (+)-pentazocine, is neuroprotective in a Brown Norway rat microbead model of glaucoma. Exp. Eye Res. 2023, 226, 109308. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-Y.A.; Pokrass, M.J.; Klauer, N.R.; Nohara, H.; Su, T.-P. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc. Natl. Acad. Sci. USA 2015, 112, 6742–6747. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lagier-Tourenne, C. Nuclear pores: The gate to neurodegeneration. Nat. Neurosci. 2018, 21, 156–158. [Google Scholar] [CrossRef]
- Lee, P.-T.; Liévens, J.-C.; Wang, S.-M.; Chuang, J.-Y.; Khalil, B.; Wu, H.-E.; Chang, W.-C.; Maurice, T.; Su, T.-P. Sigma-1 receptor chaperones rescue nucleocytoplasmic transport deficit seen in cellular and Drosophila ALS/FTD models. Nat. Commun. 2020, 11, 5580. [Google Scholar] [CrossRef]
- Wang, S.M.; Wu, H.E.; Yasui, Y.; Geva, M.; Hayden, M.; Maurice, T.; Cozzolino, M.; Su, T.P. Nucleoporin POM121 signals TFEB-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine. Autophagy 2023, 19, 126–151. [Google Scholar] [CrossRef]
- Liu, D.; Yang, L.; Liu, P.; Ji, X.; Qi, X.; Wang, Z.; Chi, T.; Zou, L. Sigma–1 receptor activation alleviates blood–brain barrier disruption post cerebral ischemia stroke by stimulating the GDNF–GFRα1–RET pathway. Exp. Neurol. 2022, 347, 113867. [Google Scholar] [CrossRef]
- Hall, A.A.; Herrera, Y.; Ajmo, C.T.; Cuevas, J.; Pennypacker, K.R. Sigma receptors suppress multiple aspects of microglial activation. Glia 2009, 57, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gonsalvez, G.B.; Mysona, B.A.; Smith, S.B.; Bollinger, K.E. Sigma 1 Receptor Contributes to Astrocyte-Mediated Retinal Ganglion Cell Protection. Investig. Opthalmol. Vis. Sci. 2022, 63, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gonsalvez, G.; Bartoli, M.; Mysona, B.A.; Smith, S.B.; Bollinger, K.E. Sigma 1 Receptor Modulates Optic Nerve Head Astrocyte Reactivity. Investig. Opthalmol. Vis. Sci. 2021, 62, 5. [Google Scholar] [CrossRef]
- Weng, T.-Y.; Hung, D.T.; Su, T.-P.; Tsai, S.-Y.A. Loss of Sigma-1 Receptor Chaperone Promotes Astrocytosis and Enhances the Nrf2 Antioxidant Defense. Oxidative Med. Cell. Longev. 2017, 2017, 4582135. [Google Scholar] [CrossRef] [PubMed]
- Lasbleiz, C.; Peyrel, A.; Tarot, P.; Sarniguet, J.; Crouzier, L.; Cubedo, N.; Delprat, B.; Rossel, M.; Maurice, T.; Liévens, J.-C. Sigma-1 receptor agonist PRE-084 confers protection against TAR DNA-binding protein-43 toxicity through NRF2 signalling. Redox Biol. 2022, 58, 102542. [Google Scholar] [CrossRef]
- Barwick, S.R.; Siddiq, M.S.; Wang, J.; Xiao, H.; Marshall, B.; Perry, E.; Smith, S.B. Sigma 1 Receptor Co-Localizes with NRF2 in Retinal Photoreceptor Cells. Antioxidants 2021, 10, 981. [Google Scholar] [CrossRef]
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’driscoll, C.M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc. Natl. Acad. Sci. USA 2004, 101, 14949–14954. [Google Scholar] [CrossRef]
- Decker, L.; Ffrench-Constant, C. Lipid Rafts and Integrin Activation Regulate Oligodendrocyte Survival. J. Neurosci. 2004, 24, 3816–3825. [Google Scholar] [CrossRef]
- Gielen, E.; Baron, W.; Vandeven, M.; Steels, P.; Hoekstra, D.; Ameloot, M. Rafts in oligodendrocytes: Evidence and structure–function relationship. Glia 2006, 54, 499–512. [Google Scholar] [CrossRef]

{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couly, S.; Yasui, Y.; Su, T.-P. SIGMAR1 Confers Innate Resilience against Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 7767. https://doi.org/10.3390/ijms24097767
Couly S, Yasui Y, Su T-P. SIGMAR1 Confers Innate Resilience against Neurodegeneration. International Journal of Molecular Sciences. 2023; 24(9):7767. https://doi.org/10.3390/ijms24097767
Chicago/Turabian StyleCouly, Simon, Yuko Yasui, and Tsung-Ping Su. 2023. "SIGMAR1 Confers Innate Resilience against Neurodegeneration" International Journal of Molecular Sciences 24, no. 9: 7767. https://doi.org/10.3390/ijms24097767