
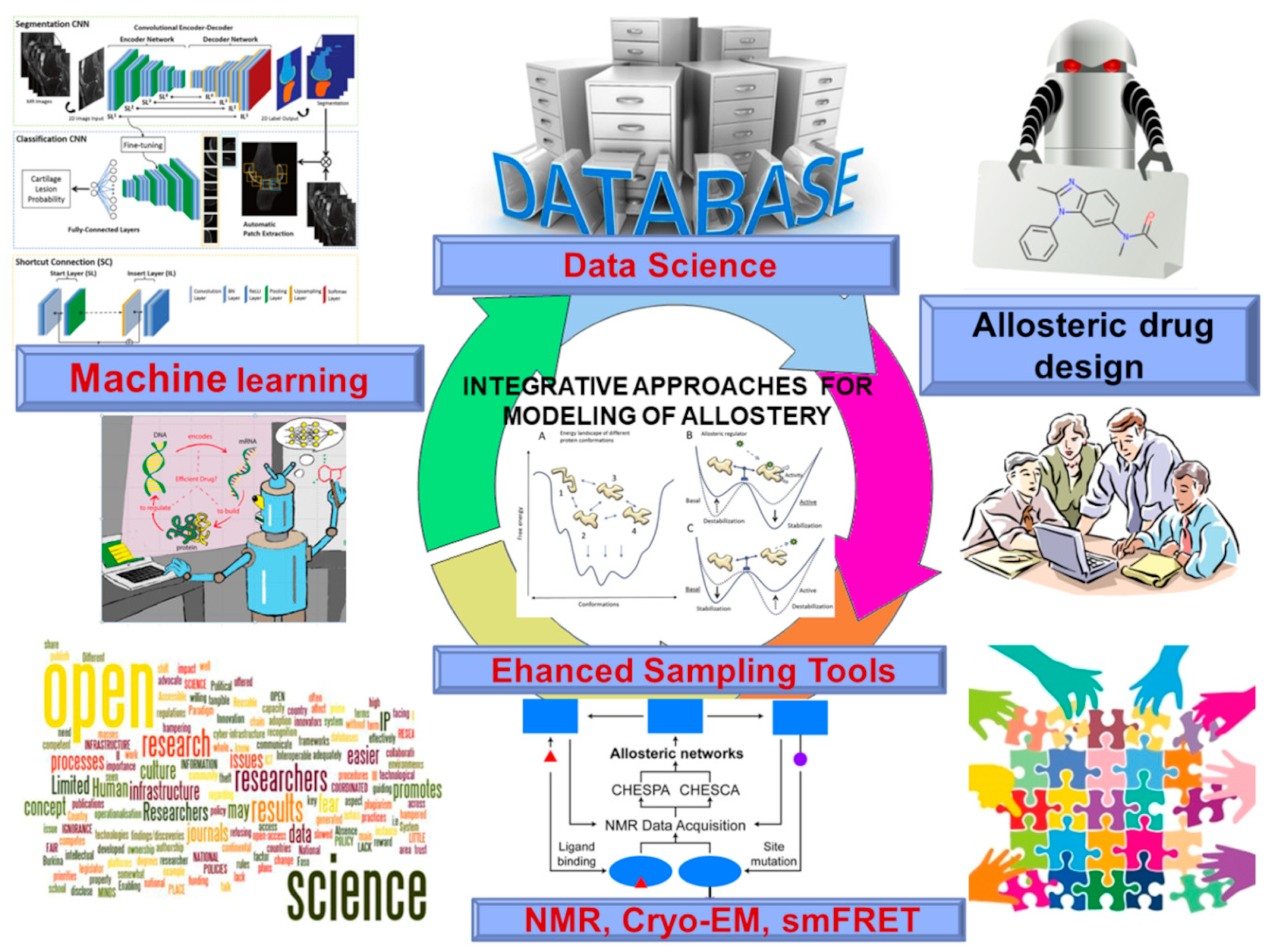
From Deep Mutational Mapping of Allosteric Protein Landscapes to Deep Learning of Allostery and Hidden Allosteric Sites: Zooming in on “Allosteric Intersection” of Biochemical and Big Data Approaches


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mining of Allosteric Protein Landscapes and Mechanisms Using Multidimensional Deep Mutational Scanning Approaches
3. Machine Learning Models Infer Genotype-Phenotype Relationships from DMS Experiments
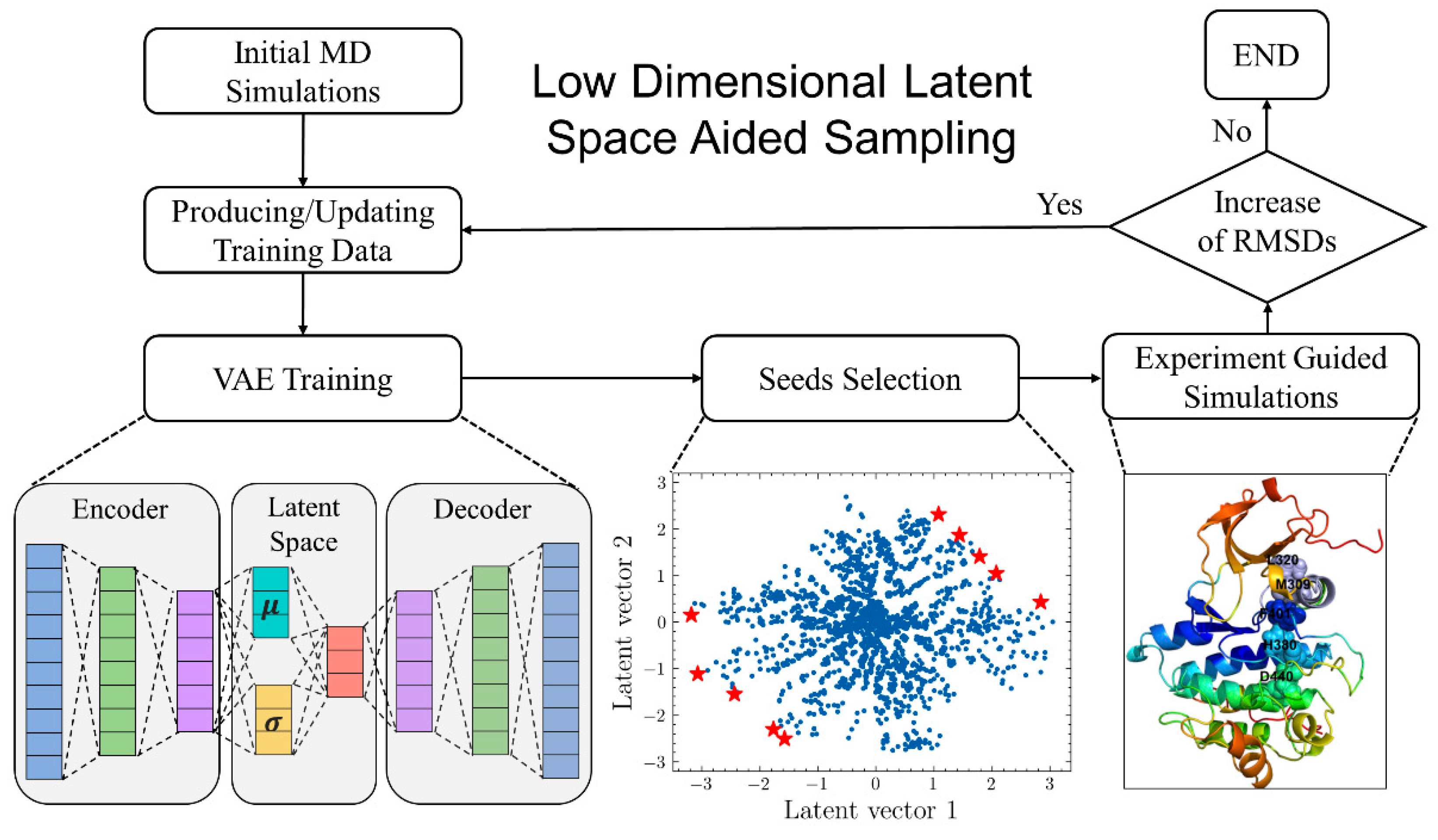
4. Learning Protein Conformational Dynamics Using AI Approaches
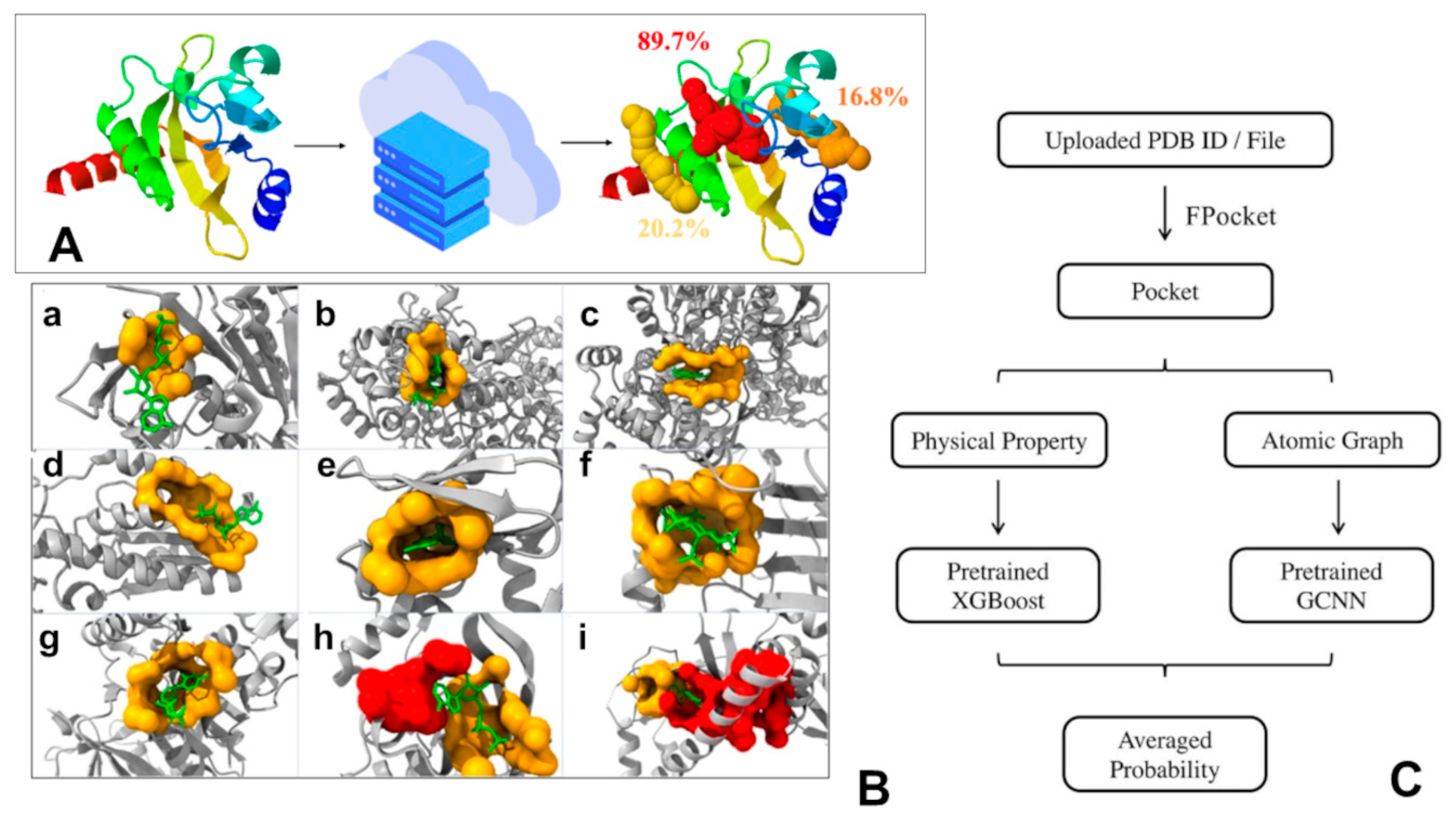
5. From AI-Enabled Mapping of Allosteric Conformational Landscapes to ML Discovery of Cryptic Allosteric Sites
6. AI-Augmented Cryo-EM and smFRET Approaches Expand the Universe of Protein Conformational Dynamics
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goh, G.B.; Hodas, N.O.; Vishnu, A. Deep learning for computational chemistry. J. Comput. Chem. 2017, 38, 1291–1307. [Google Scholar] [CrossRef]
- Dimitrov, T.; Kreisbeck, C.; Becker, J.S.; Aspuru-Guzik, A.; Saikin, S.K. Autonomous Molecular Design: Then and Now. ACS Appl. Mater. Interfaces 2019, 11, 24825–24836. [Google Scholar] [CrossRef] [PubMed]
- Mater, A.C.; Coote, M.L. Deep Learning in Chemistry. J. Chem. Inf. Model. 2019, 59, 2545–2559. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Changeux, J.-P. Allostery and the Monod-Wyman-Changeux Model after 50 Years. Annu. Rev. Biophys. 2012, 41, 103–133. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P.; Edelstein, S.J. Allosteric Mechanisms of Signal Transduction. Science 2005, 308, 1424–1428. [Google Scholar] [CrossRef]
- Popovych, N.; Sun, S.; Ebright, R.; Kalodimos, C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006, 13, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Hilser, V.J.; Wrabl, J.O.; Motlagh, H.N. Structural and Energetic Basis of Allostery. Annu. Rev. Biophys. 2012, 41, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, H.N.; Wrabl, J.O.; Li, J.; Hilser, V.J. The ensemble nature of allostery. Nature 2014, 508, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-J.; Nussinov, R. A Unified View of “How Allostery Works”. PLoS Comput. Biol. 2014, 10, e1003394. [Google Scholar] [CrossRef]
- Liu, J.; Nussinov, R. Allostery: An Overview of Its History, Concepts, Methods, and Applications. PLoS Comput. Biol. 2016, 12, e1004966. [Google Scholar] [CrossRef]
- Wodak, S.J.; Paci, E.; Dokholyan, N.V.; Berezovsky, I.N.; Horovitz, A.; Li, J.; Hilser, V.J.; Bahar, I.; Karanicolas, J.; Stock, G.; et al. Allostery in Its Many Disguises: From Theory to Applications. Structure 2019, 27, 566–578. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.-J.; Jang, H. Allostery, and how to define and measure signal transduction. Biophys. Chem. 2022, 283, 106766. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Kalodimos, C.G. Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Biophys. 2017, 46, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Kalodimos, C.G. NMR Studies of Large Proteins. J. Mol. Biol. 2017, 429, 2667–2676. [Google Scholar] [CrossRef]
- Kay, L.E. New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J. Mol. Biol. 2016, 428, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lisi, G.P.; Loria, J.P. Solution NMR Spectroscopy for the Study of Enzyme Allostery. Chem. Rev. 2016, 116, 6323–6369. [Google Scholar] [CrossRef]
- Vallurupalli, P.; Bouvignies, G.; Kay, L.E. Studying “Invisible” Excited Protein States in Slow Exchange with a Major State Conformation. J. Am. Chem. Soc. 2012, 134, 8148–8161. [Google Scholar] [CrossRef]
- Sekhar, A.; Kay, L.E. NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers. Proc. Natl. Acad. Sci. USA 2013, 110, 12867–12874. [Google Scholar] [CrossRef]
- Munte, C.E.; Erlach, M.B.; Kremer, W.; Koehler, J.; Kalbitzer, H.R. Distinct Conformational States of the Alzheimer β-Amyloid Peptide Can Be Detected by High-Pressure NMR Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 8943–8947. [Google Scholar] [CrossRef]
- Kalbitzer, H.R.; Rosnizeck, I.C.; Munte, C.E.; Narayanan, S.P.; Kropf, V.; Spoerner, P.-D.D.M. Intrinsic Allosteric Inhibition of Signaling Proteins by Targeting Rare Interaction States Detected by High-Pressure NMR Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 14242–14246. [Google Scholar] [CrossRef] [PubMed]
- ElGamacy, M.; Riss, M.; Zhu, H.; Truffault, V.; Coles, M. Mapping Local Conformational Landscapes of Proteins in Solution. Structure 2019, 27, 853–865.e5. [Google Scholar] [CrossRef] [PubMed]
- Bostock, M.J.; Solt, A.S.; Nietlispach, D. The role of NMR spectroscopy in mapping the conformational landscape of GPCRs. Curr. Opin. Struct. Biol. 2019, 57, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Usher, E.T.; Showalter, S.A. Mapping invisible epitopes by NMR spectroscopy. J. Biol. Chem. 2020, 295, 17411–17412. [Google Scholar] [CrossRef]
- Xie, T.; Saleh, T.; Rossi, P.; Kalodimos, C.G. Conformational states dynamically populated by a kinase determine its function. Science 2020, 370, eabc2754. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Saha, S.; Tvedt, N.C.; Yang, L.-W.; Bahar, I. Mutually beneficial confluence of structure-based modeling of protein dynamics and machine learning methods. Curr. Opin. Struct. Biol. 2023, 78, 102517. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.X.; Wayment-Steele, H.K.; Sultan, M.M.; Husic, B.E.; Pande, V.S. Variational encoding of complex dynamics. Phys. Rev. E 2018, 97, 062412. [Google Scholar] [CrossRef]
- Novelli, P.; Bonati, L.; Pontil, M.; Parrinello, M. Characterizing Metastable States with the Help of Machine Learning. J. Chem. Theory Comput. 2022, 18, 5195–5202. [Google Scholar] [CrossRef]
- Ribeiro, J.M.L.; Bravo, P.; Wang, Y.; Tiwary, P. Reweighted autoencoded variational Bayes for enhanced sampling (RAVE). J. Chem. Phys. 2018, 149, 072301. [Google Scholar] [CrossRef]
- Audagnotto, M.; Czechtizky, W.; De Maria, L.; Käck, H.; Papoian, G.; Tornberg, L.; Tyrchan, C.; Ulander, J. Machine learning/molecular dynamic protein structure prediction approach to investigate the protein conformational ensemble. Sci. Rep. 2022, 12, 10018. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, Z.; Cheng, K.J.; Shukla, D. Reinforcement Learning Based Adaptive Sampling: REAPing Rewards by Exploring Protein Conformational Landscapes. J. Phys. Chem. B 2018, 122, 8386–8395. [Google Scholar] [CrossRef]
- Kleiman, D.E.; Shukla, D. Multiagent Reinforcement Learning-Based Adaptive Sampling for Conformational Dynamics of Proteins. J. Chem. Theory Comput. 2022, 18, 5422–5434. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.; Sun, X.; Silva, R.R.; Bowman, G.R. Choice of Adaptive Sampling Strategy Impacts State Discovery, Transition Probabilities, and the Apparent Mechanism of Conformational Changes. J. Chem. Theory Comput. 2018, 14, 5459–5475. [Google Scholar] [CrossRef] [PubMed]
- Noé, F.; Olsson, S.; Köhler, J.; Wu, H. Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning. Science 2019, 365, eaaw1147. [Google Scholar] [CrossRef] [PubMed]
- Agajanian, S.; Alshahrani, M.; Bai, F.; Tao, P.; Verkhivker, G.M. Exploring and Learning the Universe of Protein Allostery Using Artificial Intelligence Augmented Biophysical and Computational Approaches. J. Chem. Inf. Model. 2023, 63, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Fields, S. Deep mutational scanning: A new style of protein science. Nat. Methods 2014, 11, 801–807. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef]
- Moulana, A.; Dupic, T.; Phillips, A.M.; Chang, J.; Roffler, A.; Greaney, A.J.; Starr, T.N.; Bloom, J.D.; Desai, M.M. The landscape of antibody binding affinity in SARS-CoV-2 Omicron BA.1 evolution. Elife 2023, 12, e83442. [Google Scholar] [CrossRef] [PubMed]
- Dadonaite, B.; Crawford, K.H.; Radford, C.E.; Farrell, A.G.; Yu, T.C.; Hannon, W.W.; Zhou, P.; Andrabi, R.; Burton, D.R.; Liu, L.; et al. A pseudovirus system enables deep mutational scanning of the full SARS-CoV-2 spike. Cell 2023, 186, 1263–1278.e20. [Google Scholar] [CrossRef]
- Faure, A.J.; Domingo, J.; Schmiedel, J.M.; Hidalgo-Carcedo, C.; Diss, G.; Ben Lehner, B. Mapping the energetic and allosteric landscapes of protein binding domains. Nature 2022, 604, 175–183. [Google Scholar] [CrossRef]
- Weng, C.; Faure, A.J.; Lehner, B. The energetic and allosteric landscape for KRAS inhibition. bioRxiv 2022. [Google Scholar] [CrossRef]
- Esposito, D.; Weile, J.; Shendure, J.; Starita, L.M.; Papenfuss, A.T.; Roth, F.P.; Fowler, D.M.; Rubin, A.F. MaveDB: An open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol. 2019, 20, 223. [Google Scholar] [CrossRef]
- Weile, J.; Sun, S.; Cote, A.G.; Knapp, J.; Verby, M.; Mellor, J.C.; Wu, Y.; Pons, C.; Wong, C.; van Lieshout, N.; et al. A framework for exhaustively mapping functional missense variants. Mol. Syst. Biol. 2017, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, L.; Brooks, A.N.; Noon, K.; Cialek, C.; Clark-ElSayed, A.; Schalck, T.; Krishnamurthy, N.; Versées, W.; Vranken, W.; Michiels, J. Deep mutational scanning of essential bacterial proteins can guide antibiotic development. Nat. Commun. 2023, 14, 241. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hannon, W.W.; Loes, A.N.; Hauser, K.; Dillen, J.R.; Ferri, E.; Farrell, A.G.; Dadonaite, B.; McCallum, M.; et al. Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science 2022, 377, 420–424. [Google Scholar] [CrossRef]
- Moulana, A.; Dupic, T.; Phillips, A.M.; Chang, J.; Nieves, S.; Roffler, A.A.; Greaney, A.J.; Starr, T.N.; Bloom, J.D.; Desai, M.M. Compensatory epistasis maintains ACE2 affinity in SARS-CoV-2 Omicron BA.1. Nat. Commun. 2022, 13, 7011. [Google Scholar] [CrossRef]
- Azbukina, N.; Zharikova, A.; Ramensky, V. Intragenic compensation through the lens of deep mutational scanning. Biophys. Rev. 2022, 14, 1161–1182. [Google Scholar] [CrossRef]
- Roychowdhury, H.; Romero, P.A. Microfluidic deep mutational scanning of the human executioner caspases reveals differences in structure and regulation. Cell Death Discov. 2022, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Mathy, C.J.; Mishra, P.; Flynn, J.M.; Perica, T.; Mavor, D.; Bolon, D.N.; Kortemme, T. A complete allosteric map of a GTPase switch in its native cellular network. Cell Syst. 2023, 14, 237–246.e7. [Google Scholar] [CrossRef] [PubMed]
- Tack, D.S.; Tonner, P.D.; Pressman, A.; Olson, N.D.; Levy, S.F.; Romantseva, E.F.; Alperovich, N.; Vasilyeva, O.; Ross, D. The genotype-phenotype landscape of an allosteric protein. Mol. Syst. Biol. 2021, 17, e10179. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Front. Genet. 2023, 14, 1087267. [Google Scholar] [CrossRef]
- Song, H.; Bremer, B.J.; Hinds, E.C.; Raskutti, G.; Romero, P.A. Inferring Protein Sequence-Function Relationships with Large-Scale Positive-Unlabeled Learning. Cell Syst. 2021, 12, 92–101.e8. [Google Scholar] [CrossRef]
- Leander, M.; Yuan, Y.; Meger, A.; Cui, Q.; Raman, S. Functional plasticity and evolutionary adaptation of allosteric regulation. Proc. Natl. Acad. Sci. USA 2020, 117, 25445–25454. [Google Scholar] [CrossRef]
- Leander, M.; Liu, Z.; Cui, Q.; Raman, S. Deep mutational scanning and machine learning reveal structural and molecular rules governing allosteric hotspots in homologous proteins. Elife 2022, 11, e79932. [Google Scholar] [CrossRef]
- Gelman, S.; Fahlberg, S.A.; Heinzelman, P.; Romero, P.A.; Gitter, A. Neural networks to learn protein sequence–function relationships from deep mutational scanning data. Proc. Natl. Acad. Sci. USA 2021, 118, e2104878118. [Google Scholar] [CrossRef]
- D’costa, S.; Hinds, E.C.; Freschlin, C.R.; Song, H.; Romero, P.A. Inferring protein fitness landscapes from laboratory evolution experiments. PLoS Comput. Biol. 2023, 19, e1010956. [Google Scholar] [CrossRef]
- Freschlin, C.R.; Fahlberg, S.; Romero, P. Machine learning to navigate fitness landscapes for protein engineering. Curr. Opin. Biotechnol. 2022, 75, 102713. [Google Scholar] [CrossRef] [PubMed]
- Vaishnav, E.D.; de Boer, C.G.; Molinet, J.; Yassour, M.; Fan, L.; Adiconis, X.; Thompson, D.A.; Levin, J.Z.; Cubillos, F.A.; Regev, A. The evolution, evolvability and engineering of gene regulatory DNA. Nature 2022, 603, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Tareen, A.; Kooshkbaghi, M.; Posfai, A.; Ireland, W.T.; McCandlish, D.M.; Kinney, J.B. MAVE-NN: Learning genotype-phenotype maps from multiplex assays of variant effect. Genome Biol. 2022, 23, 98. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Horovitz, A. Extending the New Generation of Structure Predictors to Account for Dynamics and Allostery. J. Mol. Biol. 2021, 433, 167007. [Google Scholar] [CrossRef]
- Nussinov, R.; Zhang, M.; Liu, Y.; Jang, H. AlphaFold, Artificial Intelligence (AI), and Allostery. J. Phys. Chem. B 2022, 126, 6372–6383. [Google Scholar] [CrossRef]
- Nussinov, R.; Zhang, M.; Liu, Y.; Jang, H. AlphaFold, allosteric, and orthosteric drug discovery: Ways forward. Drug Discov. Today 2023, 28, 103551. [Google Scholar] [CrossRef]
- del Alamo, D.; Sala, D.; Mchaourab, H.S.; Meiler, J. Sampling alternative conformational states of transporters and receptors with AlphaFold2. Elife 2022, 11, e75751. [Google Scholar] [CrossRef]
- Saldaño, T.; Escobedo, N.; Marchetti, J.; Zea, D.J.; Mac Donagh, J.; Rueda, A.J.V.; Gonik, E.; Melani, A.G.; Nechcoff, J.N.; Salas, M.N.; et al. Impact of protein conformational diversity on AlphaFold predictions. Bioinformatics 2022, 38, 2742–2748. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. Broadly Applicable and Accurate Protein Design by Integrating Structure Prediction Networks and Diffusion Generative Models, Cold Spring Harbor Laboratory. bioRxiv 2022. [Google Scholar] [CrossRef]
- Bonati, L.; Piccini, G.; Parrinello, M. Deep learning the slow modes for rare events sampling. Proc. Natl. Acad. Sci. USA 2021, 118, e2113533118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ribeiro, J.M.L.; Tiwary, P. Past–future information bottleneck for sampling molecular reaction coordinate simultaneously with thermodynamics and kinetics. Nat. Commun. 2019, 10, 3573. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tiwary, P. State predictive information bottleneck. J. Chem. Phys. 2021, 154, 134111. [Google Scholar] [CrossRef]
- Mehdi, S.; Wang, D.; Pant, S.; Tiwary, P. Accelerating All-Atom Simulations and Gaining Mechanistic Understanding of Biophysical Systems through State Predictive Information Bottleneck. J. Chem. Theory Comput. 2022, 18, 3231–3238. [Google Scholar] [CrossRef]
- Tsai, S.-T.; Fields, E.; Xu, Y.; Kuo, E.-J.; Tiwary, P. Path sampling of recurrent neural networks by incorporating known physics. Nat. Commun. 2022, 13, 7231. [Google Scholar] [CrossRef]
- Janson, G.; Valdes-Garcia, G.; Heo, L.; Feig, M. Direct generation of protein conformational ensembles via machine learning. Nat. Commun. 2023, 4, 774. [Google Scholar] [CrossRef]
- Mardt, A.; Pasquali, L.; Wu, H.; Noé, F. VAMPnets for deep learning of molecular kinetics. Nat. Commun. 2018, 9, 5. [Google Scholar] [CrossRef]
- Ghorbani, M.; Prasad, S.; Klauda, J.B.; Brooks, B.R. Variational embedding of protein folding simulations using Gaussian mixture variational autoencoders. J. Chem. Phys. 2021, 155, 194108. [Google Scholar] [CrossRef]
- Ghorbani, M.; Prasad, S.; Klauda, J.B.; Brooks, B.R. GraphVAMPNet, using graph neural networks and variational approach to Markov processes for dynamical modeling of biomolecules. J. Chem. Phys. 2022, 156, 184103. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Johannissen, L.O.; Hay, S. Predicting new protein conformations from molecular dynamics simulation conformational landscapes and machine learning. Proteins 2021, 89, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Jiang, X.; Trozzi, F.; Xiao, S.; Larson, E.C.; Tao, P. Explore Protein Conformational Space with Variational Autoencoder. Front. Mol. Biosci. 2021, 8, 781635. [Google Scholar] [CrossRef]
- Tian, H.; Jiang, X.; Xiao, S.; La Force, H.; Larson, E.C.; Tao, P. LAST: Latent Space-Assisted Adaptive Sampling for Protein Trajectories. J. Chem. Inf. Model. 2022, 63, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Uyar, A.; Karamyan, V.; Dickson, A. Long-Range Changes in Neurolysin Dynamics Upon Inhibitor Binding. J. Chem. Theory Comput. 2018, 14, 444–452. [Google Scholar] [CrossRef]
- Duro, N.; Varma, S. Role of Structural Fluctuations in Allosteric Stimulation of Paramyxovirus Hemagglutinin-Neuraminidase. Structure 2019, 27, 1601–1611.e2. [Google Scholar] [CrossRef]
- Zhou, H.; Dong, Z.; Verkhivker, G.; Zoltowski, B.D.; Tao, P. Allosteric mechanism of the circadian protein Vivid resolved through Markov state model and machine learning analysis. PLoS Comput. Biol. 2019, 15, e1006801. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Taneishi, K.; Yonezawa, Y. Autoencoder-Based Detection of Dynamic Allostery Triggered by Ligand Binding Based on Molecular Dynamics. J. Chem. Inf. Model. 2019, 59, 4043–4051. [Google Scholar] [CrossRef]
- Greener, J.G.; Sternberg, M.J. AlloPred: Prediction of allosteric pockets on proteins using normal mode perturbation analysis. BMC Bioinform. 2015, 16, 335. [Google Scholar] [CrossRef]
- Huang, W.; Lu, S.; Huang, Z.; Liu, X.; Mou, L.; Luo, Y.; Zhao, Y.; Liu, Y.; Chen, Z.; Hou, T.; et al. Allosite: A method for predicting allosteric sites. Bioinformatics 2013, 29, 2357–2359. [Google Scholar] [CrossRef]
- Song, K.; Liu, X.; Huang, W.; Lu, S.; Shen, Q.; Zhang, L.; Zhang, J. Improved Method for the Identification and Validation of Allosteric Sites. J. Chem. Inf. Model. 2017, 57, 2358–2363. [Google Scholar] [CrossRef]
- Lu, S.; Shen, Q.; Zhang, J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Accounts Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef]
- Mishra, S.K.; Kandoi, G.; Jernigan, R.L. Coupling dynamics and evolutionary information with structure to identify protein regulatory and functional binding sites. Proteins Struct. Funct. Bioinform. 2019, 87, 850–868. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, D.; Sanchez, R. EasyMIFs and SiteHound: A toolkit for the identification of ligand-binding sites in protein structures. Bioinformatics 2009, 25, 3185–3186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Lin, B.; Schroeder, M.; Huang, B. Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 2011, 27, 2083–2088. [Google Scholar] [CrossRef]
- Jian, J.-W.; Elumalai, P.; Pitti, T.; Wu, C.Y.; Tsai, K.-C.; Chang, J.-Y.; Peng, H.-P.; Yang, A.-S. Predicting Ligand Binding Sites on Protein Surfaces by 3-Dimensional Probability Density Distributions of Interacting Atoms. PLoS ONE 2016, 11, e0160315. [Google Scholar] [CrossRef]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Z.; Zhang, S.; Wang, S.; Wang, X.; Li, Z. FRSite: Protein drug binding site prediction based on faster R–CNN. J. Mol. Graph. Model. 2019, 93, 107454. [Google Scholar] [CrossRef]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Improving detection of protein-ligand binding sites with 3D segmentation. Sci. Rep. 2020, 10, 5035. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, J.; Han, W.; Xu, D. Neural relational inference to learn long-range allosteric interactions in proteins from molecular dynamics simulations. Nat. Commun. 2022, 13, 1661. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.D.; Zimmerman, M.I.; Meller, A.; Chung, M.; Swamidass, S.J.; Bowman, G.R. Deep learning the structural determinants of protein biochemical properties by comparing structural ensembles with DiffNets. Nat. Commun. 2021, 12, 3023. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, S.K.; Axenopoulos, A.; Daras, P. DeepSurf: A surface-based deep learning approach for the prediction of ligand binding sites on proteins. Bioinformatics 2021, 37, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Krivák, R.; Hoksza, D. P2Rank: Machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J. Cheminform. 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Jakubec, D.; Skoda, P.; Krivak, R.; Novotny, M.; Hoksza, D. PrankWeb 3: Accelerated ligand-binding site predictions for experimental and modelled protein structures. Nucleic Acids Res. 2022, 50, W593–W597. [Google Scholar] [CrossRef] [PubMed]
- Kozlovskii, I.; Popov, P. Spatiotemporal identification of druggable binding sites using deep learning. Commun. Biol. 2020, 3, 618. [Google Scholar] [CrossRef]
- Schmidtke, P.; Souaille, C.; Estienne, F.; Baurin, N.; Kroemer, R.T. Large-Scale Comparison of Four Binding Site Detection Algorithms. J. Chem. Inf. Model. 2010, 50, 2191–2200. [Google Scholar] [CrossRef]
- Tian, H.; Jiang, X.; Tao, P. PASSer: Prediction of allosteric sites server. Mach. Learn. Sci. Technol. 2021, 2, 035015. [Google Scholar] [CrossRef]
- Xiao, S.; Tian, H.; Tao, P. PASSer2.0: Accurate Prediction of Protein Allosteric Sites through Automated Machine Learning. Front. Mol. Biosci. 2022, 9, 879251. [Google Scholar] [CrossRef]
- Tian, H.; Xiao, S.; Jiang, X.; Tao, P. PASSerRank: Prediction of Allosteric Sites with Learning to Rank. arXiv 2023, arXiv:2302.01117v1. [Google Scholar]
- Meller, A.; Ward, M.; Borowsky, J.; Kshirsagar, M.; Lotthammer, J.M.; Oviedo, F.; Ferres, J.L.; Bowman, G.R. Predicting locations of cryptic pockets from single protein structures using the PocketMiner graph neural network. Nat. Commun. 2023, 14, 1177. [Google Scholar] [CrossRef]
- Meller, A.; Lotthammer, J.M.; Smith, L.G.; Novak, B.; Lee, L.; Kuhn, C.C.; Greenberg, L.; Leinwand, L.; Greenberg, M.J.; Bowman, G.R. Drug specificity and affinity are encoded in the probability of cryptic pocket opening in myosin motor domains. Elife 2023, 12, e83602. [Google Scholar] [CrossRef] [PubMed]
- Merk, A.; Bartesaghi, A.; Banerjee, S.; Falconieri, V.; Rao, P.; Davis, M.I.; Pragani, R.; Boxer, M.B.; Earl, L.A.; Milne, J.L.; et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell 2016, 165, 1698–1707. [Google Scholar] [CrossRef]
- Coffino, P.; Cheng, Y. Allostery Modulates Interactions between Proteasome Core Particles and Regulatory Particles. Biomolecules 2022, 12, 764. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Palczewski, K.; Engel, A.; Stahlberg, H.; Kovacik, L. Cryo-EM structure of phosphodiesterase 6 reveals insights into the allosteric regulation of type I phosphodiesterases. Sci. Adv. 2019, 5, eaav4322. [Google Scholar] [CrossRef]
- Chung, J.M.; Durie, C.L.; Lee, J. Artificial Intelligence in Cryo-Electron Microscopy. Life 2022, 12, 1267. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gong, H.; Liu, G.; Li, M.; Yan, C.; Xia, T.; Li, X.; Zeng, J. DeepPicker: A deep learning approach for fully automated particle picking in cryo-EM. J. Struct. Biol. 2016, 195, 325–336. [Google Scholar] [CrossRef]
- Nguyen, N.P.; Ersoy, I.; Gotberg, J.; Bunyak, F.; White, T.A. DRPnet: Automated particle picking in cryo-electron micrographs using deep regression. BMC Bioinform. 2021, 22, 55. [Google Scholar] [CrossRef]
- Al-Azzawi, A.; Ouadou, A.; Max, H.; Duan, Y.; Tanner, J.J.; Cheng, J. DeepCryoPicker: Fully automated deep neural network for single protein particle picking in cryo-EM. BMC Bioinform. 2020, 21, 509. [Google Scholar] [CrossRef]
- Zhu, Y.; Ouyang, Q.; Mao, Y. A deep convolutional neural network approach to single-particle recognition in cryo-electron microscopy. BMC Bioinform. 2017, 18, 1–10. [Google Scholar] [CrossRef]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Kinman, L.F.; Powell, B.M.; Zhong, E.D.; Berger, B.; Davis, J.H. Uncovering structural ensembles from single-particle cryo-EM data using cryoDRGN. Nat. Protoc. 2023, 18, 319–339. [Google Scholar] [CrossRef] [PubMed]
- Hamitouche, I.; Jonic, S. DeepHEMNMA: ResNet-based hybrid analysis of continuous conformational heterogeneity in cryo-EM single particle images. Front. Mol. Biosci. 2022, 9, 965645. [Google Scholar] [CrossRef] [PubMed]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Förster resonance energy transfer. Science 2018, 359, eaan1133. [Google Scholar] [CrossRef] [PubMed]
- Lerner, E.; Barth, A.; Hendrix, J.; Ambrose, B.; Birkedal, V.; Blanchard, S.C.; Börner, R.; Chung, H.S.; Cordes, T.; Craggs, T.D.; et al. FRET-based dynamic structural biology: Challenges, perspectives and an appeal for open-science practices. Elife 2021, 10, e60416. [Google Scholar] [CrossRef]
- Agam, G.; Gebhardt, C.; Popara, M.; Mächtel, R.; Folz, J.; Ambrose, B.; Chamachi, N.; Chung, S.Y.; Craggs, T.D.; de Boer, M.; et al. Reliability and accuracy of single-molecule FRET studies for characterization of structural dynamics and distances in proteins. Nat. Methods 2023, 20, 523–535. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Sugita, Y. Linking time-series of single-molecule experiments with molecular dynamics simulations by machine learning. Elife 2018, 7, e32668. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Sugita, Y. Use of single-molecule time-series data for refining conformational dynamics in molecular simulations. Curr. Opin. Struct. Biol. 2020, 61, 153–159. [Google Scholar] [CrossRef]
- Zheng, Y.; Cui, Q. Multiple Pathways and Time Scales for Conformational Transitions in apo-Adenylate Kinase. J. Chem. Theory Comput. 2018, 14, 1716–1726. [Google Scholar] [CrossRef]
- Dimura, M.; Peulen, T.-O.; Sanabria, H.; Rodnin, D.; Hemmen, K.; Hanke, C.A.; Seidel, C.A.M.; Gohlke, H. Automated and optimally FRET-assisted structural modeling. Nat. Commun. 2020, 11, 5394. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, Y.; Cui, Y.; Yuan, J.; Fang, X. Deep learning in single-molecule imaging and analysis: Recent advances and prospects. Chem. Sci. 2022, 13, 11964–11980. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.; Sletfjerding, M.B.; Jensen, S.B.; Stella, S.; Paul, B.; Malle, M.G.; Montoya, G.; Petersen, T.C.; Hatzakis, N.S. DeepFRET, a software for rapid and automated single-molecule FRET data classification using deep learning. Elife 2020, 9, e60404. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, L.; Johnson-Buck, A.; Walter, N.G. Automatic classification and segmentation of single-molecule fluorescence time traces with deep learning. Nat. Commun. 2020, 11, 5833. [Google Scholar] [CrossRef]
- Durham, R.J.; Paudyal, N.; Carrillo, E.; Bhatia, N.K.; Maclean, D.M.; Berka, V.; Dolino, D.M.; Gorfe, A.A.; Jayaraman, V. Conformational spread and dynamics in allostery of NMDA receptors. Proc. Natl. Acad. Sci. USA 2020, 117, 3839–3847. [Google Scholar] [CrossRef]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. Elife 2022, 11, e75433. [Google Scholar] [CrossRef]
- Wolf, S.; Sohmen, B.; Hellenkamp, B.; Thurn, J.; Stock, G.; Hugel, T. Hierarchical dynamics in allostery following ATP hydrolysis monitored by single molecule FRET measurements and MD simulations. Chem. Sci. 2021, 12, 3350–3359. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.-M.; Quast, R.B.; Fatemi, F.; Rondard, P.; Pin, J.-P.; Margeat, E. Allosteric modulators enhance agonist efficacy by increasing the residence time of a GPCR in the active state. Nat. Commun. 2021, 12, 5426. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkhivker, G.; Alshahrani, M.; Gupta, G.; Xiao, S.; Tao, P. From Deep Mutational Mapping of Allosteric Protein Landscapes to Deep Learning of Allostery and Hidden Allosteric Sites: Zooming in on “Allosteric Intersection” of Biochemical and Big Data Approaches. Int. J. Mol. Sci. 2023, 24, 7747. https://doi.org/10.3390/ijms24097747
Verkhivker G, Alshahrani M, Gupta G, Xiao S, Tao P. From Deep Mutational Mapping of Allosteric Protein Landscapes to Deep Learning of Allostery and Hidden Allosteric Sites: Zooming in on “Allosteric Intersection” of Biochemical and Big Data Approaches. International Journal of Molecular Sciences. 2023; 24(9):7747. https://doi.org/10.3390/ijms24097747
Chicago/Turabian StyleVerkhivker, Gennady, Mohammed Alshahrani, Grace Gupta, Sian Xiao, and Peng Tao. 2023. "From Deep Mutational Mapping of Allosteric Protein Landscapes to Deep Learning of Allostery and Hidden Allosteric Sites: Zooming in on “Allosteric Intersection” of Biochemical and Big Data Approaches" International Journal of Molecular Sciences 24, no. 9: 7747. https://doi.org/10.3390/ijms24097747