
Probing Mechanisms of Binding and Allostery in the SARS-CoV-2 Spike Omicron Variant Complexes with the Host Receptor: Revealing Functional Roles of the Binding Hotspots in Mediating Epistatic Effects and Communication with Allosteric Pockets


Abstract
:1. Introduction
2. Results and Discussion
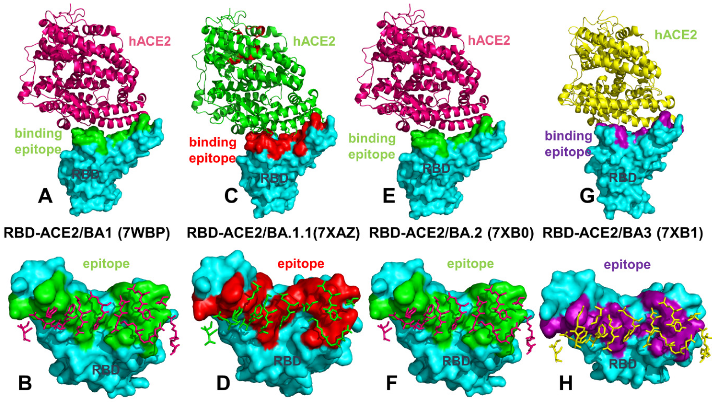
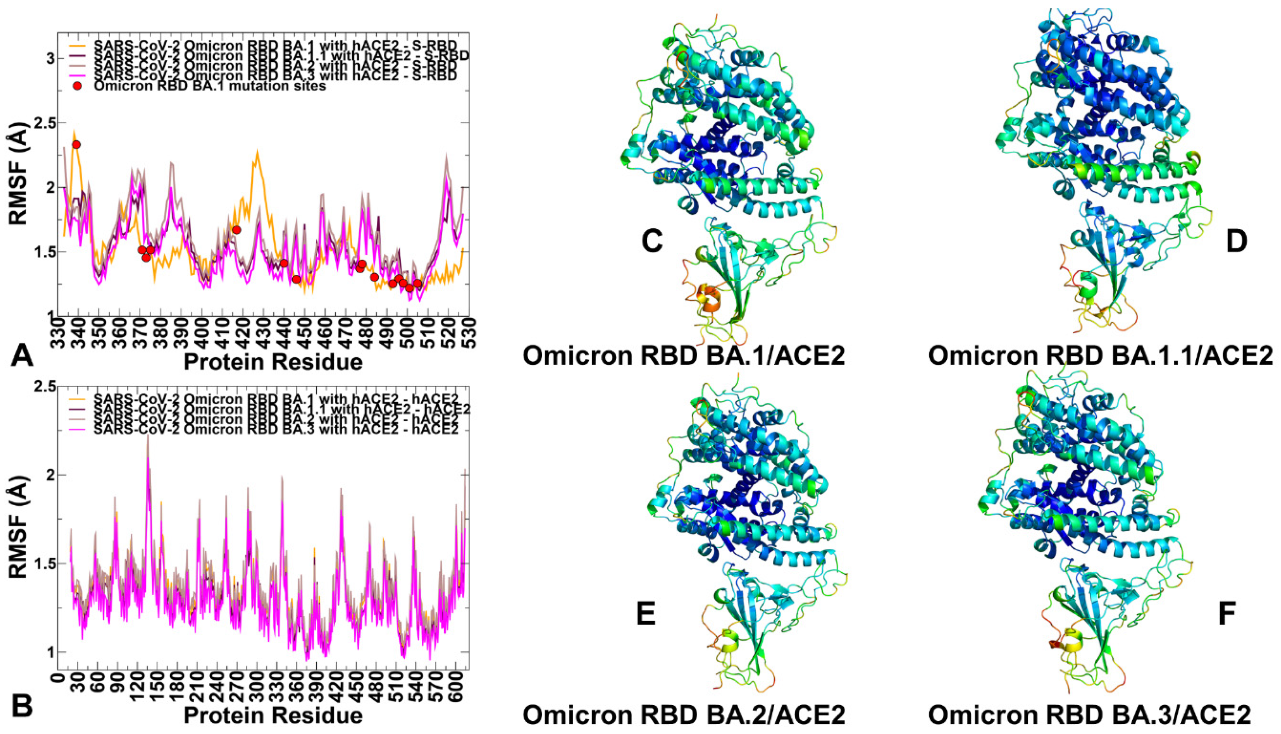
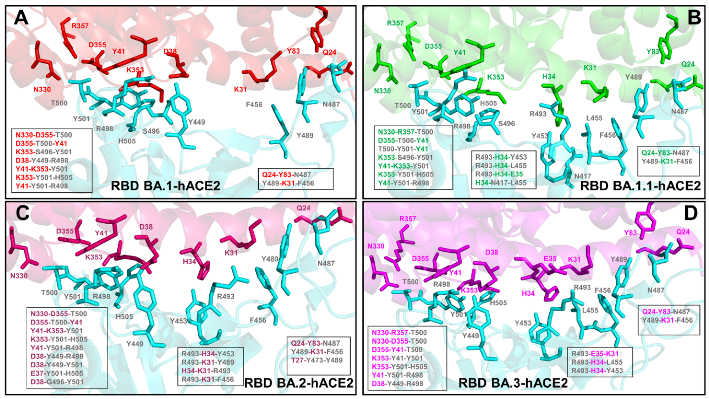
2.1. Atomistic MD Simulations Reveal Common and Distinct Signatures of Conformational Dynamics and Interaction Patterns in the ACE2 Complexes with the Omicron RBD Variants
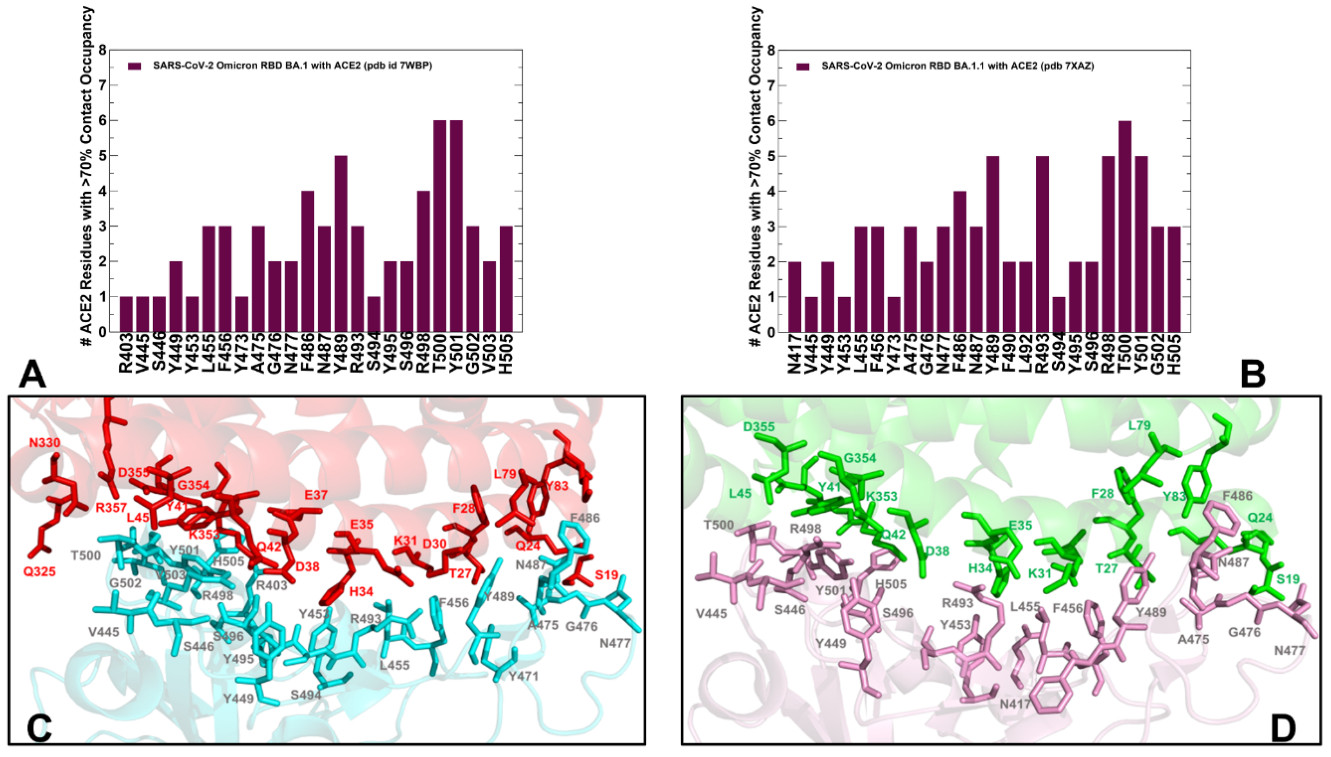
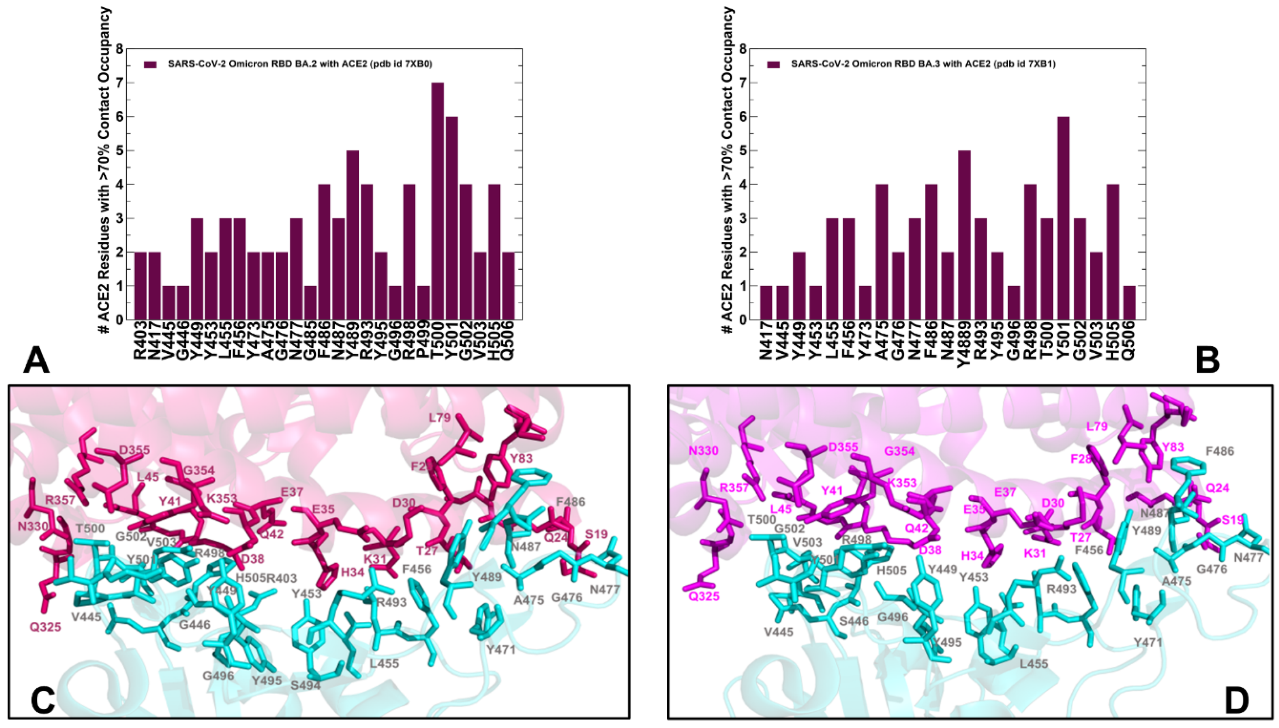
2.2. Ensemble-Based Analysis of the Intermolecular Interactions in the Omicron RBD-hACE2 Complexes: Quantifying Common and Unique Energetic Signatures of the Omicron RBD Variants
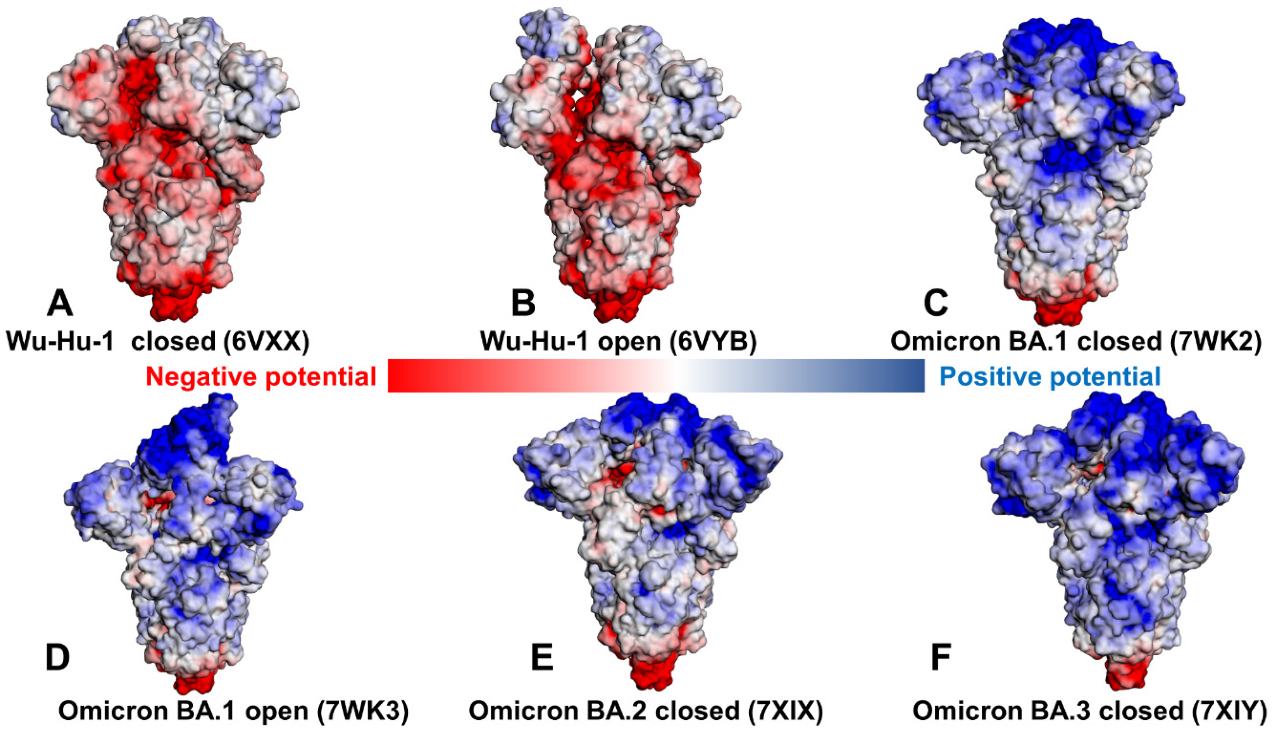
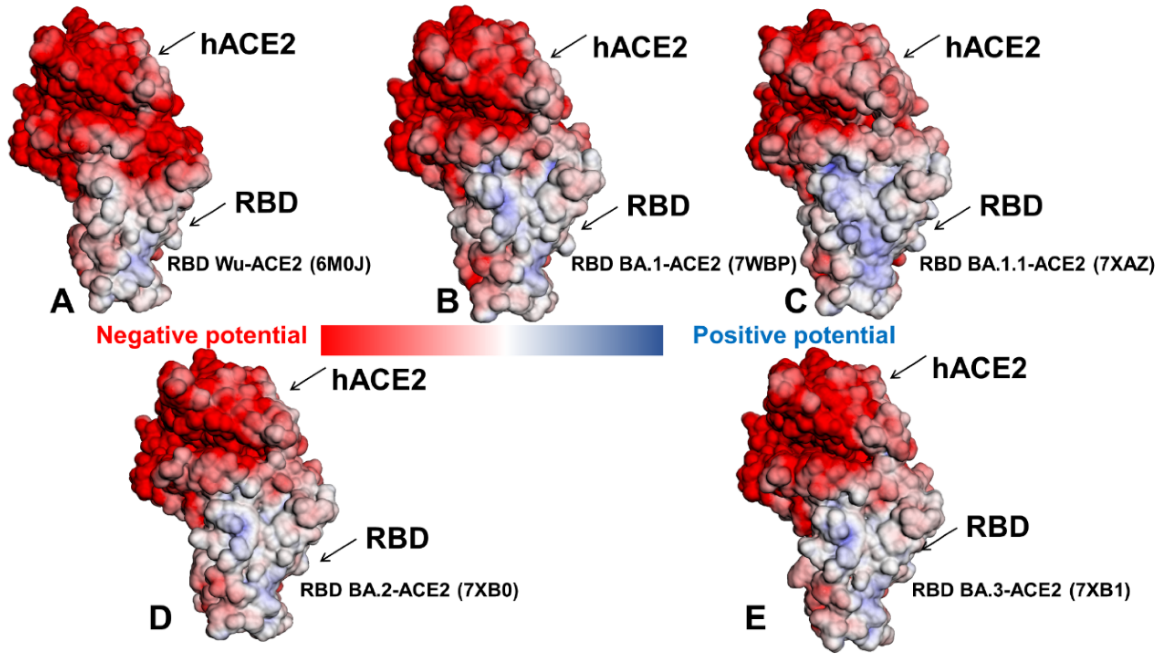
2.3. Binding Free Energy Analysis and Electrostatic Interaction Potentials in the Omicron RBD-hACE2 Complexes
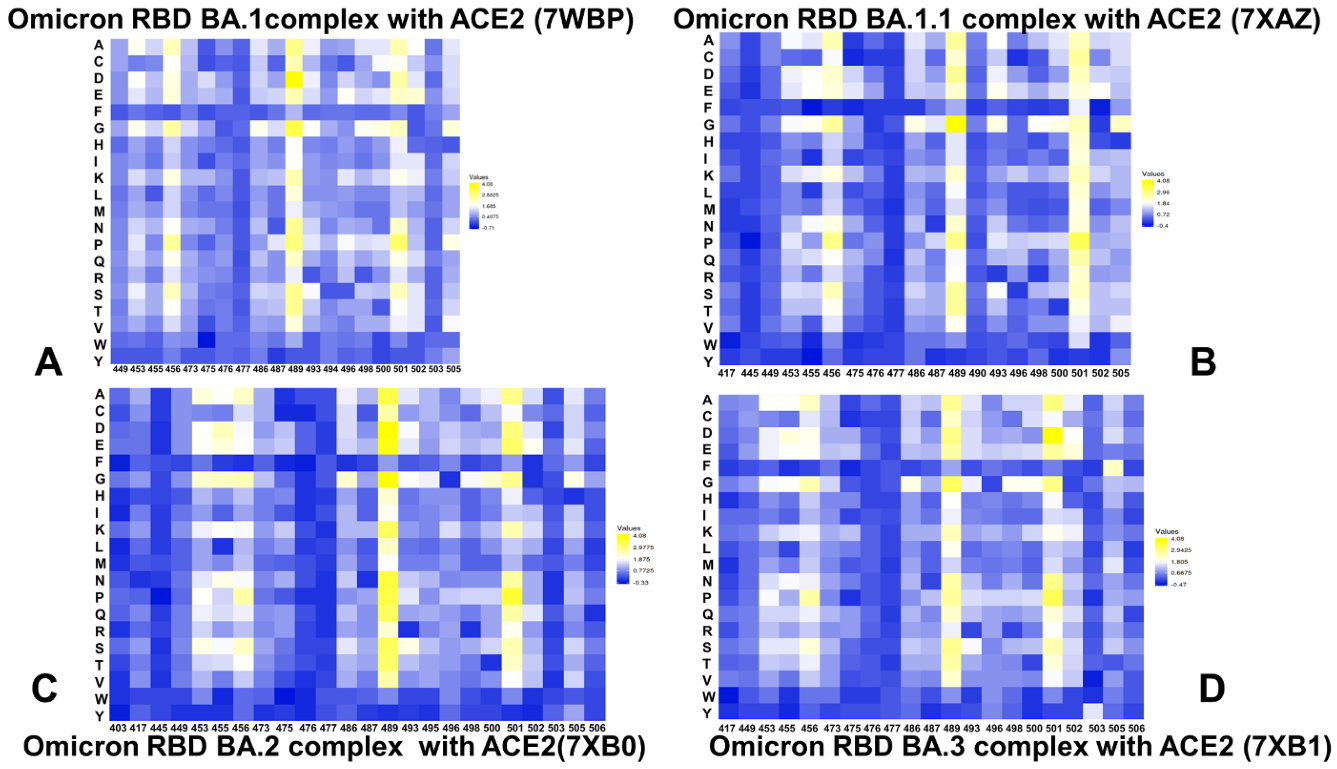
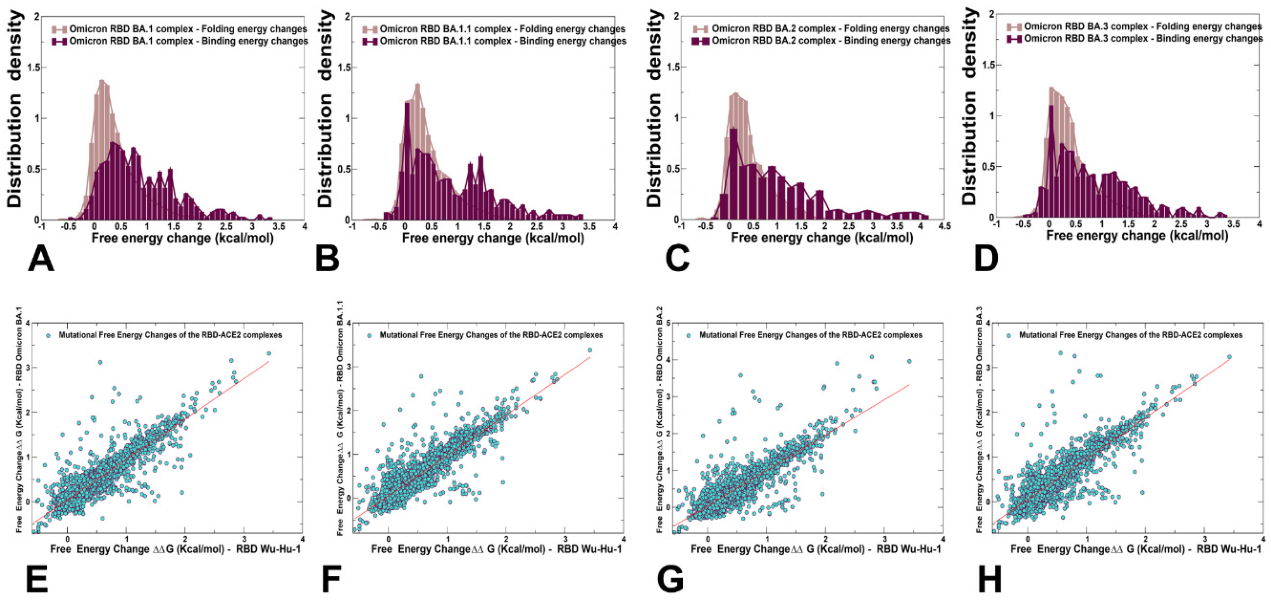
2.4. Ensemble-Based Mutational Sensitivity Analysis Identifies Key Structural Stability and Binding Affinity Hotspots in the SARS-CoV-2 RBD Complexes with ACE2
2.5. Dynamic Modeling of the Residue Interaction Networks and Community Analysis of the Omicron RBD Complexes Detail Role of Binding Hotspots in Mediating Epistatic Interaction Effects
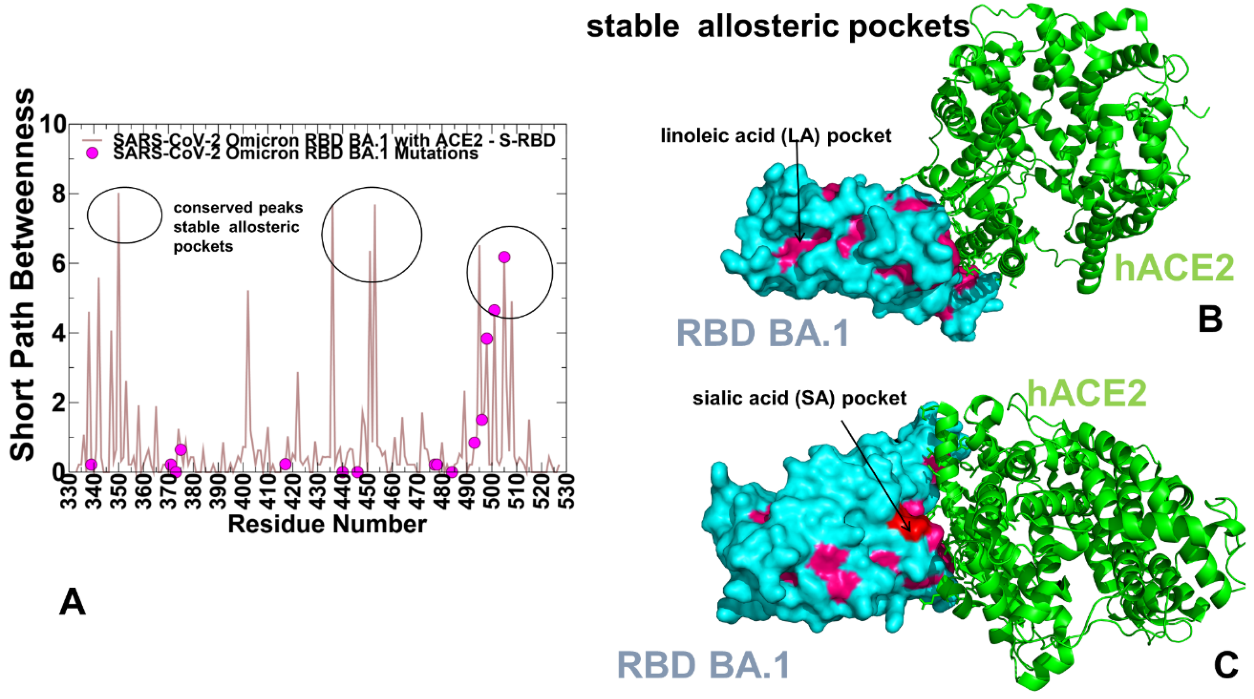
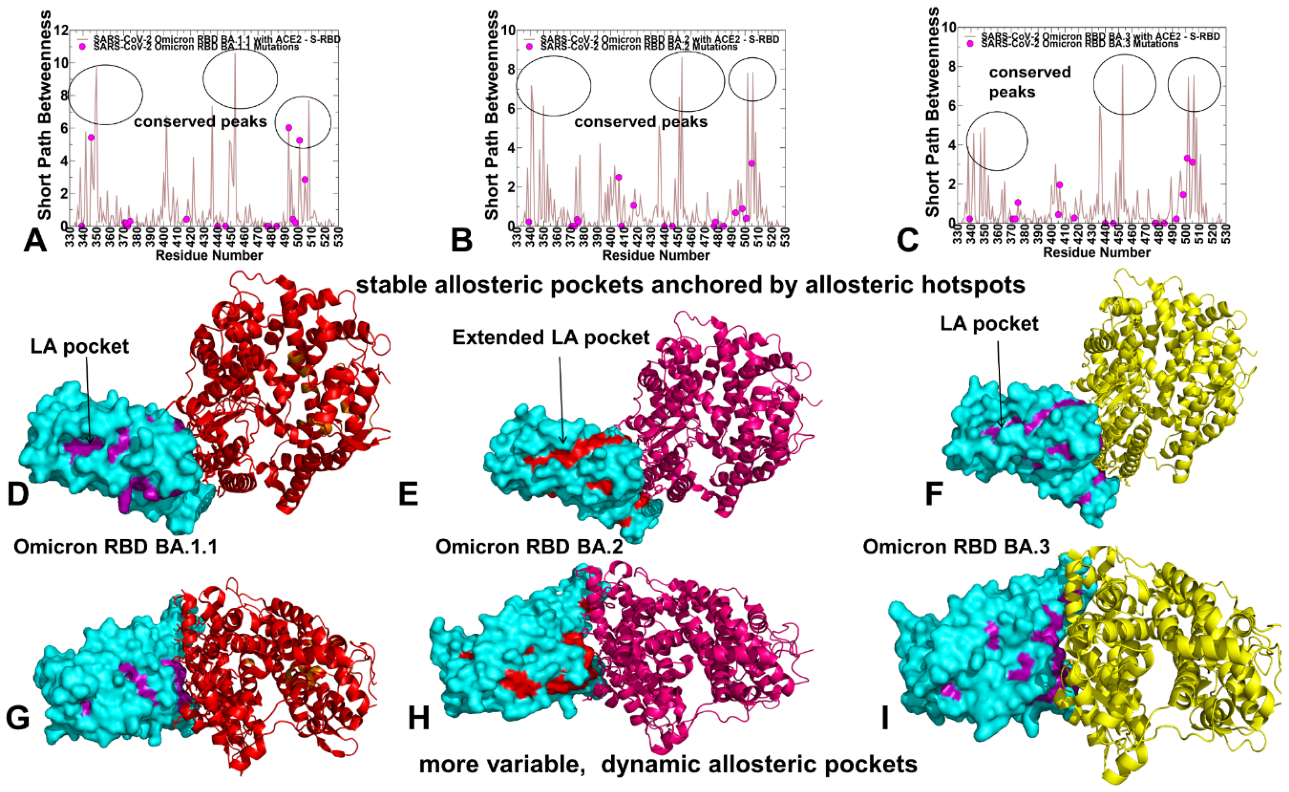
2.6. Reversed Allosteric Communication Analysis of the Omicron RBD Complexes Identifies Conserved Allosteric Binding Site and Suggests That Allosteric Hotspots Anchor Allosteric Pockets in the RBD
3. Materials and Methods
3.1. Molecular Dynamics Simulations
3.2. Electrostatic Calculations
3.3. Binding Free Energy Computations
3.4. Mutational Scanning and Sensitivity Analysis
3.5. Dynamic Network Analysis and Topological Clique-Based Model for Assessment of Non-Additivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Bowen, J.E.; Corti, D.; Veesler, D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020, 27, 942–949. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Costello, S.M.; Shoemaker, S.R.; Hobbs, H.T.; Nguyen, A.W.; Hsieh, C.L.; Maynard, J.A.; McLellan, J.S.; Pak, J.E.; Marqusee, S. The SARS-CoV-2 spike reversibly samples an open-trimer conformation exposing novel epitopes. Nat. Struct. Mol. Biol. 2022, 27, 229–238. [Google Scholar] [CrossRef]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The emerging plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, D.; Han, Y.; Lu, M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses 2022, 14, 1255. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Yang, Z.; Han, Y.; Ding, S.; Shi, W.; Zhou, T.; Finzi, A.; Kwong, P.D.; Mothes, W.; Lu, M. SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy. mBio 2022, 13, e0322721. [Google Scholar] [CrossRef]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. Elife 2022, 11, e75433. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Li, X.; Peng, Q.; Su, C.; Huang, B.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef]
- Saville, J.W.; Mannar, D.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.P.; Zhou, S.; Tuttle, K.S.; Sekirov, I.; Kim, A.; et al. Structural and biochemical rationale for enhanced spike protein fitness in delta and kappa SARS-CoV-2 variants. Nat. Commun. 2022, 13, 742. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular basis of receptor binding and antibody neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural diversity of the SARS-CoV-2 Omicron spike. bioRxiv 2022. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484.e415. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife 2021, 10, e70658. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 2022, 602, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; Rits-Volloch, S.; et al. Structural and functional impact by SARS-CoV-2 Omicron spike mutations. Cell Rep. 2022, 39, 110729. [Google Scholar] [CrossRef] [PubMed]
- Koehler, M.; Ray, A.; Moreira, R.A.; Juniku, B.; Poma, A.B.; Alsteens, D. Molecular insights into receptor binding energetics and neutralization of SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6977. [Google Scholar] [CrossRef]
- Bauer, M.S.; Gruber, S.; Hausch, A.; Gomes, P.S.F.C.; Milles, L.F.; Nicolaus, T.; Schendel, L.C.; Navajas, P.L.; Procko, E.; Lietha, D.; et al. A tethered ligand assay to probe SARS-CoV-2: ACE2 interactions. Proc. Natl. Acad. Sci. USA 2022, 119, e2114397119. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, Y.; Fei, P.; Zhang, T.; Yao, D.; Gao, Y.; Liu, J.; Chen, H.; Lu, Q.; Mudianto, T.; et al. Mechanical activation of spike fosters SARS-CoV-2 viral infection. Cell Res. 2021, 31, 1047–1060. [Google Scholar] [CrossRef]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L.; et al. Structural basis of human ACE2 higher binding affinity to currently circulating Omicron SARS-CoV-2 sub-variants BA.2 and BA.1.1. Cell 2022, 185, 2952–2960.e10. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, C.; Cao, X.; Gu, C.; Liu, H.; Jiang, M.; Wang, X.; Yuan, Q.; Wu, K.; Liu, J.; et al. Structural and biochemical mechanism for increased infectivity and immune evasion of Omicron BA.2 variant compared to BA.1 and their possible mouse origins. Cell Res. 2022, 32, 609–620. [Google Scholar] [CrossRef]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433.e13. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.E.; Dijokaite-Guraliuc, A.; et al. Potent cross-reactive antibodies following Omicron breakthrough in vaccinees. Cell 2022, 185, 2116–2131.e18. [Google Scholar] [CrossRef]
- Bowen, J.E.; Addetia, A.; Dang, H.V.; Stewart, C.; Brown, J.T.; Sharkey, W.K.; Sprouse, K.R.; Walls, A.C.; Mazzitelli, I.G.; Logue, J.K.; et al. Omicron spike function and neutralizing activity elicited by a comprehensive panel of vaccines. Science 2022, 377, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Pak, A.J.; He, P.; Monje-Galvan, V.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Amaro, R.E.; Voth, G.A. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys. J. 2021, 120, 1097–1104. [Google Scholar] [CrossRef]
- Sztain, T.; Ahn, S.H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef]
- Sikora, M.; von Bülow, S.; Blanc, F.E.C.; Gecht, M.; Covino, R.; Hummer, G. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput. Biol. 2021, 17, e1008790. [Google Scholar] [CrossRef] [PubMed]
- Taka, E.; Yilmaz, S.Z.; Golcuk, M.; Kilinc, C.; Aktas, U.; Yildiz, A.; Gur, M. Critical Interactions between the SARS-CoV-2 Spike Glycoprotein and the Human ACE2 Receptor. J. Phys. Chem. B 2021, 125, 5537–5548. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2100943118. [Google Scholar] [CrossRef] [PubMed]
- Fallon, L.; Belfon, K.A.A.; Raguette, L.; Wang, Y.; Stepanenko, D.; Cuomo, A.; Guerra, J.; Budhan, S.; Varghese, S.; Corbo, C.P.; et al. Free Energy Landscapes from SARS-CoV-2 Spike Glycoprotein Simulations Suggest that RBD Opening can be Modulated via Interactions in an Allosteric Pocket. J. Am. Chem. Soc. 2021, 143, 11349–11360. [Google Scholar] [CrossRef] [PubMed]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci. Adv. 2021, 7, eabf3671. [Google Scholar] [CrossRef]
- Peng, C.; Zhu, Z.; Shi, Y.; Wang, X.; Mu, K.; Yang, Y.; Zhang, X.; Xu, Z.; Zhu, W. Computational Insights into the Conformational Accessibility and Binding Strength of SARS-CoV-2 Spike Protein to Human Angiotensin-Converting Enzyme 2. J. Phys. Chem. Lett. 2020, 11, 10482–10488. [Google Scholar] [CrossRef] [PubMed]
- Golcuk, M.; Yildiz, A.; Gur, M. Omicron BA.1 and BA.2 variants increase the interactions of SARS-CoV-2 spike glycoprotein with ACE2. J. Mol. Graph. Model. 2022, 117, 108286. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Jung, J.; Kobayashi, C.; Dokainish, H.M.; Re, S.; Sugita, Y. Elucidation of interactions regulating conformational stability and dynamics of SARS-CoV-2 S-protein. Biophys. J. 2021, 120, 1060–1071. [Google Scholar] [CrossRef]
- Dokainish, H.M.; Re, S.; Mori, T.; Kobayashi, C.; Jung, J.; Sugita, Y. The inherent flexibility of receptor binding domains in SARS-CoV-2 spike protein. Elife 2022, 11, e75720. [Google Scholar] [CrossRef]
- Hossen, M.L.; Baral, P.; Sharma, T.; Gerstman, B.; Chapagain, P. Significance of the RBD mutations in the SARS-CoV-2 omicron: From spike opening to antibody escape and cell attachment. Phys. Chem. Chem. Phys. 2022, 24, 9123–9129. [Google Scholar] [CrossRef]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef]
- Gan, H.H.; Zinno, J.; Piano, F.; Gunsalus, K.C. Omicron Spike protein has a positive electrostatic surface that promotes ACE2 recognition and antibody escape. Front. Virol. 2022, 2, 894531. [Google Scholar] [CrossRef]
- Fazekas, Z.; Menyhárd, D.K.; Perczel, A. Omicron Binding Mode: Contact Analysis and Dynamics of the Omicron Receptor-Binding Domain in Complex with ACE2. J. Chem. Inf. Model. 2022, 62, 3844–3853. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Binding Interactions between Receptor-Binding Domain of Spike Protein and Human Angiotensin Converting Enzyme-2 in Omicron Variant. J. Phys. Chem. Lett. 2022, 13, 3915–3921. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Spratt, A.N.; Sharma, K.; Goyal, R.; Sönnerborg, A.; Apparsundaram, S.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Complex Mutation Pattern of Omicron BA.2: Evading Antibodies without Losing Receptor Interactions. Int. J. Mol. Sci. 2022, 23, 5534. [Google Scholar] [CrossRef]
- Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Computer Simulations and Network-Based Profiling of Binding and Allosteric Interactions of SARS-CoV-2 Spike Variant Complexes and the Host Receptor: Dissecting the Mechanistic Effects of the Delta and Omicron Mutations. Int. J. Mol. Sci. 2022, 23, 4376. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.H.; Twaddle, A.; Marchand, B.; Gunsalus, K.C. Structural Modeling of the SARS-CoV-2 Spike/Human ACE2 Complex Interface can Identify High-Affinity Variants Associated with Increased Transmissibility. J. Mol. Biol. 2021, 433, 167051. [Google Scholar] [CrossRef]
- Rochman, N.D.; Faure, G.; Wolf, Y.I.; Freddolino, P.L.; Zhang, F.; Koonin, E.V. Epistasis at the SARS-CoV-2 Receptor-Binding Domain Interface and the Propitiously Boring Implications for Vaccine Escape. mBio 2022, 13, e0013522. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hannon, W.W.; Loes, A.N.; Hauser, K.; Dillen, J.R.; Ferri, E.; Farrell, A.G.; Dadonaite, B.; McCallum, M.; et al. Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science 2022, 377, 420–424. [Google Scholar] [CrossRef]
- Martin, D.P.; Lytras, S.; Lucaci, A.G.; Maier, W.; Grüning, B.; Shank, S.D.; Weaver, S.; MacLean, O.A.; Orton, R.J.; Lemey, P.; et al. Selection analysis identifies clusters of unusual mutational changes in Omicron lineage BA.1 that likely impact Spike function. Mol. Biol. Evol. 2022, 39, msac061. [Google Scholar] [CrossRef]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target. Ther. 2022, 7, 138. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef]
- Verkhivker, G.M. Coevolution, dynamics and allostery conspire in shaping cooperative binding and signal transmission of the SARS-CoV-2 spike protein with human angiotensin-converting enzyme 2. Int. J. Mol. Sci. 2020, 21, 8268. [Google Scholar] [CrossRef]
- Verkhivker, G.M. Molecular simulations and network modeling reveal an allosteric signaling in the SARS-CoV-2 spike proteins. J. Proteome Res. 2020, 19, 4587–4608. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Dynamic Network Modeling of Allosteric Interactions and Communication Pathways in the SARS-CoV-2 Spike Trimer Mutants: Differential Modulation of Conformational Landscapes and Signal Transmission via Cascades of Regulatory Switches. J Phys. Chem. B 2021, 125, 850–873. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies. J. Phys. Chem. B 2021, 125, 4596–4619. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Comparative Perturbation-Based Modeling of the SARS-CoV-2 Spike Protein Binding with Host Receptor and Neutralizing Antibodies: Structurally Adaptable Allosteric Communication Hotspots Define Spike Sites Targeted by Global Circulating Mutations. Biochemistry 2021, 60, 1459–1484. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots. J. Chem. Theory Comput. 2021, 17, 4578–4598. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Allosteric Control of Structural Mimicry and Mutational Escape in the SARS-CoV-2 Spike Protein Complexes with the ACE2 Decoys and Miniprotein Inhibitors: A Network-Based Approach for Mutational Profiling of Binding and Signaling. J. Chem. Inf. Model. 2021, 61, 5172–5191. [Google Scholar] [CrossRef]
- Huang, M.; Wu, L.; Zheng, A.; Xie, Y.; He, Q.; Rong, X.; Han, P.; Du, P.; Han, P.; Zhang, Z.; et al. Atlas of currently available human neutralizing antibodies against SARS-CoV-2 and escape by Omicron sub-variants BA.1/BA.1.1/BA.2/BA.3. Immunity 2022, 55, 1501–1514.e3. [Google Scholar] [CrossRef]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Liu, H.; Hou, T. CaFE: A tool for binding affinity prediction using end-point free energy methods. Bioinformatics 2016, 32, 2216–2218. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein-protein complexes. Elife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup, Execution, and Analysis of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, M.; Alexov, E. DelPhiPKa Web Server: Predicting pKa of proteins, RNAs and DNAs. Bioinformatics 2016, 32, 614–615. [Google Scholar] [CrossRef]
- Pahari, S.; Sun, L.; Basu, S.; Alexov, E. DelPhiPKa: Including salt in the calculations and enabling polar residues to titrate. Proteins 2018, 86, 1277–1283. [Google Scholar] [CrossRef]
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef]
- Ali, F.; Kasry, A.; Amin, M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov. 2021, 10, 100086. [Google Scholar] [CrossRef]
- Kumar, R.; Murugan, N.A.; Srivastava, V. Improved Binding Affinity of Omicron’s Spike Protein for the Human Angiotensin-Converting Enzyme 2 Receptor Is the Key behind Its Increased Virulence. Int. J. Mol. Sci. 2022, 23, 3409. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Gilis, D.; Rooman, M. A new generation of statistical potentials for proteins. Biophys. J. 2006, 90, 4010–4017. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef]
- Collier, D.A.; De Marco, A.; Ferreira, I.; Meng, B.; Datir, R.P.; Walls, A.C.; Kemp, S.A.; Bassi, J.; Pinto, D.; Silacci-Fregni, C.; et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 2021, 593, 136–141. [Google Scholar] [CrossRef]
- Tsai, K.C.; Lee, Y.C.; Tseng, T.S. Comprehensive Deep Mutational Scanning Reveals the Immune-Escaping Hotspots of SARS-CoV-2 Receptor-Binding Domain Targeting Neutralizing Antibodies. Front. Microbiol. 2021, 12, 698365. [Google Scholar] [CrossRef]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [Green Version]
- Stetz, G.; Verkhivker, G.M. Computational analysis of residue interaction networks and coevolutionary relationships in the Hsp70 chaperones: A community-hopping model of allosteric regulation and communication. PLoS Comput. Biol. 2017, 13, e1005299. [Google Scholar] [CrossRef]
- Czemeres, J.; Buse, K.; Verkhivker, G.M. Atomistic simulations and network-based modeling of the Hsp90-Cdc37 chaperone binding with Cdk4 client protein: A mechanism of chaperoning kinase clients by exploiting weak spots of intrinsically dynamic kinase domains. PLoS ONE 2017, 12, e0190267. [Google Scholar] [CrossRef]
- Stetz, G.; Verkhivker, G.M. Dancing through life: Molecular dynamics simulations and network-centric modeling of allosteric mechanisms in Hsp70 and Hsp110 chaperone proteins. PLoS ONE 2015, 10, e0143752.61. [Google Scholar] [CrossRef]
- Fan, J.; Liu, Y.; Kong, R.; Ni, D.; Yu, Z.; Lu, S.; Zhang, J. Harnessing Reversed Allosteric Communication: A Novel Strategy for Allosteric Drug Discovery. J. Med. Chem. 2021, 64, 17728–17743. [Google Scholar] [CrossRef]
- Lu, S.; Shen, Q.; Zhang, J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2020, 12, 464–476. [Google Scholar] [CrossRef]
- Tee, W.-V.; Guarnera, E.; Berezovsky, I.N. Reversing allosteric communication: From detecting allosteric sites to inducing and tuning targeted allosteric response. PLoS Comput. Biol. 2018, 14, e1006228. [Google Scholar] [CrossRef]
- Mersmann, S.F.; Strömich, L.; Song, F.J.; Wu, N.; Vianello, F.; Barahona, M.; Yaliraki, S.N. ProteinLens: A web-based application for the analysis of allosteric signalling on atomistic graphs of biomolecules. Nucleic Acids Res. 2021, 49, W551–W558. [Google Scholar] [CrossRef]
- Amor, B.R.; Schaub, M.T.; Yaliraki, S.N.; Barahona, M. Prediction of allosteric sites and mediating interactions through bond-to-bond propensities. Nat. Commun. 2016, 7, 12477. [Google Scholar] [CrossRef]
- Tse, A.; Verkhivker, G.M. Exploring Molecular Mechanisms of Paradoxical Activation in the BRAF Kinase Dimers: Atomistic Simulations of Conformational Dynamics and Modeling of Allosteric Communication Networks and Signaling Pathways. PLoS ONE 2016, 11, e0166583. [Google Scholar] [CrossRef]
- Astl, L.; Verkhivker, G.M. Atomistic modeling of the ABL kinase regulation by allosteric modulators using structural perturbation analysis and community-based network reconstruction of allosteric communications. J. Chem. Theory Comput. 2019, 15, 3362–3380. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Y.; Ni, D.; Huang, Z.; Wei, J.; Feng, L.; Su, J.C.; Wei, Y.; Ning, S.; Yang, X.; et al. Targeting a cryptic allosteric site of SIRT6 with small-molecule inhibitors that inhibit the migration of pancreatic cancer cells. Acta Pharm. Sin. B 2022, 12, 876–889. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Li, B.; Wang, L.; Ge, H.; Zhang, X.; Ren, P.; Guo, Y.; Chen, W.; Li, J.; Zhu, W.; Chen, W.; et al. Identification of Potential Binding Sites of Sialic Acids on the RBD Domain of SARS-CoV-2 Spike Protein. Front. Chem. 2021, 9, 659764. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; Te Beek, T.A.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A protein structure bioinformatics web service collection. Nucleic Acids Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef]
- Fernandez-Fuentes, N.; Zhai, J.; Fiser, A. ArchPRED: A template based loop structure prediction server. Nucleic Acids Res. 2006, 34, W173–W176. [Google Scholar] [CrossRef]
- Ko, J.; Lee, D.; Park, H.; Coutsias, E.A.; Lee, J.; Seok, C. The FALC-Loop web server for protein loop modeling. Nucleic Acids Res. 2011, 39, W210–W214. [Google Scholar] [CrossRef]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Fernandes, H.S.; Ramos, M.J.; Cerqueira, N.M.F.S.A. molUP: A VMD plugin to handle QM and ONIOM calculations using the gaussian software. J Comput Chem. 2018, 39, 1344–1353. [Google Scholar] [CrossRef]
- Di Pierro, M.; Elber, R.; Leimkuhler, B. A Stochastic Algorithm for the Isobaric-Isothermal Ensemble with Ewald Summations for All Long Range Forces. J. Chem. Theory Comput. 2015, 11, 5624–5637. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef]
- Mackerell, A.D., Jr.; Feig, M.; Brooks, C.L., III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. VMD Store-A VMD Plugin to Browse, Discover, and Install VMD Extensions. J. Chem. Inf. Model. 2019, 59, 4519–4523. [Google Scholar] [CrossRef]
- Brinda, K.V.; Vishveshwara, S. A network representation of protein structures: Implications for protein stability. Biophys. J. 2005, 89, 4159–4170. [Google Scholar] [CrossRef] [Green Version]
- Vijayabaskar, M.S.; Vishveshwara, S. Interaction energy based protein structure networks. Biophys. J. 2010, 99, 3704–3715. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Minervini, G.; Tosatto, S.C. The RING 2.0 Web Server for High Quality Residue Interaction Networks. Nucleic Acids Res. 2016, 44, W367–W374. [Google Scholar] [CrossRef]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; Tosatto, S.C.E. RING 3.0: Fast generation of probabilistic residue interaction networks from structural ensembles. Nucleic Acids Res. 2022, 50, gkac365. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008; Varoquaux, G., Vaught, T., Millman, J., Eds.; Scientific Research: Atlanta, GA, USA, 2011; pp. 11–15. [Google Scholar]
- Newman, M.E. Finding community structure in networks using the eigenvectors of matrices. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2006, 74, 36104. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E. Modularity and Community Structure in Networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed]
- Palla, G.; Derényi, I.; Farkas, I.; Vicsek, T. Uncovering the overlapping community structure of complex networks in nature and society. Nature 2005, 435, 814–818. [Google Scholar] [CrossRef]
- Adamcsek, B.; Palla, G.; Farkas, I.J.; Derényi, I.; Vicsek, T. CFinder: Locating cliques and overlapping modules in biological networks. Bioinformatics 2006, 22, 1021–1023. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Omicron Variant | Mutational Landscape of the RBD |
|---|---|
| BA.1 | G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H |
| BA.1.1 | G339D, R346K, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H |
| BA.2 | G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H |
| BA.3 | G339D, S371F, S373P, S375F, D405N, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H |
| BA.4/BA.5 | G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q493R, Q498R, N501Y, Y505H |
| PDB | System | Per Simulation | # Simulations |
|---|---|---|---|
| 7WBP | Omicron RBD BA.1-hACE2 | 500 ns | 10 |
| 7XAZ | Omicron RBD BA.1.1-hACE2 | 500 ns | 10 |
| 7XB0 | Omicron RBD BA.2-hACE2 | 500 ns | 10 |
| 7XB1 | Omicron RBD BA.3-hACE2 | 500 ns | 10 |
| Systems | MM-PBSA Average Binding Free Energy (kcal/mol) | Standard Deviation (kcal/mol) | Experimental Binding Affinity |
|---|---|---|---|
| RBD Omicron BA.1-hACE2 | −48.785 | 6.345 | 19.5 nM |
| RBD Omicron BA.1.1-hACE2 | −50.565 | 7.235 | 5.9 nM |
| RBD Omicron BA.2-hACE2 | −53.125 | 6.454 | 10.0 nM |
| RBD Omicron BA.3-hACE2 | −49.778 | 6.976 | 22.1 nM |
| Interactions | RBD Omicron BA.1-hACE2 | RBD Omicron BA.1.1-hACE2 | RBD Omicron BA.2-hACE2 | RBD Omicron BA.3-hACE2 |
|---|---|---|---|---|
| Charged-charged | 7 | 8 | 9 | 6 |
| Charged-polar | 7 | 9 | 8 | 5 |
| Charged-apolar | 18 | 20 | 20 | 18 |
| Polar-polar | 5 | 5 | 6 | 6 |
| Polar-apolar | 17 | 16 | 20 | 18 |
| Apolar-apolar | 11 | 12 | 16 | 11 |
| ΔG computed (kcal/mol) | −11.2 | −11.8 | −12.3 | −11.2 |
| Kd (nM) experiment | 19.5 nM | 5.9 nM | 10.0 nM | 22.1 nM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Probing Mechanisms of Binding and Allostery in the SARS-CoV-2 Spike Omicron Variant Complexes with the Host Receptor: Revealing Functional Roles of the Binding Hotspots in Mediating Epistatic Effects and Communication with Allosteric Pockets. Int. J. Mol. Sci. 2022, 23, 11542. https://doi.org/10.3390/ijms231911542
Verkhivker G, Agajanian S, Kassab R, Krishnan K. Probing Mechanisms of Binding and Allostery in the SARS-CoV-2 Spike Omicron Variant Complexes with the Host Receptor: Revealing Functional Roles of the Binding Hotspots in Mediating Epistatic Effects and Communication with Allosteric Pockets. International Journal of Molecular Sciences. 2022; 23(19):11542. https://doi.org/10.3390/ijms231911542
Chicago/Turabian StyleVerkhivker, Gennady, Steve Agajanian, Ryan Kassab, and Keerthi Krishnan. 2022. "Probing Mechanisms of Binding and Allostery in the SARS-CoV-2 Spike Omicron Variant Complexes with the Host Receptor: Revealing Functional Roles of the Binding Hotspots in Mediating Epistatic Effects and Communication with Allosteric Pockets" International Journal of Molecular Sciences 23, no. 19: 11542. https://doi.org/10.3390/ijms231911542