
Connecting Neurobiological Features with Interregional Dysconnectivity in Social-Cognitive Impairments of Schizophrenia

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Macroscale Knowledge—Brain Structures and Networks That Regulate Social Cognition Are Affected in SZ
2.1. Impairments in the Recruitment of the Social Brain during Social-Cognitive Behavior in SZ
2.2. Reduced Structural Connectivity within the Social Brain Affects Social Cognition in SZ
2.3. Abnormal Properties of Functional Social Brain Networks in SZ
3. Microscale Knowledge—Molecular and Cellular Mechanisms Underlying Social-Cognitive Dysfunction in SZ
3.1. Oxidative Stress Is Associated with Social-Cognitive Impairments in SZ
3.2. Immune Dysregulation Is Associated with Social-Cognitive Impairments in SZ
3.3. NMDA Receptor Hypofunction Is Associated with Social-Cognitive Impairments in SZ
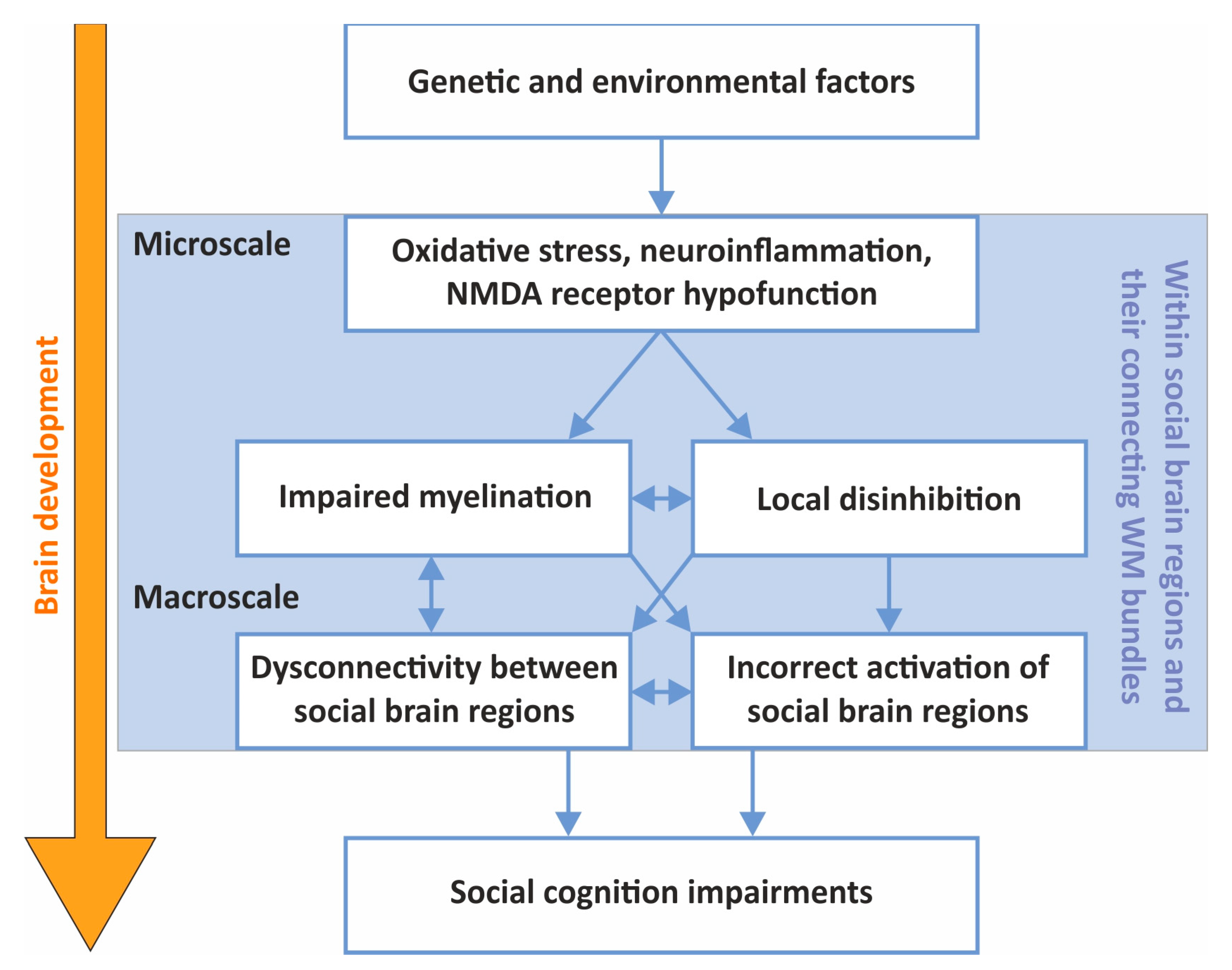
4. Connecting the Macro- and Microscales in SZ Social-Cognitive Research
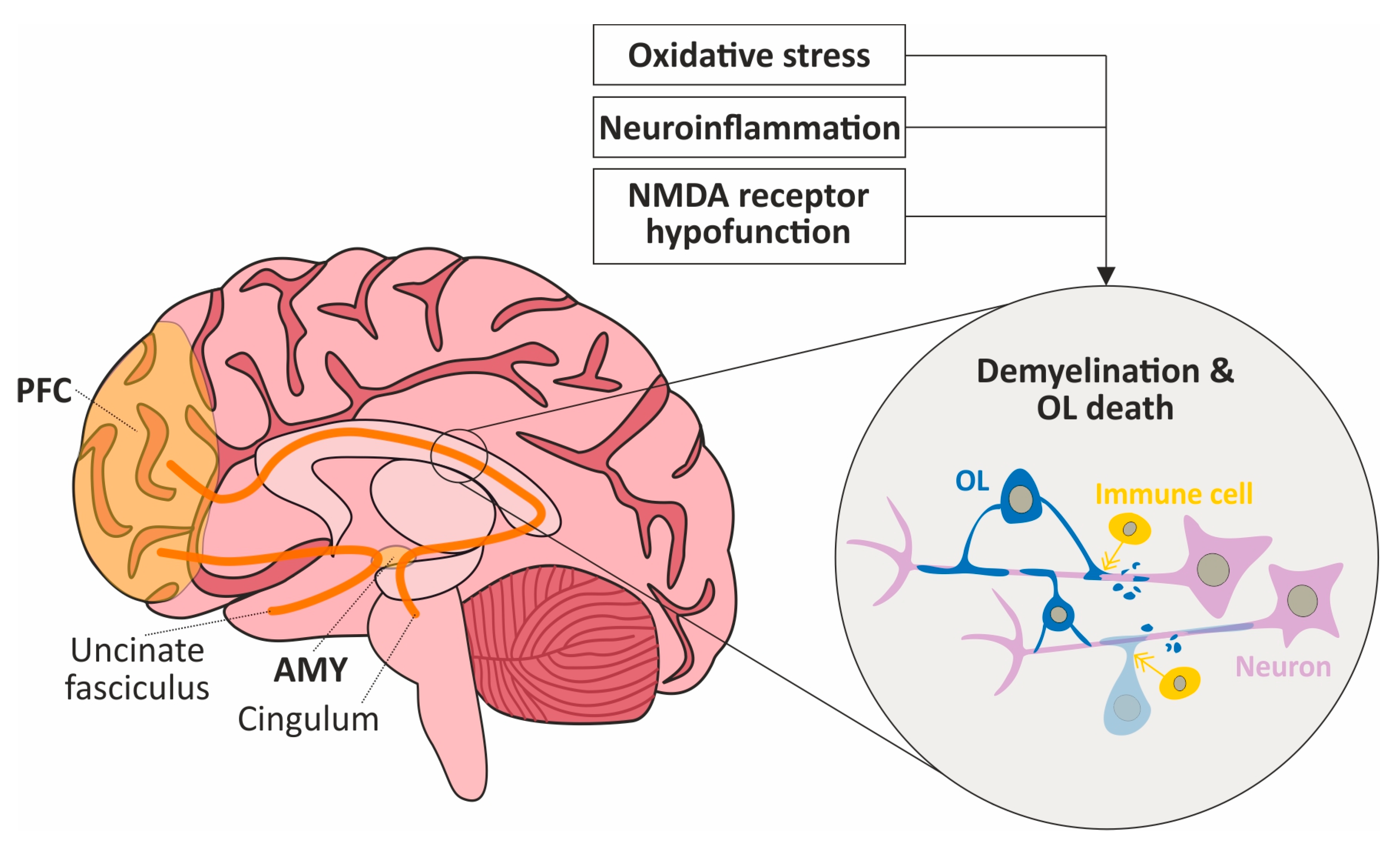
4.1. The Effects of Oxidative Stress, Neuroinflammation and NMDA Receptor Hypofunction on Local Brain Circuits
4.2. Oxidative Stress, Neuroinflammation and NMDA Receptor Hypofunction Might Impact Interregional Connectivity through WM Damage within the Social Brain
5. Implications for Translational Research and Drug Development
5.1. Promising Pharmacological Candidates for the Treatment of Social-Cognitive Impairments in SZ
5.2. Better Translational Methods Could Improve Drug Development for Social-Cognitive Impairments in SZ: EEG as an Example
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Green, M.F.; Horan, W.P.; Lee, J. Social cognition in schizophrenia. Nat. Rev. Neurosci. 2015, 16, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Rodgers, B.; Murray, R.; Marmot, M. Child development risk factors for adult schizophrenia in the British 1946 birth cohort. Lancet 1994, 344, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Murray, R.M. Schizophrenia: An integrated sociodevelopmental-cognitive model. Lancet 2014, 383, 1677–1687. [Google Scholar] [CrossRef]
- Keskinen, E.; Marttila, A.; Jones, P.; Murray, G.; Moilanen, K.; Koivumaa-Honkanen, H.; Mäki, P.; Isohanni, M.; Jääskeläinen, E.; Miettunen, J. Interaction between parental psychosis and early motor development and the risk of schizophrenia in a general population birth cohort. Eur. Psychiatry 2015, 30, 719–727. [Google Scholar] [CrossRef]
- Chong, H.Y.; Teoh, S.L.; Wu, D.B.; Kotirum, S.; Chiou, C.F.; Chaiyakunapruk, N. Global economic burden of schizophrenia: A systematic review. Neuropsychiatr. Dis. Treat. 2016, 12, 357–373. [Google Scholar] [CrossRef]
- Dunbar, R.I. The social brain hypothesis and its implications for social evolution. Ann. Hum. Biol. 2009, 36, 562–572. [Google Scholar] [CrossRef]
- Bickart, K.C.; Dickerson, B.C.; Barrett, L.F. The amygdala as a hub in brain networks that support social life. Neuropsychologia 2014, 63, 235–248. [Google Scholar] [CrossRef]
- Porcelli, S.; Van Der Wee, N.; van der Werff, S.; Aghajani, M.; Glennon, J.C.; van Heukelum, S.; Mogavero, F.; Lobo, A.; Olivera, F.J.; Lobo, E.; et al. Social brain, social dysfunction and social withdrawal. Neurosci. Biobehav. Rev. 2019, 97, 10–33. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.; Monin, A.; Dwir, D.; O’Donnell, P.; Cuenod, M.; Do, K. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A “central hub” in schizophrenia pathophysiology? Schizophr. Res. 2016, 176, 41–51. [Google Scholar] [CrossRef]
- Maas, D.A.; Vallès, A.; Martens, G.J.M. Oxidative stress, prefrontal cortex hypomyelination and cognitive symptoms in schizophrenia. Transl. Psychiatry 2017, 7, e1171. [Google Scholar] [CrossRef]
- Uranova, N.A.; Vikhreva, O.V.; Rachmanova, V.I.; Orlovskaya, D.D. Ultrastructural alterations of myelinated fibers and oligodendrocytes in the prefrontal cortex in schizophrenia: A postmortem morphometric study. Schizophr. Res. Treat. 2011, 2011, 325789. [Google Scholar] [CrossRef]
- Bobilev, A.M.; Perez, J.M.; Tamminga, C.A. Molecular alterations in the medial temporal lobe in schizophrenia. Schizophr. Res. 2020, 217, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.A.; Norris, A.L.; Yang, K.; Cheshire, M.; Newkirk, I.; Chen, X.; Ishizuka, K.; Jaffe, A.E.; Sawa, A.; Pevsner, J. Dysfunction of mitochondria and GABAergic interneurons in the anterior cingulate cortex of individuals with schizophrenia. Neurosci. Res. 2022, 185, 67–72. [Google Scholar] [CrossRef]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Alavijeh, M.S. Translational CNS medicines research. Drug Discov. Today 2012, 17, 1068–1078. [Google Scholar] [CrossRef]
- Pangalos, M.N.; Schechter, L.E.; Hurko, O. Drug development for CNS disorders: Strategies for balancing risk and reducing attrition. Nat. Rev. Drug Discov. 2007, 6, 521–532. [Google Scholar] [CrossRef]
- Kas, M.J.; Penninx, B.; Sommer, B.; Serretti, A.; Arango, C.; Marston, H. A quantitative approach to neuropsychiatry: The why and the how. Neurosci. Biobehav. Rev. 2019, 97, 3–9. [Google Scholar] [CrossRef]
- Morris, R.W.; Sparks, A.; Mitchell, P.B.; Weickert, C.S.; Green, M.J. Lack of cortico-limbic coupling in bipolar disorder and schizophrenia during emotion regulation. Transl. Psychiatry 2012, 2, e90. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meer, L.; Swart, M.; Van Der Velde, J.; Pijnenborg, G.; Wiersma, D.; Bruggeman, R.; Aleman, A. Neural correlates of emotion regulation in patients with schizophrenia and non-affected siblings. PLoS ONE 2014, 9, e99667. [Google Scholar] [CrossRef]
- Thakkar, K.N.; Peterman, J.S.; Park, S. Altered brain activation during action imitation and observation in schizophrenia: A translational approach to investigating social dysfunction in schizophrenia. Am. J. Psychiatry 2014, 171, 539–548. [Google Scholar] [CrossRef]
- Eack, S.M.; Wojtalik, J.A.; Newhill, C.E.; Keshavan, M.S.; Phillips, M.L. Prefrontal cortical dysfunction during visual perspective-taking in schizophrenia. Schizophr. Res. 2013, 150, 491–497. [Google Scholar] [CrossRef]
- Lee, J.; Quintana, J.; Nori, P.; Green, M.F. Theory of mind in schizophrenia: Exploring neural mechanisms of belief attribution. Soc. Neurosci. 2011, 6, 569–581. [Google Scholar] [CrossRef]
- Dodell-Feder, D.; Tully, L.M.; Lincoln, S.H.; Hooker, C.I. The neural basis of theory of mind and its relationship to social functioning and social anhedonia in individuals with schizophrenia. NeuroImage Clin. 2013, 4, 154–163. [Google Scholar] [CrossRef]
- Li, H.; Chan, R.C.; McAlonan, G.M.; Gong, Q.Y. Facial emotion processing in schizophrenia: A meta-analysis of functional neuroimaging data. Schizophr. Bull. 2010, 36, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, G.; Sugranyes, G.; Frangou, S. Evidence of diagnostic specificity in the neural correlates of facial affect processing in bipolar disorder and schizophrenia: A meta-analysis of functional imaging studies. Psychol. Med. 2013, 43, 553–569. [Google Scholar] [CrossRef]
- Taylor, S.F.; Kang, J.; Brege, I.S.; Tso, I.F.; Hosanagar, A.; Johnson, T.D. Meta-analysis of functional neuroimaging studies of emotion perception and experience in schizophrenia. Biol. Psychiatry 2012, 71, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Anticevic, A.; Van Snellenberg, J.X.; Cohen, R.E.; Repovs, G.; Dowd, E.C.; Barch, D.M. Amygdala recruitment in schizophrenia in response to aversive emotional material: A meta-analysis of neuroimaging studies. Schizophr. Bull. 2012, 38, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.L.; Elliott, R.; Barry, M.; Cruttenden, A.; Woodruff, P.W. Neural response to emotional prosody in schizophrenia and in bipolar affective disorder. Br. J. Psychiatry 2004, 184, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Leitman, D.I.; Wolf, D.H.; Laukka, P.; Ragland, J.D.; Valdez, J.N.; Turetsky, B.I.; Gur, R.E.; Gur, R.C. Not pitch perfect: Sensory contributions to affective communication impairment in schizophrenia. Biol. Psychiatry 2011, 70, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-M.; Kim, J.-J.; Ku, J.; Kim, S.Y.; Lee, H.R.; Kim, S.I.; Yoon, K.-J. Neural basis of attributional style in schizophrenia. Neurosci. Lett. 2009, 459, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Olson, I.R. The Original Social Network: White Matter and Social Cognition. Trends Cogn. Sci. 2018, 22, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Metoki, A.; Alm, K.H.; Olson, I.R. White matter pathways and social cognition. Neurosci. Biobehav. Rev. 2018, 90, 350–370. [Google Scholar] [CrossRef] [PubMed]
- Samartzis, L.; Dima, D.; Fusar-Poli, P.; Kyriakopoulos, M. White matter alterations in early stages of schizophrenia: A systematic review of diffusion tensor imaging studies. J. Neuroimaging 2014, 24, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, P.; Tansella, M. The role of white matter for the pathophysiology of schizophrenia. Int. Rev. Psychiatry 2007, 19, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Crocker, C.E.; Tibbo, P.G. Confused Connections? Targeting White Matter to Address Treatment Resistant Schizophrenia. Front. Pharmacol. 2018, 9, 1172. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.L.; Voineskos, A.N. A review of structural neuroimaging in schizophrenia: From connectivity to connectomics. Front. Hum. Neurosci. 2014, 8, 653. [Google Scholar] [CrossRef]
- Kanaan, R.; Barker, G.; Brammer, M.; Giampietro, V.; Shergill, S.; Woolley, J.; Picchioni, M.; Toulopoulou, T.; McGuire, P. White matter microstructure in schizophrenia: Effects of disorder, duration and medication. Br. J. Psychiatry 2009, 194, 236–242. [Google Scholar] [CrossRef]
- Liu, X.; Lai, Y.; Wang, X.; Hao, C.; Chen, L.; Zhou, Z.; Yu, X.; Hong, N. A combined DTI and structural MRI study in medicated-naïve chronic schizophrenia. Magn. Reson. Imaging 2014, 32, 1–8. [Google Scholar] [CrossRef]
- Bloemen, O.J.N.; de Koning, M.B.; Schmitz, N.; Nieman, D.H.; Becker, H.E.; de Haan, L.; Dingemans, P.; Linszen, D.H.; van Amelsvoort, T.A.M.J. White-matter markers for psychosis in a prospective ultra-high-risk cohort. Psychol. Med. 2010, 40, 1297–1304. [Google Scholar] [CrossRef]
- Friedman, J.I.; Tang, C.; Carpenter, D.; Buchsbaum, M.; Schmeidler, J.; Flanagan, L.; Golembo, S.; Kanellopoulou, I.; Ng, J.; Hof, P.R.; et al. Diffusion tensor imaging findings in first-episode and chronic schizophrenia patients. Am. J. Psychiatry 2008, 165, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Lui, S.; Liao, Y.; Du, M.-Y.; Hu, N.; Thomas, J.A.; Gong, Q.-Y. White matter deficits in first episode schizophrenia: An activation likelihood estimation meta-analysis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 45, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Holleran, L.; Ahmed, M.; Anderson-Schmidt, H.; McFarland, J.; Emsell, L.; Leemans, A.; Scanlon, C.; Dockery, P.; McCarthy, P.; Barker, G.J.; et al. Altered interhemispheric and temporal lobe white matter microstructural organization in severe chronic schizophrenia. Neuropsychopharmacology 2014, 39, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Matsuzawa, H.; Shioiri, T.; Someya, T.; Kwee, I.L.; Nakada, T. Diffusion tensor analysis in chronic schizophrenia. A preliminary study on a high-field (3.0T) system. Eur. Arch. Psychiatry Clin. Neurosci. 2005, 255, 313–318. [Google Scholar] [CrossRef]
- Tordesillas-Gutiérrez, D.; McGuire, P.K.; Barker, G.J.; Roiz-Santiañez, R.; Mata, I.; de Lucas, E.M.; Rodríguez-Sánchez, J.M.; Ayesa-Arriola, R.; Vazquez-Barquero, J.L.; Crespo-Facorro, B. White matter integrity and cognitive impairment in first-episode psychosis. Am. J. Psychiatry 2010, 167, 451–458. [Google Scholar] [CrossRef]
- Voineskos, A.N.; Foussias, G.; Lerch, J.; Felsky, D.; Remington, G.; Rajji, T.K.; Lobaugh, N.; Pollock, B.G.; Mulsant, B.H. Neuroimaging evidence for the deficit subtype of schizophrenia. JAMA Psychiatry 2013, 70, 472–480. [Google Scholar] [CrossRef]
- Roalf, D.R.; Ruparel, K.; Verma, R.; Elliott, M.A.; Gur, R.E.; Gur, R.C. White matter organization and neurocognitive performance variability in schizophrenia. Schizophr. Res. 2013, 143, 172–178. [Google Scholar] [CrossRef]
- Dusi, N.; Bellani, M.; Perlini, C.; Squarcina, L.; Marinelli, V.; Finos, L.; Altamura, C.; Ruggeri, M.; Brambilla, P. Progressive disability and prefrontal shrinkage in schizophrenia patients with poor outcome: A 3-year longitudinal study. Schizophr. Res. 2017, 179, 104–111. [Google Scholar] [CrossRef]
- Viher, P.V.; Stegmayer, K.; Giezendanner, S.; Federspiel, A.; Bohlhalter, S.; Vanbellingen, T.; Wiest, R.; Strik, W.; Walther, S. Cerebral white matter structure is associated with DSM-5 schizophrenia symptom dimensions. NeuroImage Clin. 2016, 12, 93–99. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hirao, K.; Namiki, C.; Yamada, M.; Shimizu, M.; Fukuyama, H.; Hayashi, T.; Murai, T. Anterior cingulate pathology and social cognition in schizophrenia: A study of gray matter, white matter and sulcal morphometry. Neuroimage 2007, 36, 1236–1245. [Google Scholar] [CrossRef]
- Ohtani, T.; Bouix, S.; Hosokawa, T.; Saito, Y.; Eckbo, R.; Ballinger, T.; Rausch, A.; Melonakos, E.; Kubicki, M. Abnormalities in white matter connections between orbitofrontal cortex and anterior cingulate cortex and their associations with negative symptoms in schizophrenia: A DTI study. Schizophr. Res. 2014, 157, 190–197. [Google Scholar] [CrossRef]
- Adamczyk, P.; Płonka, O.; Kruk, D.; Jáni, M.; Błądziński, P.; Kalisz, A.; Castelein, S.; Cechnicki, A.; Wyczesany, M. On the relation of white matter brain abnormalities and the asociality symptoms in schizophrenia outpatients—A DTI study. Acta Neurobiol. Exp. Wars 2021, 81, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Karlsgodt, K.H.; Niendam, T.A.; Bearden, C.E.; Cannon, T.D. White matter integrity and prediction of social and role functioning in subjects at ultra-high risk for psychosis. Biol. Psychiatry 2009, 66, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Miyata, J.; Yamada, M.; Namiki, C.; Hirao, K.; Saze, T.; Fujiwara, H.; Shimizu, M.; Kawada, R.; Fukuyama, H.; Sawamoto, N.; et al. Reduced white matter integrity as a neural correlate of social cognition deficits in schizophrenia. Schizophr. Res. 2010, 119, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, D.; Fukunaga, M.; Okada, N.; Morita, K.; Nemoto, K.; Yamashita, F.; Yamamori, H.; Yasuda, Y.; Fujimoto, M.; Kelly, S.; et al. Role of frontal white matter and corpus callosum on social function in schizophrenia. Schizophr. Res. 2018, 202, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kubicki, M.; Koerte, I.; Otsuka, T.; Rathi, Y.; Pasternak, O.; Bouix, S.; Eckbo, R.; Kikinis, Z.; von Hohenberg, C.C.; et al. Impaired white matter connectivity between regions containing mirror neurons, and relationship to negative symptoms and social cognition, in patients with first-episode schizophrenia. Brain Imaging Behav. 2018, 12, 229–237. [Google Scholar] [CrossRef]
- Olszewski, A.K.; Kikinis, Z.; Gonzalez, C.S.; Coman, I.L.; Makris, N.; Gong, X.; Rathi, Y.; Zhu, A.; Antshel, K.M.; Fremont, W.; et al. The social brain network in 22q11.2 deletion syndrome: A diffusion tensor imaging study. Behav. Brain Funct. 2017, 13, 4. [Google Scholar] [CrossRef]
- Kim, N.S.; Lee, T.Y.; Hwang, W.J.; Bin Kwak, Y.; Kim, S.; Moon, S.-Y.; Lho, S.K.; Oh, S.; Kwon, J.S. White Matter Correlates of Theory of Mind in Patients With First-Episode Psychosis. Front. Psychiatry 2021, 12, 617683. [Google Scholar] [CrossRef]
- Singh, S.; Singh, K.; Trivedi, R.; Goyal, S.; Kaur, P.; Singh, N.; Bhatia, T.; Deshpande, S.N.; Khushu, S. Microstructural abnormalities of uncinate fasciculus as a function of impaired cognition in schizophrenia: A DTI study. J. Biosci. 2016, 41, 419–426. [Google Scholar] [CrossRef]
- Ho, N.F.; Li Hui Chong, P.; Lee, D.R.; Chew, Q.H.; Chen, G.; Sim, K. The Amygdala in Schizophrenia and Bipolar Disorder: A Synthesis of Structural MRI, Diffusion Tensor Imaging, and Resting-State Functional Connectivity Findings. Harv. Rev. Psychiatry 2019, 27, 150–164. [Google Scholar] [CrossRef]
- Jung, S.; Kim, J.-H.; Sung, G.; Ko, Y.-G.; Bang, M.; Park, C.-I.; Lee, S.-H. Uncinate fasciculus white matter connectivity related to impaired social perception and cross-sectional and longitudinal symptoms in patients with schizophrenia spectrum psychosis. Neurosci. Lett. 2020, 737, 135144. [Google Scholar] [CrossRef]
- Erdeniz, B.; Serin, E.; İbadi, Y.; Taş, C. Decreased functional connectivity in schizophrenia: The relationship between social functioning, social cognition and graph theoretical network measures. Psychiatry Res. Neuroimaging 2017, 270, 22–31. [Google Scholar] [CrossRef]
- Bitsch, F.; Berger, P.; Nagels, A.; Falkenberg, I.; Straube, B. Characterizing the theory of mind network in schizophrenia reveals a sparser network structure. Schizophr. Res. 2021, 228, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Farrow, T.F.; Spence, S.A.; Woodruff, P.W. Social cognition, brain networks and schizophrenia. Psychol. Med. 2004, 34, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Li, Y.; Wang, Y.-M.; Wang, S.-K.; Pu, C.-C.; Zhou, S.-Z.; Ma, Y.-T.; Wang, Y.; Lui, S.S.Y.; Yu, X.; et al. Hub-connected functional connectivity within social brain network weakens the association with real-life social network in schizophrenia patients. Eur. Arch. Psychiatry Clin. Neurosci. 2022, 272, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Wölwer, W.; Frommann, N. Social-cognitive remediation in schizophrenia: Generalization of effects of the Training of Affect Recognition (TAR). Schizophr. Bull. 2011, 37, S63–S70. [Google Scholar] [CrossRef]
- Horan, W.P.; Green, M.F. Treatment of social cognition in schizophrenia: Current status and future directions. Schizophr. Res. 2019, 203, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Cuenod, M.; Steullet, P.; Cabungcal, J.-H.; Dwir, D.; Khadimallah, I.; Klauser, P.; Conus, P.; Do, K.Q. Caught in vicious circles: A perspective on dynamic feed-forward loops driving oxidative stress in schizophrenia. Mol. Psychiatry 2022, 27, 1886–1897. [Google Scholar] [CrossRef]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Washizuka, S.; Kametani, M.; Sasaki, T.; Tochigi, M.; Umekage, T.; Kohda, K.; Kato, T. Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with schizophrenia in the Japanese population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 301–304. [Google Scholar] [CrossRef]
- Bošković, M.; Vovk, T.; Plesničar, K.B.; Grabnar, I. Oxidative stress in schizophrenia. Curr. Neuropharmacol. 2011, 9, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Rollins, B.L.; Morgan, L.; Hjelm, B.E.; Sequeira, A.; Schatzberg, A.F.; Barchas, J.D.; Lee, F.S.; Myers, R.M.; Watson, S.J.; Akil, H.; et al. Mitochondrial Complex I Deficiency in Schizophrenia and Bipolar Disorder and Medication Influence. Mol. Neuropsychiatry 2018, 3, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, V.F.; Cappi, C.; Hagen, C.M.; Sequeira, A.; Vawter, M.P.; Derkach, A.; Zai, C.C.; Hedley, P.L.; Bybjerg-Grauholm, J.; Pouget, J.G.; et al. A Comprehensive Analysis of Nuclear-Encoded Mitochondrial Genes in Schizophrenia. Biol. Psychiatry 2018, 83, 780–789. [Google Scholar] [CrossRef]
- Tosic, M.; Ott, J.; Barral, S.; Bovet, P.; Deppen, P.; Gheorghita, F.; Matthey, M.-L.; Parnas, J.; Preisig, M.; Saraga, M.; et al. Schizophrenia and oxidative stress: Glutamate cysteine ligase modifier as a susceptibility gene. Am. J. Hum. Genet. 2006, 79, 586–592. [Google Scholar] [CrossRef]
- Gysin, R.; Kraftsik, R.; Sandell, J.; Bovet, P.; Chappuis, C.; Conus, P.; Deppen, P.; Preisig, M.; Ruiz, V.; Steullet, P.; et al. Impaired glutathione synthesis in schizophrenia: Convergent genetic and functional evidence. Proc. Natl. Acad. Sci. USA 2007, 104, 16621–16626. [Google Scholar] [CrossRef]
- Rodríguez-Santiago, B.; Brunet, A.; Sobrino, B.; Serra-Juhé, C.; Flores, R.; Armengol, L.; Vilella, E.; Gabau, E.; Guitart, M.; Guillamat, R.; et al. Association of common copy number variants at the glutathione S-transferase genes and rare novel genomic changes with schizophrenia. Mol. Psychiatry 2010, 15, 1023–1033. [Google Scholar] [CrossRef]
- Chowdari, K.V.; Bamne, M.N.; Nimgaonkar, V.L.; Ivanova, S.A.; Smirnova, L.P.; Shchigoreva, Y.G.; Semke, A.V.; Bokhan, N.A.; Smesny, S.; Milleit, B.; et al. Genetic association studies of antioxidant pathway genes and schizophrenia. Antioxid. Redox Signal. 2011, 15, 2037–2045. [Google Scholar] [CrossRef]
- Gravina, P.; Spoletini, I.; Masini, S.; Valentini, A.; Vanni, D.; Paladini, E.; Bossù, P.; Caltagirone, C.; Federici, G.; Spalletta, G.; et al. Genetic polymorphisms of glutathione S-transferases GSTM1, GSTT1, GSTP1 and GSTA1 as risk factors for schizophrenia. Psychiatry Res. 2011, 187, 454–456. [Google Scholar] [CrossRef]
- Oskvig, D.B.; Elkahloun, A.G.; Johnson, K.R.; Phillips, T.M.; Herkenham, M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav. Immun. 2012, 26, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Clelland, J.D.; Read, L.L.; Drouet, V.; Kaon, A.; Kelly, A.; Duff, K.E.; Nadrich, R.H.; Rajparia, A.; Clelland, C.L. Vitamin D insufficiency and schizophrenia risk: Evaluation of hyperprolinemia as a mediator of association. Schizophr. Res. 2014, 156, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Zugno, A.I.; Pacheco, F.D.; Budni, J.; de Oliveira, M.B.; Canever, L.; Heylmann, A.S.; Wessler, P.G.; Silveira, F.D.R.; Mastella, G.A.; Gonçalves, C.L.; et al. Maternal deprivation disrupts mitochondrial energy homeostasis in the brain of rats subjected to ketamine-induced schizophrenia. Metab. Brain Dis. 2015, 30, 1043–1053. [Google Scholar] [CrossRef]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Krüger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef]
- Wang, J.F.; Shao, L.; Sun, X.; Young, L.T. Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord. 2009, 11, 23–529. [Google Scholar] [CrossRef]
- Michel, T.M.; Sheldrick, A.J.; Camara, S.; Grünblatt, E.; Schneider, F.; Riederer, P. Alteration of the pro-oxidant xanthine oxidase (XO) in the thalamus and occipital cortex of patients with schizophrenia. World J. Biol. Psychiatry 2011, 12, 588–597. [Google Scholar] [CrossRef]
- Das, T.K.; Javadzadeh, A.; Dey, A.; Sabesan, P.; Theberge, J.; Radua, J.; Palaniyappan, L. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 91, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, J.; Yang, H.; Yan, L.; Liu, D.; Zhu, L.; Zhang, X. Cognitive Impairment and Psychopathology Are Related to Plasma Oxidative Stress in Long Term Hospitalized Patients With Chronic Schizophrenia. Front. Psychiatry 2022, 13, 896694. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Do, K.Q. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat. Rev. Neurosci. 2016, 17, 125–134. [Google Scholar] [CrossRef]
- Lin, C.H.; Lane, H.Y. Early Identification and Intervention of Schizophrenia: Insight From Hypotheses of Glutamate Dysfunction and Oxidative Stress. Front. Psychiatry 2019, 10, 93. [Google Scholar] [CrossRef]
- Xie, T.; Li, Q.; Luo, X.; Tian, L.; Wang, Z.; Tan, S.; Chen, S.; Yang, G.; An, H.; Yang, F.; et al. Plasma total antioxidant status and cognitive impairments in first-episode drug-naïve patients with schizophrenia. Cogn. Neurodyn. 2019, 13, 357–365. [Google Scholar] [CrossRef]
- Matsuzawa, D.; Hashimoto, K. Magnetic resonance spectroscopy study of the antioxidant defense system in schizophrenia. Antioxid. Redox Signal. 2011, 15, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, D.; Obata, T.; Shirayama, Y.; Nonaka, H.; Kanazawa, Y.; Yoshitome, E.; Takanashi, J.; Matsuda, T.; Shimizu, E.; Ikehira, H.; et al. Negative correlation between brain glutathione level and negative symptoms in schizophrenia: A 3T 1H-MRS study. PLoS ONE 2008, 3, e1944. [Google Scholar] [CrossRef] [PubMed]
- Więdłocha, M.; Zborowska, N.; Marcinowicz, P.; Dębowska, W.; Dębowska, M.; Zalewska, A.; Maciejczyk, M.; Waszkiewicz, N.; Szulc, A. Oxidative Stress Biomarkers among Schizophrenia Inpatients. Brain Sci. 2023, 13, 490. [Google Scholar] [CrossRef]
- Cruz, B.F.; de Campos-Carli, S.M.; de Oliveira, A.M.; de Brito, C.B.; Garcia, Z.M.; Arifa, R.D.D.N.; Souza, D.D.G.D.; Teixeira, A.L.; Salgado, J.V. Investigating potential associations between neurocognition/social cognition and oxidative stress in schizophrenia. Psychiatry Res. 2021, 298, 113832. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Liencres, C.; Tas, C.; Brown, E.C.; Erdin, S.; Onur, E.; Cubukcoglu, Z.; Aydemir, O.; Esen-Danaci, A.; Brüne, M. Oxidative stress in schizophrenia: A case-control study on the effects on social cognition and neurocognition. BMC Psychiatry 2014, 14, 268. [Google Scholar] [CrossRef]
- Berk, M.; Copolov, D.; Dean, O.; Lu, K.; Jeavons, S.; Schapkaitz, I.; Anderson-Hunt, M.; Judd, F.; Katz, F.; Katz, P.; et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—A double-blind, randomized, placebo-controlled trial. Biol Psychiatry 2008, 64, 361–368. [Google Scholar] [CrossRef]
- Farokhnia, M.; Azarkolah, A.; Adinehfar, F.; Khodaie-Ardakani, M.R.; Hosseini, S.M.; Yekehtaz, H.; Tabrizi, M.; Rezaei, F.; Salehi, B.; Sadeghi, S.M.; et al. N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: A randomized, double-blind, placebo-controlled study. Clin. Neuropharmacol. 2013, 36, 185–192. [Google Scholar] [CrossRef]
- Breier, A.; Liffick, E.; Hummer, T.A.; Vohs, J.L.; Yang, Z.; Mehdiyoun, N.F.; Visco, A.C.; Metzler, E.; Zhang, Y.; Francis, M.M. Effects of 12-month, double-blind N-acetyl cysteine on symptoms, cognition and brain morphology in early phase schizophrenia spectrum disorders. Schizophr. Res. 2018, 199, 395–402. [Google Scholar] [CrossRef]
- Sepehrmanesh, Z.; Heidary, M.; Akasheh, N.; Akbari, H.; Heidary, M. Therapeutic effect of adjunctive N-acetyl cysteine (NAC) on symptoms of chronic schizophrenia: A double-blind, randomized clinical trial. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 82, 289–296. [Google Scholar] [CrossRef]
- Kawakubo, Y.; Kasai, K. Support for an association between mismatch negativity and social functioning in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1367–1368. [Google Scholar] [CrossRef] [PubMed]
- Wynn, J.K.; Sugar, C.; Horan, W.P.; Kern, R.; Green, M.F. Mismatch negativity, social cognition, and functioning in schizophrenia patients. Biol. Psychiatry 2010, 67, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, D.; Miyakoshi, M.; Thomas, M.L.; Joshi, Y.B.; Molina, J.L.; Tanaka-Koshiyama, K.; Sprock, J.; Braff, D.L.; Swerdlow, N.R.; Light, G.A. Unique contributions of sensory discrimination and gamma synchronization deficits to cognitive, clinical, and psychosocial functional impairments in schizophrenia. Schizophr. Res. 2021, 228, 280–287. [Google Scholar] [CrossRef]
- Lee, M.; Sehatpour, P.; Hoptman, M.J.; Lakatos, P.; Dias, E.C.; Kantrowitz, J.T.; Martinez, A.M.; Javitt, D.C. Neural mechanisms of mismatch negativity dysfunction in schizophrenia. Mol. Psychiatry 2017, 22, 1585–1593. [Google Scholar] [CrossRef]
- Lavoie, S.; Murray, M.M.; Deppen, P.; Knyazeva, M.G.; Berk, M.; Boulat, O.; Bovet, P.; I Bush, A.; Conus, P.; Copolov, D.; et al. Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology 2008, 33, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Górny, M.; Wnuk, A.; Kamińska, A.; Kamińska, K.; Chwatko, G.; Bilska-Wilkosz, A.; Iciek, M.; Kajta, M.; Rogóż, Z.; Lorenc-Koci, E. Glutathione Deficiency and Alterations in the Sulfur Amino Acid Homeostasis during Early Postnatal Development as Potential Triggering Factors for Schizophrenia-Like Behavior in Adult Rats. Molecules 2019, 24, 4253. [Google Scholar] [CrossRef]
- Lech, M.A.; Leśkiewicz, M.; Kamińska, K.; Rogóż, Z.; Lorenc-Koci, E. Glutathione Deficiency during Early Postnatal Development Causes Schizophrenia-Like Symptoms and a Reduction in BDNF Levels in the Cortex and Hippocampus of Adult Sprague-Dawley Rats. Int. J. Mol. Sci. 2021, 22, 6171. [Google Scholar] [CrossRef] [PubMed]
- Rogóż, Z.; Kamińska, K.; Lech, M.A.; Lorenc-Koci, E. N-Acetylcysteine and Aripiprazole Improve Social Behavior and Cognition and Modulate Brain BDNF Levels in a Rat Model of Schizophrenia. Int. J. Mol. Sci. 2022, 23, 2125. [Google Scholar] [CrossRef] [PubMed]
- Krabbendam, L.; van Os, J. Schizophrenia and urbanicity: A major environmental influence—Conditional on genetic risk. Schizophr. Bull. 2005, 31, 795–799. [Google Scholar] [CrossRef]
- Mizrahi, R. Social Stress and Psychosis Risk: Common Neurochemical Substrates? Neuropsychopharmacology 2016, 41, 666–674. [Google Scholar] [CrossRef]
- Möller, M.; Du Preez, J.L.; Emsley, R.; Harvey, B.H. Isolation rearing-induced deficits in sensorimotor gating and social interaction in rats are related to cortico-striatal oxidative stress, and reversed by sub-chronic clozapine administration. Eur. Neuropsychopharmacol. 2011, 21, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Neill, J.C.; Barnes, S.; Cook, S.; Grayson, B.; Idris, N.F.; McLean, S.L.; Snigdha, S.; Rajagopal, L.; Harte, M.K. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: Focus on NMDA receptor antagonism. Pharmacol. Ther. 2010, 128, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Araújo, T.D.S.; Filho, A.J.M.C.; Monte, A.S.; Queiroz, A.I.D.G.; Cordeiro, R.C.; Machado, M.D.J.S.; Lima, R.D.F.; de Lucena, D.F.; Maes, M.; Macêdo, D. Reversal of schizophrenia-like symptoms and immune alterations in mice by immunomodulatory drugs. J. Psychiatr. Res. 2017, 84, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Onaolapo, A.Y.; Aina, O.A.; Onaolapo, O.J. Melatonin attenuates behavioural deficits and reduces brain oxidative stress in a rodent model of schizophrenia. Biomed. Pharmacother. 2017, 92, 373–383. [Google Scholar] [CrossRef]
- Akosman, M.S.; Türkmen, R.; Demirel, H.H. Investigation of the protective effect of resveratrol in an MK-801-induced mouse model of schizophrenia. Environ. Sci. Pollut. Res. Int. 2021, 28, 65872–65884. [Google Scholar] [CrossRef]
- Baker, D.A.; Madayag, A.; Kristiansen, L.V.; Meador-Woodruff, J.H.; Haroutunian, V.; Raju, I. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology 2008, 33, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Phensy, A.; Duzdabanian, H.E.; Brewer, S.; Panjabi, A.; Driskill, C.; Berz, A.; Peng, G.; Kroener, S. Antioxidant Treatment with N-acetyl Cysteine Prevents the Development of Cognitive and Social Behavioral Deficits that Result from Perinatal Ketamine Treatment. Front. Behav. Neurosci. 2017, 11, 106. [Google Scholar] [CrossRef]
- Swanepoel, T.; Möller, M.; Harvey, B.H. N-acetyl cysteine reverses bio-behavioural changes induced by prenatal inflammation, adolescent methamphetamine exposure and combined challenges. Psychopharmacology 2018, 235, 351–368. [Google Scholar] [CrossRef]
- Depino, A.M. Perinatal inflammation and adult psychopathology: From preclinical models to humans. Semin. Cell Dev. Biol. 2018, 77, 104–114. [Google Scholar] [CrossRef]
- Hagberg, H.; Gressens, P.; Mallard, C. Inflammation during fetal and neonatal life: Implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 2012, 71, 444–457. [Google Scholar] [CrossRef]
- Grant, K.M.; LeVan, T.D.; Wells, S.M.; Li, M.; Stoltenberg, S.F.; Gendelman, H.E.; Carlo, G.; Bevins, R. Methamphetamine-associated psychosis. J. Neuroimmune Pharmacol. 2012, 7, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, B.; Miller, M.L.; Hurd, Y.L. Cannabis Use during Adolescent Development: Susceptibility to Psychiatric Illness. Front. Psychiatry 2013, 4, 129. [Google Scholar] [CrossRef]
- Monte, A.S.; da Silva, F.E.R.; Lima, C.N.D.C.; Vasconcelos, G.S.; Gomes, N.S.; Miyajima, F.; Vasconcelos, S.M.M.; Gama, C.S.; Seeman, M.V.; de Lucena, D.F.; et al. Sex influences in the preventive effects of N-acetylcysteine in a two-hit animal model of schizophrenia. J. Psychopharmacol. 2020, 34, 125–136. [Google Scholar] [CrossRef]
- Romero-Miguel, D.; Casquero-Veiga, M.; MacDowell, K.S.; Torres-Sanchez, S.; Garcia-Partida, J.A.; Lamanna-Rama, N.; Romero-Miranda, A.; Berrocoso, E.; Leza, J.C.; Desco, M.; et al. A Characterization of the Effects of Minocycline Treatment During Adolescence on Structural, Metabolic, and Oxidative Stress Parameters in a Maternal Immune Stimulation Model of Neurodevelopmental Brain Disorders. Int. J. Neuropsychopharmacol. 2021, 24, 734–748. [Google Scholar] [CrossRef] [PubMed]
- Buckley, P.F. Neuroinflammation and Schizophrenia. Curr. Psychiatry Rep. 2019, 21, 72. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef]
- Pandey, G.N.; Rizavi, H.S.; Zhang, H.; Ren, X. Abnormal gene and protein expression of inflammatory cytokines in the postmortem brain of schizophrenia patients. Schizophr. Res. 2018, 192, 247–254. [Google Scholar] [CrossRef]
- Gallego, J.A.; Blanco, E.A.; Husain-Krautter, S.; Fagen, E.M.; Moreno-Merino, P.; del Ojo-Jiménez, J.A.; Ahmed, A.; Rothstein, T.L.; Lencz, T.; Malhotra, A.K. Cytokines in cerebrospinal fluid of patients with schizophrenia spectrum disorders: New data and an updated meta-analysis. Schizophr. Res. 2018, 202, 64–71. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Schwarz, J.M. The immune system and developmental programming of brain and behavior. Front. Neuroendocrinol. 2012, 33, 267–286. [Google Scholar] [CrossRef]
- Hammond, T.R.; Robinton, D.; Stevens, B. Microglia and the Brain: Complementary Partners in Development and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 523–544. [Google Scholar] [CrossRef]
- Bar, E.; Barak, B. Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia 2019, 67, 2125–2141. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef]
- Mokhtari, R.; Lachman, H.M. The Major Histocompatibility Complex (MHC) in Schizophrenia: A Review. J. Clin. Cell Immunol. 2016, 7, 479. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Leschak, C.J.; Eisenberger, N.I. Two Distinct Immune Pathways Linking Social Relationships With Health: Inflammatory and Antiviral Processes. Psychosom. Med. 2019, 81, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, N.I.; Moieni, M.; Inagaki, T.K.; Muscatell, K.A.; Irwin, M.R. In Sickness and in Health: The Co-Regulation of Inflammation and Social Behavior. Neuropsychopharmacology 2017, 42, 242–253. [Google Scholar] [CrossRef]
- Barcik, W.; Chiacchierini, G.; Bimpisidis, Z.; Papaleo, F. Immunology and microbiology: How do they affect social cognition and emotion recognition? Curr. Opin. Immunol. 2021, 71, 46–54. [Google Scholar] [CrossRef]
- Garés-Caballer, M.; Sánchez-Ortí, J.V.; Correa-Ghisays, P.; Balanzá-Martínez, V.; Selva-Vera, G.; Vila-Francés, J.; Magdalena-Benedito, R.; San-Martin, C.; Victor, V.M.; Escribano-Lopez, I.; et al. Immune-Inflammatory Biomarkers Predict Cognition and Social Functioning in Patients With Type 2 Diabetes Mellitus, Major Depressive Disorder, Bipolar Disorder, and Schizophrenia: A 1-Year Follow-Up Study. Front. Neurol. 2022, 13, 883927. [Google Scholar] [CrossRef]
- Gonzalez-Blanco, L.; Garcia-Portilla, M.P.; Santo, F.D.; Garcia-Alvarez, L.; de la Fuente-Tomas, L.; Menendez-Miranda, I.; Bascarán, M.T.B.; Saiz, P.A.; Bobes, J. Predicting real-world functioning in outpatients with schizophrenia: Role of inflammation and psychopathology. Psychiatry Res. 2019, 280, 112509. [Google Scholar] [CrossRef]
- González-Blanco, L.; García-Portilla, M.P.; García-Álvarez, L.; de la Fuente-Tomás, L.; García, C.I.; Sáiz, P.A.; Rodríguez-González, S.; Coto-Montes, A.; Bobes, J. Can interleukin-2 and interleukin-1β be specific biomarkers of negative symptoms in schizophrenia? ¿Pueden ser la interleucina-2 y la interleucina-1β biomarcadores específicos de la sintomatología negativa en la esquizofrenia? Rev. Psiquiatr. Salud Ment. Engl. Ed. 2019, 12, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.-H.; Kim, H.; Kim, J.-W.; Ryu, S.; Lee, J.-Y.; Kim, J.-M.; Shin, I.-S.; Kim, S.-W. Association between Peripheral Inflammatory Cytokines and Cognitive Function in Patients with First-Episode Schizophrenia. J. Pers. Med. 2022, 12, 1137. [Google Scholar] [CrossRef] [PubMed]
- Levkovitz, Y.; Mendlovich, S.; Riwkes, S.; Braw, Y.; Levkovitch-Verbin, H.; Gal, G.; Fennig, S.; Treves, I.; Kron, S. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J. Clin. Psychiatry 2010, 71, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, I.B.; Hallak, J.; Husain, N.; Minhas, F.; Stirling, J.; Richardson, P.; Dursun, S.; Dunn, G.; Deakin, B. Minocycline benefits negative symptoms in early schizophrenia: A andomized double-blind placebo-controlled clinical trial in patients on standard treatment. J. Psychopharmacol. 2012, 26, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Guo, X.; Wu, R.; Ou, J.; Zheng, Y.; Zhang, B.; Xie, L.; Zhang, L.; Yang, L.; Yang, S.; et al. Minocycline supplementation for treatment of negative symptoms in early-phase schizophrenia: A double blind, randomized, controlled trial. Schizophr. Res. 2014, 153, 169–176. [Google Scholar] [CrossRef]
- Khodaie-Ardakani, M.-R.; Mirshafiee, O.; Farokhnia, M.; Tajdini, M.; Hosseini, S.-M.; Modabbernia, A.; Rezaei, F.; Salehi, B.; Yekehtaz, H.; Ashrafi, M.; et al. Minocycline add-on to risperidone for treatment of negative symptoms in patients with stable schizophrenia: Randomized double-blind placebo-controlled study. Psychiatry Res. 2014, 215, 540–546. [Google Scholar] [CrossRef]
- Oya, K.; Kishi, T.; Iwata, N. Efficacy and tolerability of minocycline augmentation therapy in schizophrenia: A systematic review and meta-analysis of randomized controlled trials. Hum. Psychopharmacol. 2014, 29, 483–491. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, H.; Wu, R.; Zhu, F.; Kosten, T.R.; Zhang, X.-Y.; Zhao, J. Minocycline adjunctive treatment to risperidone for negative symptoms in schizophrenia: Association with pro-inflammatory cytokine levels. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 85, 69–76. [Google Scholar] [CrossRef]
- Turano, A.; McAuley, E.M.; Muench, M.C.; Schwarz, J.M. Examining the impact of neuroimmune dysregulation on social behavior of male and female juvenile rats. Behav. Brain Res. 2021, 415, 113449. [Google Scholar] [CrossRef]
- Vojtechova, I.; Maleninska, K.; Kutna, V.; Klovrza, O.; Tuckova, K.; Petrasek, T.; Stuchlik, A. Behavioral Alterations and Decreased Number of Parvalbumin-Positive Interneurons in Wistar Rats after Maternal Immune Activation by Lipopolysaccharide: Sex Matters. Int. J. Mol. Sci. 2021, 22, 3274. [Google Scholar] [CrossRef]
- O’Loughlin, E.; Pakan, J.M.P.; Yilmazer-Hanke, D.; McDermott, K.W. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J. Neuroinflammation 2017, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.K.; Peleg-Raibstein, D.; Mouttet, F.; Feldon, J.; Meyer, U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology 2010, 35, 2462–2478. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Taricano, M.; Maiorka, P.C.; Palermo-Neto, J.; Bernardi, M.M. Prenatal lipopolysaccharide reduces social behavior in male offspring. Neuroimmunomodulation 2010, 17, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.; Schwarting, R.K.; Fuchs, E.; Wöhr, M. Increased affective ultrasonic communication during fear learning in adult male rats exposed to maternal immune activation. J. Psychiatr. Res. 2012, 46, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Javitt, D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 2022, 604, 509–516. [Google Scholar] [CrossRef]
- Hu, W.; MacDonald, M.L.; Elswick, D.E.; Sweet, R.A. The glutamate hypothesis of schizophrenia: Evidence from human brain tissue studies. Ann. N. Y. Acad. Sci. 2015, 1338, 38–57. [Google Scholar] [CrossRef]
- Janhunen, S.K.; Svärd, H.; Talpos, J.; Kumar, G.; Steckler, T.; Plath, N.; Lerdrup, L.; Ruby, T.; Haman, M.; Wyler, R.; et al. The subchronic phencyclidine rat model: Relevance for the assessment of novel therapeutics for cognitive impairment associated with schizophrenia. Psychopharmacology 2015, 232, 4059–4083. [Google Scholar] [CrossRef]
- Aoyama, N.; Théberge, J.; Drost, D.J.; Manchanda, R.; Northcott, S.; Neufeld, R.W.J.; Menon, R.S.; Rajakumar, N.; Pavlosky, W.F.; Densmore, M.; et al. Grey matter and social functioning correlates of glutamatergic metabolite loss in schizophrenia. Br. J. Psychiatry 2011, 198, 448–456. [Google Scholar] [CrossRef]
- Dempster, K.; Norman, R.; Théberge, J.; Densmore, M.; Schaefer, B.; Williamson, P. Glutamatergic metabolite correlations with neuropsychological tests in first episode schizophrenia. Psychiatry Res. 2015, 233, 180–185. [Google Scholar] [CrossRef]
- Dracheva, S.; Marras, S.A.; Elhakem, S.L.; Kramer, F.R.; Davis, K.L.; Haroutunian, V. N-methyl-D-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am. J. Psychiatry 2001, 158, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Knable, M.B.; Barci, B.M.; Bartko, J.J.; Webster, M.J.; Torrey, E.F. Molecular abnormalities in the major psychiatric illnesses: Classification and Regression Tree (CRT) analysis of post-mortem prefrontal markers. Mol. Psychiatry 2002, 7, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, L.V.; Beneyto, M.; Haroutunian, V.; Meador-Woodruff, J.H. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol. Psychiatry 2006, 11, 705–737. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, B.F.; Vohs, J.L.; Krishnan, G.P.; Rass, O.; Hetrick, W.P.; Morzorati, S.L. The auditory steady-state response (ASSR): A translational biomarker for schizophrenia. Suppl. Clin. Neurophysiol. 2013, 62, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Kawakubo, Y.; Kamio, S.; Nose, T.; Iwanami, A.; Nakagome, K.; Fukuda, M.; Kato, N.; Rogers, M.A.; Kasai, K. Phonetic mismatch negativity predicts social skills acquisition in schizophrenia. Psychiatry Res. 2007, 152, 261–265. [Google Scholar] [CrossRef]
- Light, G.A.; Braff, D.L. Mismatch negativity deficits are associated with poor functioning in schizophrenia patients. Arch. Gen. Psychiatry 2005, 62, 127–136. [Google Scholar] [CrossRef]
- Zhou, T.-H.; Mueller, N.E.; Spencer, K.M.; Mallya, S.G.; Lewandowski, K.; Norris, L.; Levy, D.L.; Cohen, B.M.; Öngür, D.; Hall, M.-H. Auditory steady state response deficits are associated with symptom severity and poor functioning in patients with psychotic disorder. Schizophr. Res. 2018, 201, 278–286. [Google Scholar] [CrossRef]
- Leishman, E.; O’donnell, B.F.; Millward, J.B.; Vohs, J.L.; Rass, O.; Krishnan, G.P.; Bolbecker, A.R.; Morzorati, S.L. Phencyclidine Disrupts the Auditory Steady State Response in Rats. PLoS ONE 2015, 10, e0134979. [Google Scholar] [CrossRef]
- Plourde, G.; Baribeau, J.; Bonhomme, V. Ketamine increases the amplitude of the 40-Hz auditory steady-state response in humans. Br. J. Anaesth. 1997, 78, 524–529. [Google Scholar] [CrossRef]
- Sivarao, D.V.; Chen, P.; Senapati, A.; Yang, Y.; Fernandes, A.; Benitex, Y.; Whiterock, V.; Li, Y.-W.; Ahlijanian, M.K. 40 Hz Auditory Steady-State Response Is a Pharmacodynamic Biomarker for Cortical NMDA Receptors. Neuropsychopharmacology 2016, 41, 2232–2240. [Google Scholar] [CrossRef]
- Rosburg, T.; Kreitschmann-Andermahr, I. The effects of ketamine on the mismatch negativity (MMN) in humans—A meta-analysis. Clin. Neurophysiol. 2016, 127, 1387–1394. [Google Scholar] [CrossRef]
- Ahnaou, A.; Huysmans, H.; Biermans, R.; Manyakov, N.V.; Drinkenburg, W.H.I.M. Ketamine: Differential neurophysiological dynamics in functional networks in the rat brain. Transl. Psychiatry 2017, 7, e1237. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Balla, A.; Sershen, H.; Sehatpour, P.; Lakatos, P.; Javitt, D.C. Rodent Mismatch Negativity/theta Neuro-Oscillatory Response as a Translational Neurophysiological Biomarker for N-Methyl-D-Aspartate Receptor-Based New Treatment Development in Schizophrenia. Neuropsychopharmacology 2018, 43, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, T.; Noda, Y.; Nabeshima, T. Phencyclidine and genetic animal models of schizophrenia developed in relation to the glutamate hypothesis. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 291–301. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kamegaya, E.; Hagino, Y.; Takamatsu, Y.; Sawada, W.; Matsuzawa, M.; Ide, S.; Yamamoto, T.; Mishina, M.; Ikeda, K. Loss of GluN2D subunit results in social recognition deficit, social stress, 5-HT2C receptor dysfunction, and anhedonia in mice. Neuropharmacology 2017, 112, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; Liu, H.; Xu, J.; Tan, Z.; Dong, H.; Tian, B.; Wu, S.; Wang, W. Activation of basolateral amygdala to anterior cingulate cortex circuit alleviates MK-801 induced social and cognitive deficits of schizophrenia. Front. Cell. Neurosci. 2022, 16, 1070015. [Google Scholar] [CrossRef]
- Dogra, S.; Conn, P.J. Metabotropic Glutamate Receptors As Emerging Targets for the Treatment of Schizophrenia. Mol. Pharmacol. 2022, 101, 275–285. [Google Scholar] [CrossRef]
- Harich, S.; Gross, G.; Bespalov, A. Stimulation of the metabotropic glutamate 2/3 receptor attenuates social novelty discrimination deficits induced by neonatal phencyclidine treatment. Psychopharmacology 2007, 192, 511–519. [Google Scholar] [CrossRef]
- Shimazaki, T.; Kaku, A.; Chaki, S. D-Serine and a glycine transporter-1 inhibitor enhance social memory in rats. Psychopharmacology 2010, 209, 263–270. [Google Scholar] [CrossRef]
- Clifton, N.E.; Morisot, N.; Girardon, S.; Millan, M.J.; Loiseau, F. Enhancement of social novelty discrimination by positive allosteric modulators at metabotropic glutamate 5 receptors: Adolescent administration prevents adult-onset deficits induced by neonatal treatment with phencyclidine. Psychopharmacology 2013, 225, 579–594. [Google Scholar] [CrossRef]
- Chaki, S.; Shimazaki, T.; Karasawa, J.-I.; Aoki, T.; Kaku, A.; Iijima, M.; Kambe, D.; Yamamoto, S.; Kawakita, Y.; Shibata, T.; et al. Efficacy of a glycine transporter 1 inhibitor TASP0315003 in animal models of cognitive dysfunction and negative symptoms of schizophrenia. Psychopharmacology 2015, 232, 2849–2861. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Epstein, M.L.; Lee, M.; Lehrfeld, N.; A Nolan, K.; Shope, C.; Petkova, E.; Silipo, G.; Javitt, D.C. Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: Correlation with symptoms. Schizophr. Res. 2018, 191, 70–79. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Woods, S.W.; Petkova, E.; Cornblatt, B.; Corcoran, C.; Chen, H.; Silipo, G.; Javitt, D.C. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: A pilot, double-blind, placebo-controlled, andomized parallel group mechanistic proof-of-concept trial. Lancet Psychiatry 2015, 2, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Hirayasu, Y.; Sato, S.-I.; Takahashi, H.; Iida, S.; Shuto, N.; Yoshida, S.; Funatogawa, T.; Yamada, T.; Higuchi, T. A double-blind randomized study assessing safety and efficacy following one-year adjunctive treatment with bitopertin, a glycine reuptake inhibitor, in Japanese patients with schizophrenia. BMC Psychiatry 2016, 16, 66. [Google Scholar] [CrossRef]
- Deiana, S.; Hauber, W.; Munster, A.; Sommer, S.; Ferger, B.; Marti, A.; Schmid, B.; Dorner-Ciossek, C.; Rosenbrock, H. Pro-cognitive effects of the GlyT1 inhibitor Bitopertin in rodents. Eur. J. Pharmacol. 2022, 935, 175306. [Google Scholar] [CrossRef]
- Ferguson, B.R.; Gao, W.J. PV Interneurons: Critical Regulators of E/I Balance for Prefrontal Cortex-Dependent Behavior and Psychiatric Disorders. Front. Neural Circuits 2018, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.F.; Demeter, E.; Phan, K.L.; Tso, I.F.; Welsh, R.C. Abnormal GABAergic function and negative affect in schizophrenia. Neuropsychopharmacology 2014, 39, 1000–1008. [Google Scholar] [CrossRef]
- Tso, I.F.; Fang, Y.; Phan, K.L.; Welsh, R.C.; Taylor, S.F. Abnormal GABAergic function and face processing in schizophrenia: A pharmacologic-fMRI study. Schizophr. Res. 2015, 168, 338–344. [Google Scholar] [CrossRef]
- Fang, L.-P.; Na Zhao, N.; Caudal, L.C.; Chang, H.-F.; Zhao, R.; Lin, C.-H.; Hainz, N.; Meier, C.; Bettler, B.; Huang, W.; et al. Impaired bidirectional communication between interneurons and oligodendrocyte precursor cells affects social cognitive behavior. Nat. Commun. 2022, 13, 1394. [Google Scholar] [CrossRef]
- Deng, X.; Gu, L.; Sui, N.; Guo, J.; Liang, J. Parvalbumin interneuron in the ventral hippocampus functions as a discriminator in social memory. Proc. Natl. Acad. Sci. USA 2019, 116, 16583–16592. [Google Scholar] [CrossRef]
- Soares, A.R.; Gildawie, K.R.; Honeycutt, J.A.; Brenhouse, H.C. Region-specific effects of maternal separation on oxidative stress accumulation in parvalbumin neurons of male and female rats. Behav. Brain Res. 2020, 388, 112658. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.J.; Webster, M.J.; Sivagnanasundaram, S.; Duncan, C.; Elashoff, M.; Weickert, C.S. Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am. J. Psychiatry 2010, 167, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Hoftman, G.D.; Volk, D.W.; Bazmi, H.H.; Li, S.; Sampson, A.R.; Lewis, D.A. Altered cortical expression of GABA-related genes in schizophrenia: Illness progression vs developmental disturbance. Schizophr. Bull. 2015, 41, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Avilés, J.G.; Curley, A.A.; Hashimoto, T.; Morrow, A.L.; Ramsey, A.J.; O’donnell, P.; Volk, D.W.; Lewis, D.A. Altered markers of tonic inhibition in the dorsolateral prefrontal cortex of subjects with schizophrenia. Am. J. Psychiatry 2009, 166, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Burgos, G.; Hashimoto, T.; Lewis, D.A. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr. Psychiatry Rep. 2010, 12, 335–344. [Google Scholar] [CrossRef]
- Hashimoto, T.; Arion, D.; Unger, T.; Maldonado-Avilés, J.G.; Morris, H.M.; Volk, D.W.; Mirnics, K.; Lewis, D.A. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2008, 13, 147–161. [Google Scholar] [CrossRef]
- Woo, T.U.; Miller, J.L.; Lewis, D.A. Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am. J. Psychiatry 1997, 154, 1013–1015. [Google Scholar] [CrossRef]
- Beasley, C.L.; Zhang, Z.J.; Patten, I.; Reynolds, G.P. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol. Psychiatry 2002, 52, 708–715. [Google Scholar] [CrossRef]
- Tooney, P.A.; Chahl, L.A. Neurons expressing calcium-binding proteins in the prefrontal cortex in schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2004, 28, 273–278. [Google Scholar] [CrossRef]
- Glausier, J.R.; Fish, K.N.; Lewis, D.A. Altered parvalbumin basket cell inputs in the dorsolateral prefrontal cortex of schizophrenia subjects. Mol. Psychiatry 2014, 19, 30–36. [Google Scholar] [CrossRef]
- Maas, D.A.; Eijsink, V.D.; Spoelder, M.; van Hulten, J.A.; De Weerd, P.; Homberg, J.R.; Vallès, A.; Nait-Oumesmar, B.; Martens, G.J.M. Interneuron hypomyelination is associated with cognitive inflexibility in a rat model of schizophrenia. Nat. Commun. 2020, 11, 2329. [Google Scholar] [CrossRef]
- Maas, D.A.; Eijsink, V.D.; van Hulten, J.A.; Panic, R.; De Weerd, P.; Homberg, J.R.; Vallès, A.; Nait-Oumesmar, B.; Martens, G.J.M. Antioxidant treatment ameliorates prefrontal hypomyelination and cognitive deficits in a rat model of schizophrenia. Neuropsychopharmacology 2021, 46, 1161–1171. [Google Scholar] [CrossRef]
- Selten, M.M.; Meyer, F.; Ba, W.; Vallès, A.; Maas, D.A.; Negwer, M.; Eijsink, V.D.; van Vugt, R.W.M.; van Hulten, J.A.; van Bakel, N.H.M.; et al. Increased GABAB receptor signaling in a rat model for schizophrenia. Sci. Rep. 2016, 6, 34240. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.-H.; Kulak, A.; Kraftsik, R.; Chen, Y.; Dalton, T.P.; Cuenod, M.; Do, K.Q. Redox dysregulation affects the ventral but not dorsal hippocampus: Impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J. Neurosci. 2010, 30, 2547–2558. [Google Scholar] [CrossRef] [PubMed]
- Ferrarelli, F.; Massimini, M.; Peterson, M.J.; Riedner, B.A.; Lazar, M.; Murphy, M.J.; Huber, R.; Rosanova, M.; Alexander, A.L.; Kalin, N.H.; et al. Reduced evoked gamma oscillations in the frontal cortex in schizophrenia patients: A TMS/EEG study. Am. J. Psychiatry 2008, 165, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Burgos, G.; Cho, R.Y.; Lewis, D.A. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol. Psychiatry 2015, 77, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.H.; Nicolas, D.; Kraftsik, R.; Cuénod, M.; Do, K.Q.; Hornung, J.P. Glutathione deficit during development induces anomalies in the rat anterior cingulate GABAergic neurons: Relevance to schizophrenia. Neurobiol. Dis. 2006, 22, 624–637. [Google Scholar] [CrossRef]
- Ferguson, B.R.; Gao, W.J. Thalamic control of cognition and social behavior via regulation of g-aminobutyric acidergic signaling and excitation/inhibition balance in the medial prefrontal cortex. Biol. Psychiatry 2018, 83, 657–669. [Google Scholar] [CrossRef]
- Perez, S.M.; Boley, A.; Lodge, D.J. Region specific knockdown of Parvalbumin or Somatostatin produces neuronal and behavioral deficits consistent with those observed in schizophrenia. Transl. Psychiatry 2019, 9, 264. [Google Scholar] [CrossRef]
- Uranova, N.A.; Vostrikov, V.M.; Orlovskaya, D.D.; Rachmanova, V.I. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: A study from the Stanley Neuropathology Consortium. Schizophr. Res. 2004, 67, 269–275. [Google Scholar] [CrossRef]
- Vostrikov, V.M.; Uranova, N.A.; Rakhmanova, V.I.; Orlovskaia, D.D. Snizhennaia chislennaia plotnost’ oligodendrogliotsitov v prefrontal’noĭ kore pri shizofrenii [Lowered oligodendroglial cell density in the prefrontal cortex in schizophrenia]. Zhurnal Nevrol. I Psikhiatrii Im. S.S. Korsakova 2004, 104, 47–51. [Google Scholar]
- Hof, P.R.; Haroutunian, V.; Friedrich, V.L., Jr.; Byne, W.; Buitron, C.; Perl, D.P.; Davis, K.L. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol. Psychiatry 2003, 53, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.K.; Uylings, H.B.; Sanz-Arigita, E.; Pakkenberg, B. Glial cell loss in the anterior cingulate cortex, a subregion of the prefrontal cortex, in subjects with schizophrenia. Am. J. Psychiatry 2004, 161, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Flynn, S.W.; Lang, D.; Mackay, A.L.; Goghari, V.; Vavasour, I.M.; Whittall, K.P.; Smith, G.N.; Arango, V.; Mann, J.J.; Dwork, A.J.; et al. Abnormalities of myelination in schizophrenia detected in vivo with MRI, and post-mortem with analysis of oligodendrocyte proteins. Mol. Psychiatry 2003, 8, 811–820. [Google Scholar] [CrossRef]
- Hakak, Y.; Walker, J.R.; Li, C.; Wong, W.H.; Davis, K.L.; Buxbaum, J.D.; Haroutunian, V.; Fienberg, A.A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4746–4751. [Google Scholar] [CrossRef]
- Stedehouder, J.; Kushner, S.A. Myelination of parvalbumin interneurons: A parsimonious locus of pathophysiological convergence in schizophrenia. Mol. Psychiatry 2017, 22, 4–12. [Google Scholar] [CrossRef]
- Nakamura, J.P.; Schroeder, A.; Gibbons, A.; Sundram, S.; Hill, R.A. Timing of maternal immune activation and sex influence schizophrenia-relevant cognitive constructs and neuregulin and GABAergic pathways. Brain Behav. Immun. 2022, 100, 70–82. [Google Scholar] [CrossRef]
- Nakamura, J.P.; Schroeder, A.; Hudson, M.; Jones, N.; Gillespie, B.; Du, X.; Notaras, M.; Swaminathan, V.; Reay, W.R.; Atkins, J.R.; et al. The maternal immune activation model uncovers a role for the Arx gene in GABAergic dysfunction in schizophrenia. Brain Behav. Immun. 2019, 81, 161–171. [Google Scholar] [CrossRef]
- Canetta, S.; Bolkan, S.; Padilla-Coreano, N.; Song, L.J.; Sahn, R.; Harrison, N.L.; Gordon, J.A.; Brown, A.; Kellendonk, C. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol. Psychiatry 2016, 21, 956–968. [Google Scholar] [CrossRef]
- Okamoto, K.; Hitora-Imamura, N.; Hioki, H.; Ikegaya, Y. GABAergic malfunction in the anterior cingulate cortex underlying maternal immune activation-induced social deficits. J. Neuroimmunol. 2018, 321, 92–96. [Google Scholar] [CrossRef]
- Dickerson, D.D.; Overeem, K.A.; Wolff, A.R.; Williams, J.M.; Abraham, W.C.; Bilkey, D.K. Association of aberrant neural synchrony and altered GAD67 expression following exposure to maternal immune activation, a risk factor for schizophrenia. Transl. Psychiatry 2014, 4, e418. [Google Scholar] [CrossRef]
- Park, G.-H.; Noh, H.; Shao, Z.; Ni, P.; Qin, Y.; Liu, D.; Beaudreault, C.P.; Park, J.S.; Abani, C.P.; Park, J.M.; et al. Activated microglia cause metabolic disruptions in developmental cortical interneurons that persist in interneurons from individuals with schizophrenia. Nat. Neurosci. 2020, 23, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- De Felice, M.; Melis, M.; Aroni, S.; Muntoni, A.L.; Fanni, S.; Frau, R.; Devoto, P.; Pistis, M. The PPARα agonist fenofibrate attenuates disruption of dopamine function in a maternal immune activation rat model of schizophrenia. CNS Neurosci. Ther. 2019, 25, 549–561. [Google Scholar] [CrossRef]
- Luchicchi, A.; Lecca, S.; Melis, M.; De Felice, M.; Cadeddu, F.; Frau, R.; Muntoni, A.L.; Fadda, P.; Devoto, P.; Pistis, M. Maternal Immune Activation Disrupts Dopamine System in the Offspring. Int. J. Neuropsychopharmacol. 2016, 19, pyw007. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Kapur, S. The dopamine hypothesis of schizophrenia: Version III—The final common pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef]
- Nakazawa, K.; Jeevakumar, V.; Nakao, K. Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr. 2017, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.J.; Yang, S.S.; Mack, N.R.; Chamberlin, L.A. Aberrant maturation and connectivity of prefrontal cortex in schizophrenia-contribution of NMDA receptor development and hypofunction. Mol. Psychiatry 2022, 27, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Parellada, E.; Gassó, P. Glutamate and microglia activation as a driver of dendritic apoptosis: A core pathophysiological mechanism to understand schizophrenia. Transl. Psychiatry 2021, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Osimo, E.F.; Beck, K.; Marques, T.R.; Howes, O.D. Synaptic loss in schizophrenia: A meta-analysis and systematic review of synaptic protein and mRNA measures. Mol. Psychiatry 2019, 24, 549–561. [Google Scholar] [CrossRef]
- Flores, C.; Wen, X.; Labelle-Dumais, C.; Kolb, B. Chronic phencyclidine treatment increases dendritic spine density in prefrontal cortex and nucleus accumbens neurons. Synapse 2007, 61, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Hajszan, T.; Leranth, C.; Roth, R.H. Subchronic phencyclidine treatment decreases the number of dendritic spine synapses in the rat prefrontal cortex. Biol. Psychiatry 2006, 60, 639–644. [Google Scholar] [CrossRef]
- Jami, S.A.; Cameron, S.; Wong, J.M.; Daly, E.R.; McAllister, A.K.; Gray, J.A. Increased excitation-inhibition balance and loss of GABAergic synapses in the serine racemase knockout model of NMDA receptor hypofunction. J. Neurophysiol. 2021, 126, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Corcoba, A.; Steullet, P.; Duarte, J.M.N.; Van de Looij, Y.; Monin, A.; Cuenod, M.; Gruetter, R.; Do, K.Q. Glutathione Deficit Affects the Integrity and Function of the Fimbria/Fornix and Anterior Commissure in Mice: Relevance for Schizophrenia. Int. J. Neuropsychopharmacol. 2015, 19, pyv110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, S.; Huang, Q.; Xu, H. N-acetylcysteine attenuates the cuprizone-induced behavioral changes and oligodendrocyte loss in male C57BL/7 mice via its anti-inflammation actions. J. Neurosci. Res. 2018, 96, 803–816. [Google Scholar] [CrossRef]
- Xu, H.; Yang, H.J.; McConomy, B.; Browning, R.; Li, X.M. Behavioral and neurobiological changes in C57BL/6 mouse exposed to cuprizone: Effects of antipsychotics. Front. Behav. Neurosci. 2010, 4, 8. [Google Scholar] [CrossRef]
- Xu, H.; Yang, H.J.; Rose, G.M.; Li, X.M. Recovery of behavioral changes and compromised white matter in C57BL/6 mice exposed to cuprizone: Effects of antipsychotic drugs. Front. Behav. Neurosci. 2011, 5, 31. [Google Scholar] [CrossRef]
- Xu, H.; Yang, H.J.; Zhang, Y.; Clough, R.; Browning, R.; Li, X.M. Behavioral and neurobiological changes in C57BL/6 mice exposed to cuprizone. Behav. Neurosci. 2009, 123, 418–429. [Google Scholar] [CrossRef]
- Makinodan, M.; Yamauchi, T.; Tatsumi, K.; Okuda, H.; Takeda, T.; Kiuchi, K.; Sadamatsu, M.; Wanaka, A.; Kishimoto, T. Demyelination in the juvenile period, but not in adulthood, leads to long-lasting cognitive impairment and deficient social interaction in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 978–985. [Google Scholar] [CrossRef]
- Mullier, E.; Roine, T.; Griffa, A.; Xin, L.; Baumann, P.S.; Klauser, P.; Cleusix, M.; Jenni, R.; Alemàn-Gómez, Y.; Gruetter, R.; et al. N-Acetyl-Cysteine Supplementation Improves Functional Connectivity Within the Cingulate Cortex in Early Psychosis: A Pilot Study. Int. J. Neuropsychopharmacol. 2019, 22, 478–487. [Google Scholar] [CrossRef]
- Klauser, P.; Xin, L.; Fournier, M.; Griffa, A.; Cleusix, M.; Jenni, R.; Cuenod, M.; Gruetter, R.; Hagmann, P.; Conus, P.; et al. N-acetylcysteine add-on treatment leads to an improvement of fornix white matter integrity in early psychosis: A double-blind randomized placebo-controlled trial. Transl. Psychiatry 2018, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, A.; Tyburski, E.; Podwalski, P.; Waszczuk, K.; Rudkowski, K.; Kucharska-Mazur, J.; Mak, M.; Rek-Owodziń, K.; Plichta, P.; Bielecki, M.; et al. Serum inflammatory markers and their associations with white matter integrity of the corpus callosum in schizophrenia patients and healthy controls. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 116, 110510. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, Y.; Edmiston, E.K.; Womer, F.Y.; Zhang, X.; Duan, J.; Zhu, Y.; Zhang, R.; Yin, Z.; Zhang, Y.; et al. Altered structural connectivity and cytokine levels in Schizophrenia and Genetic high-risk individuals: Associations with disease states and vulnerability. Schizophr. Res. 2020, 223, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Zhang, W.; Dai, J.; Liu, J.; Li, F.; Wu, D.; Xiao, Y.; Shah, C.; Sweeney, J.A.; Wu, M.; et al. Increased Peripheral Interleukin 10 Relate to White Matter Integrity in Schizophrenia. Front. Neurosci. 2019, 13, 52. [Google Scholar] [CrossRef]
- Prasad, K.M.; Upton, C.H.; Nimgaonkar, V.L.; Keshavan, M.S. Differential susceptibility of white matter tracts to inflammatory mediators in schizophrenia: An integrated DTI study. Schizophr. Res. 2015, 161, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Chew, L.J.; Fusar-Poli, P.; Schmitz, T. Oligodendroglial alterations and the role of microglia in white matter injury: Relevance to schizophrenia. Dev. Neurosci. 2013, 35, 102–129. [Google Scholar] [CrossRef]
- Li, Q.; Cheung, C.; Wei, R.; Cheung, V.; Hui, E.S.; You, Y.; Wong, P.; Chua, S.E.; McAlonan, G.M.; Wu, E.X. Voxel-based analysis of postnatal white matter microstructure in mice exposed to immune challenge in early or late pregnancy. Neuroimage 2010, 52, 1–8. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Chen, T.; Yan, A.; Xiao, J.; Xie, Y.-L.; Yuan, J.; Chen, P.; Wong, A.O.-L.; Zhang, Y.; Wong, N.-K. Poly(I:C) Challenge Alters Brain Expression of Oligodendroglia-Related Genes of Adult Progeny in a Mouse Model of Maternal Immune Activation. Front. Mol. Neurosci. 2020, 13, 115. [Google Scholar] [CrossRef]
- Makinodan, M.; Tatsumi, K.; Manabe, T.; Yamauchi, T.; Makinodan, E.; Matsuyoshi, H.; Shimoda, S.; Noriyama, Y.; Kishimoto, T.; Wanaka, A. Maternal immune activation in mice delays myelination and axonal development in the hippocampus of the offspring. J. Neurosci. Res. 2008, 86, 2190–2200. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M. Neuroinflammation and white matter pathology in schizophrenia: Systematic review. Schizophr. Res. 2015, 161, 102–112. [Google Scholar] [CrossRef]
- Palaniyappan, L.; Al-Radaideh, A.; Mougin, O.; Das, T.; Gowland, P.; Liddle, P.F. Aberrant myelination of the cingulum and Schneiderian delusions in schizophrenia: A 7T magnetization transfer study. Psychol. Med. 2019, 49, 1890–1896. [Google Scholar] [CrossRef]
- Saia-Cereda, V.M.; Cassoli, J.S.; Schmitt, A.; Falkai, P.; Nascimento, J.M.; Martins-de-Souza, D. Proteomics of the corpus callosum unravel pivotal players in the dysfunction of cell signaling, structure, and myelination in schizophrenia brains. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Xiu, Y.; Kong, X.-R.; Zhang, L.; Qiu, X.; Gao, Y.; Huang, C.-X.; Chao, F.-L.; Wang, S.-R.; Tang, Y. The myelinated fiber loss in the corpus callosum of mouse model of schizophrenia induced by MK-801. J. Psychiatr. Res. 2015, 63, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Xiu, Y.; Kong, X.-R.; Zhang, L.; Qiu, X.; Chao, F.-L.; Peng, C.; Gao, Y.; Huang, C.-X.; Wang, S.-R.; Tang, Y. White matter injuries induced by MK-801 in a mouse model of schizophrenia based on NMDA antagonism. Anat. Rec. Hoboken 2014, 297, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Tsunoda, M.; Sumiyoshi, T.; Takasaki, I.; Tabuchi, Y.; Seo, T.; Tanaka, K.; Uehara, T.; Itoh, H.; Suzuki, M.; et al. Effect of MK-801 on gene expressions in the amygdala of rats. Synapse 2008, 62, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Alberdi, E.; Domercq, M.; Pérez-Cerdá, F.; Pérez-Samartín, A.; Sánchez-Gómez, M.V. The link between excitotoxic oligodendroglial death and demyelinating diseases. Trends Neurosci. 2001, 24, 224–230. [Google Scholar] [CrossRef]
- Gandal, M.J.; Sisti, J.; Klook, K.; Ortinski, P.I.; Leitman, V.; Liang, Y.; Thieu, T.; Anderson, R.; Pierce, R.C.; Jonak, G.; et al. GABAB-mediated rescue of altered excitatory-inhibitory balance, gamma synchrony and behavioral deficits following constitutive NMDAR-hypofunction. Transl. Psychiatry 2012, 2, e142. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Mouri, A.; Kubota, H.; Lee, H.-J.; Chang, M.-H.; Wu, C.-Y.; Knutson, D.E.; Mihovilovic, M.; Cook, J.; Sieghart, W.; et al. Targeting α6GABAA receptors as a novel therapy for schizophrenia: A proof-of-concept preclinical study using various animal models. Biomed Pharmacother. 2022, 150, 113022. [Google Scholar] [CrossRef]
- Sehatpour, P.; Javitt, D.C.; De Baun, H.M.; Carlson, M.; Beloborodova, A.; Margolin, D.H.; Carlton, M.B.L.; Brice, N.L.; Kantrowitz, J.T. Mismatch negativity as an index of target engagement for excitation/inhibition-based treatment development: A double-blind, placebo-controlled, randomized, single-dose cross-over study of the serotonin type-3 receptor antagonist CVN058. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2022, 47, 711–718. [Google Scholar] [CrossRef]
- Marx, C.; Bradford, D.; Hamer, R.; Naylor, J.; Allen, T.; Lieberman, J.; Strauss, J.; Kilts, J. Pregnenolone as a novel therapeutic candidate in schizophrenia: Emerging preclinical and clinical evidence. Neuroscience 2011, 191, 78–90. [Google Scholar] [CrossRef]
- Ritsner, M.S.; Bawakny, H.; Kreinin, A. Pregnenolone treatment reduces severity of negative symptoms in recent-onset schizophrenia: An 8-week, double-blind, randomized add-on two-center trial. Psychiatry Clin. Neurosci. 2014, 68, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B.M. Multiple Sclerosis and Schizophrenia. Int. J. Mol. Sci. 2017, 18, 1760. [Google Scholar] [CrossRef] [PubMed]
- Gouvêa-Junqueira, D.; Falvella, A.C.B.; Antunes, A.S.L.M.; Seabra, G.; Brandão-Teles, C.; Martins-de-Souza, D.; Crunfli, F. Novel Treatment Strategies Targeting Myelin and Oligodendrocyte Dysfunction in Schizophrenia. Front. Psychiatry 2020, 11, 379. [Google Scholar] [CrossRef] [PubMed]
- Homberg, J.R. Measuring behaviour in rodents: Towards translational neuropsychiatric research. Behav. Brain Res. 2013, 236, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Drinkenburg, W.H.; Ruigt, G.S.; Ahnaou, A. Pharmaco-EEG Studies in Animals: An Overview of Contemporary Translational Applications. Neuropsychobiology 2015, 72, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Michie, P.T.; Malmierca, M.S.; Harms, L.; Todd, J. The neurobiology of MMN and implications for schizophrenia. Biol. Psychol. 2016, 116, 90–97. [Google Scholar] [CrossRef]
- Metzner, C.; Zurowski, B.; Steuber, V. The Role of Parvalbumin-positive Interneurons in Auditory Steady-State Response Deficits in Schizophrenia. Sci. Rep. 2019, 9, 18525. [Google Scholar] [CrossRef]
- Ross, J.M.; Hamm, J.P. Cortical Microcircuit Mechanisms of Mismatch Negativity and Its Underlying Subcomponents. Front. Neural Circuits 2020, 14, 13. [Google Scholar] [CrossRef]
- Onitsuka, T.; Tsuchimoto, R.; Oribe, N.; Spencer, K.M.; Hirano, Y. Neuronal imbalance of excitation and inhibition in schizophrenia: A scoping review of gamma-band ASSR findings. Psychiatry Clin. Neurosci. 2022, 76, 610–619. [Google Scholar] [CrossRef]
- Oestreich, L.K.L.; Randeniya, R.; Garrido, M.I. Auditory prediction errors and auditory white matter microstructure associated with psychotic-like experiences in healthy individuals. Brain Struct. Funct. 2019, 224, 3277–3289. [Google Scholar] [CrossRef]
- Loiodice, S.; Drinkenburg, W.H.; Ahnaou, A.; McCarthy, A.; Viardot, G.; Cayre, E.; Rion, B.; Bertaina-Anglade, V.; Mano, M.; L’hostis, P.; et al. Mismatch negativity as EEG biomarker supporting CNS drug development: A transnosographic and translational study. Transl. Psychiatry 2021, 11, 253. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, L.-M.; Leung, S.; Michie, P.T.; Green, A.; Nathan, P.J.; Fitzgerald, P.; Johnston, P.; Solowij, N.; Kulkarni, J.; Croft, R.J. The effects of glycine on auditory mismatch negativity in schizophrenia. Schizophr. Res. 2018, 191, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.-C.; Luo, D.-Z.; Gau, S.-S.; Chang, C.-Y.; Lai, W.-S. Directly and Indirectly Targeting the Glycine Modulatory Site to Modulate NMDA Receptor Function to Address Unmet Medical Needs of Patients With Schizophrenia. Front. Psychiatry 2021, 12, 742058. [Google Scholar] [CrossRef] [PubMed]
- Umbricht, D.; Javitt, D.; Novak, G.; Bates, J.; Pollack, S.; Lieberman, J.; Kane, J. Effects of clozapine on auditory event-related potentials in schizophrenia. Biol. Psychiatry 1998, 44, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Umbricht, D.; Javitt, D.; Novak, G.; Bates, J.; Pollack, S.; Lieberman, J.; Kane, J. Effects of risperidone on auditory event-related potentials in schizophrenia. Int. J. Neuropsychopharmacol. 1999, 2, 299–304. [Google Scholar] [CrossRef]
- Korostenskaja, M.; Dapsys, K.; Siurkute, A.; Maciulis, V.; Ruksenas, O.; Kähkönen, S. Effects of olanzapine on auditory P300 and mismatch negativity (MMN) in schizophrenia spectrum disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 543–548. [Google Scholar] [CrossRef]
- Korostenskaja, M.; Kähkönen, S. What do ERPs and ERFs reveal about the effect of antipsychotic treatment on cognition in schizophrenia? Curr. Pharm. Des. 2009, 15, 2573–2593. [Google Scholar] [CrossRef]
- Horton, J.; Millar, A.; Labelle, A.; Knott, V.J. MMN responsivity to manipulations of frequency and duration deviants in chronic, clozapine-treated schizophrenia patients. Schizophr. Res. 2011, 126, 202–211. [Google Scholar] [CrossRef]
- Dawson, N.; Morris, B.J.; Pratt, J.A. Functional brain connectivity phenotypes for schizophrenia drug discovery. J. Psychopharmacol. Oxf. Engl. 2015, 29, 169–177. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adraoui, F.W.; Douw, L.; Martens, G.J.M.; Maas, D.A. Connecting Neurobiological Features with Interregional Dysconnectivity in Social-Cognitive Impairments of Schizophrenia. Int. J. Mol. Sci. 2023, 24, 7680. https://doi.org/10.3390/ijms24097680
Adraoui FW, Douw L, Martens GJM, Maas DA. Connecting Neurobiological Features with Interregional Dysconnectivity in Social-Cognitive Impairments of Schizophrenia. International Journal of Molecular Sciences. 2023; 24(9):7680. https://doi.org/10.3390/ijms24097680
Chicago/Turabian StyleAdraoui, Florian W., Linda Douw, Gerard J. M. Martens, and Dorien A. Maas. 2023. "Connecting Neurobiological Features with Interregional Dysconnectivity in Social-Cognitive Impairments of Schizophrenia" International Journal of Molecular Sciences 24, no. 9: 7680. https://doi.org/10.3390/ijms24097680