
VEGF Induces Expression of Genes That Either Promote or Limit Relaxation of the Retinal Endothelial Barrier

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
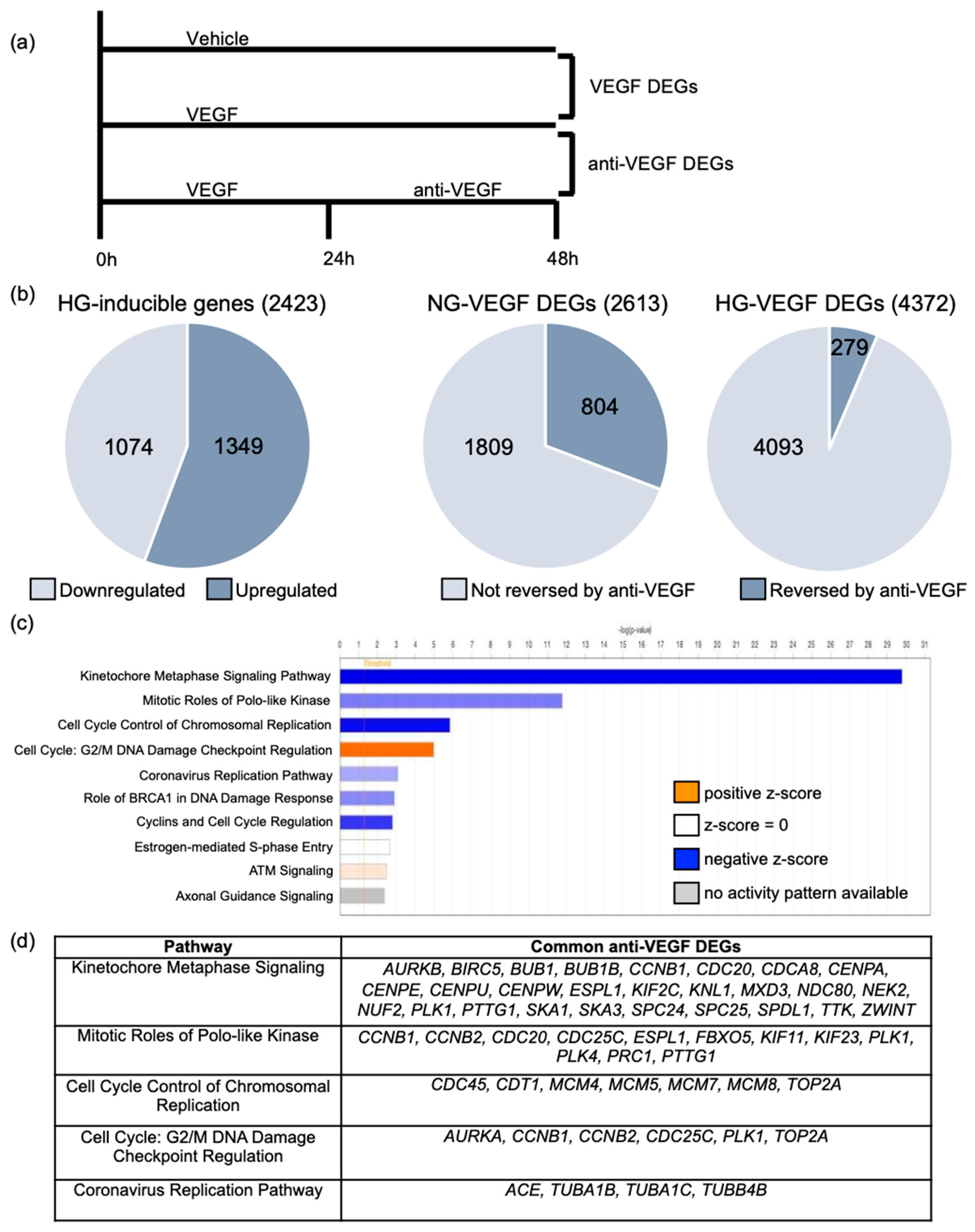
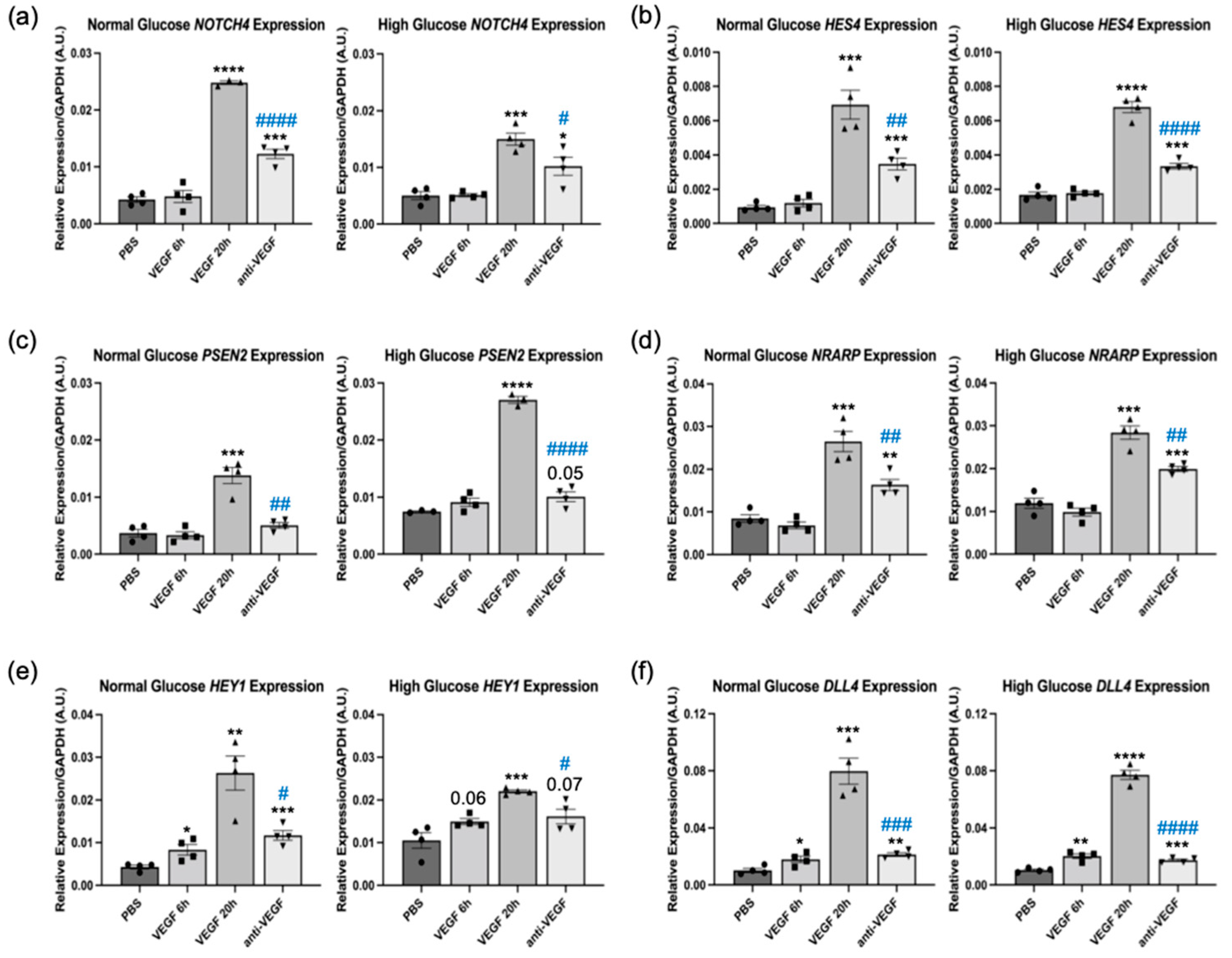
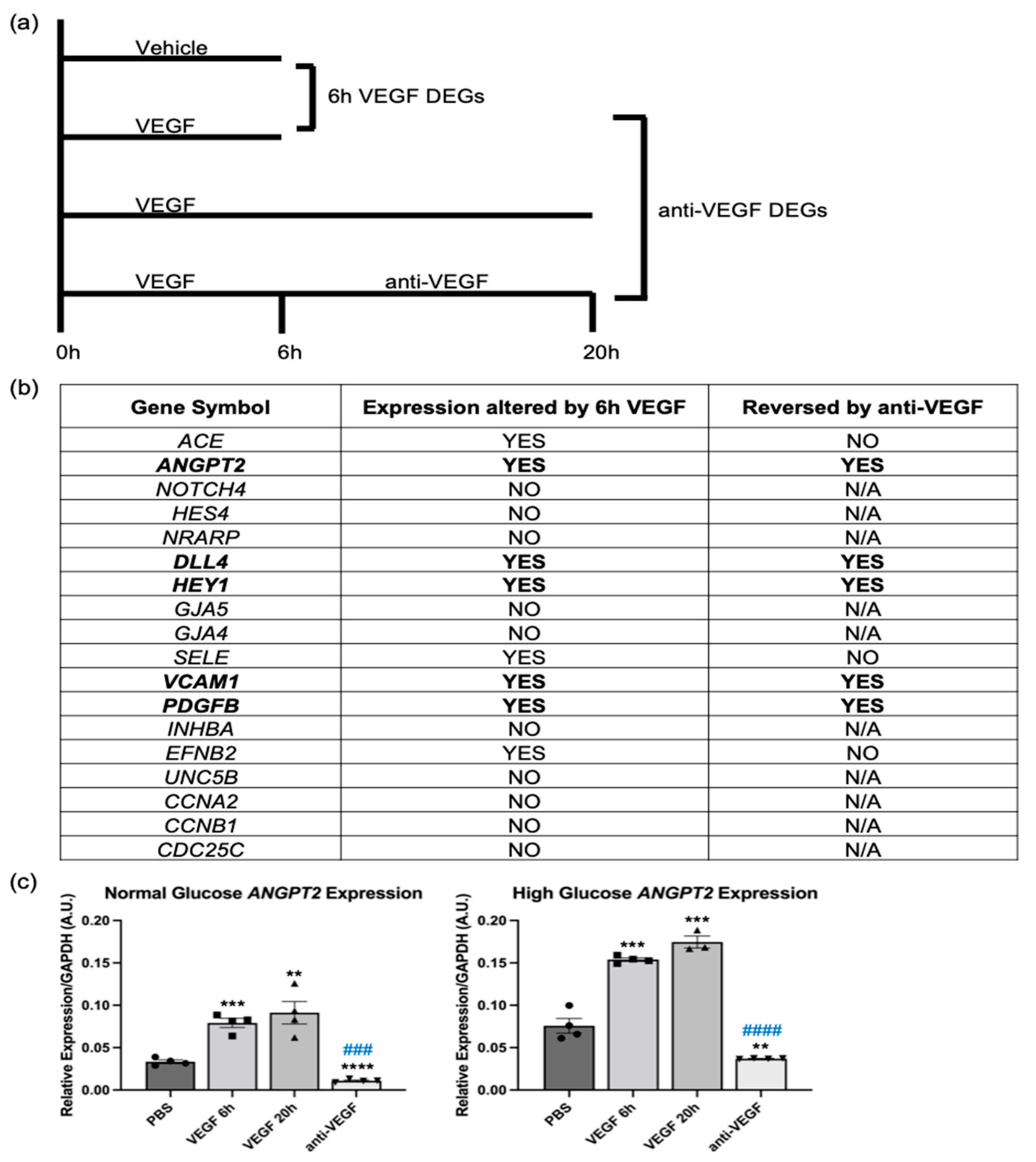
2.1. Some VEGF and Anti-VEGF-Regulated Genes Were Insensitive to Hyperglycemia
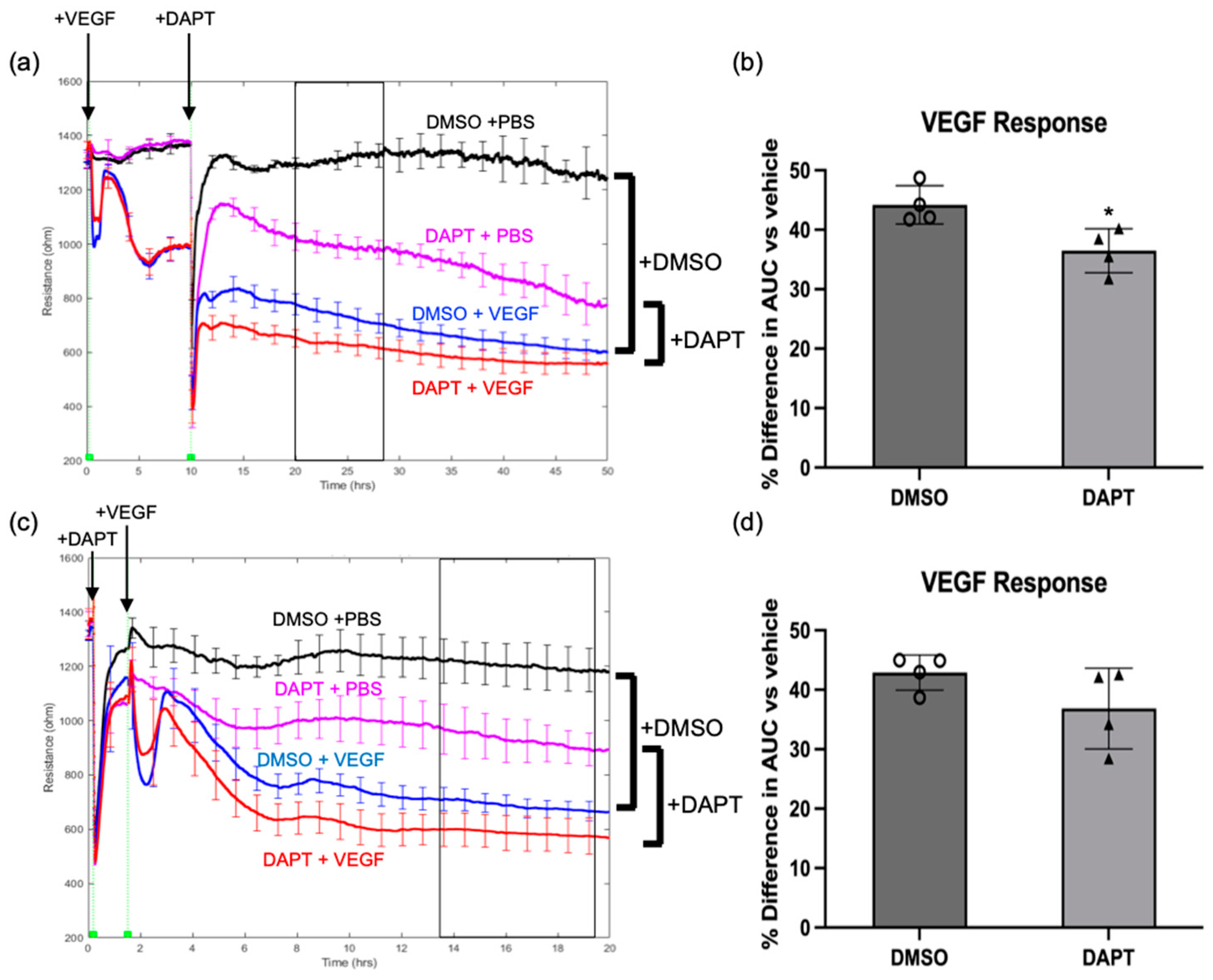
2.2. Notch Signaling Was Required for VEGF to Sustain Barrier Relaxation
2.3. VEGF and Anti-VEGF Regulated ANGPT2 Expression
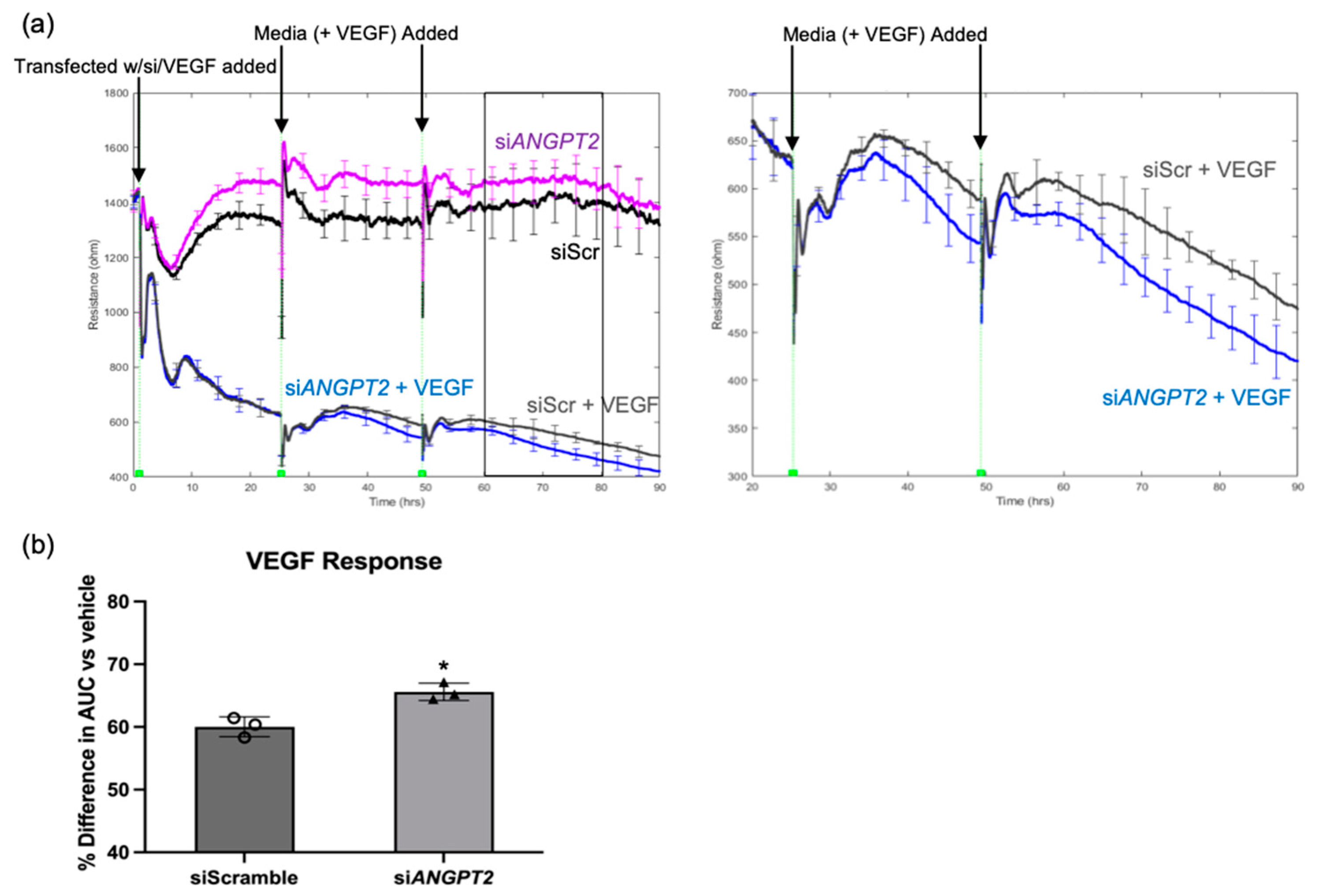
2.4. Ang2 Limited the Extent of VEGF-Induced Permeability
3. Discussion
4. Materials and Methods
4.1. Culture of Human Retinal Endothelial Cells
4.2. RNA Sequencing
4.3. RNA-Seq QC and Quantification
4.4. Western Blotting
4.5. Electric Cell-Substrate Impedance Sensing (ECIS) Assay
4.6. RNA Extraction and Real-Time PCR
4.7. Quantification and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stewart, M.W. The Expanding Role of Vascular Endothelial Growth Factor Inhibitors in Ophthalmology. Mayo Clin. Proc. 2012, 87, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Le Couter, J.; Strauss, E.C.; Ferrara, N. Vascular Endothelial Growth Factor A in Intraocular Vascular Disease. Ophthalmology 2013, 120, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yu, H.; Yu, Y.; Geng, Y.; Li, D.; Yang, C.; Lv, Q.; Lu, L.; Liu, T.; Li, G.; et al. Levels of Inflammatory Cytokines Il-1β, Il-6, Il-8, Il-17a, and Tnf-A in Aqueous Humour of Patients with Diabetic Retinopathy. J. Diabetes Res. 2018, 2018, 8546423. [Google Scholar] [CrossRef] [PubMed]
- Funk, M.; Karl, D.; Georgopoulos, M.; Benesch, T.; Sacu, S.; Polak, K.; Zlabinger, G.J.; Schmidt-Erfurth, U. Neovascular Age-related Macular Degeneration: Intraocular Cytokines and Growth Factors and the Influence of Therapy with Ranibizumab. Ophthalmology 2009, 116, 2393–2399. [Google Scholar] [CrossRef]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E.; et al. Vascular Endothelial Growth Factor in Ocular Fluid of Patients with Diabetic Retinopathy and Other Retinal Disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Nguyen, Q.D.; Marcus, D.M.; Boyer, D.S.; Patel, S.; Feiner, L.; Schlottmann, P.G.; Rundle, A.C.; Zhang, J.; Rubio, R.G.; et al. Long-Term Outcomes of Ranibizumab Therapy for Diabetic Macular Edema: The 36-Month Results from Two Phase Iii Trials: Rise and Ride. Ophthalmology 2013, 120, 2013–2022. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. Vegf in Signaling and Disease: Beyond Discovery and Devel-opment. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Claesson-Welsh, L. Vascular Permeability—The Essentials. Upsala J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef]
- Li, Y.; Yan, Z.; Chaudhry, K.; Kazlauskas, A. The Renin-Angiotensin-Aldosterone System (Raas) Is One of the Effectors by Which Vascular Endothelial Growth Factor (Vegf)/Anti-Vegf Controls the Endothelial Cell Barrier. Am. J. Pathol. 2020, 190, 1971–1981. [Google Scholar] [CrossRef]
- Li, Y.; Baccouche, B.; Olayinka, O.; Serikbaeva, A.; Kazlauskas, A. The Role of the Wnt Pathway in VEGF/Anti-VEGF-Dependent Control of the Endothelial Cell Barrier. Investig. Opthalmology Vis. Sci. 2021, 62, 17. [Google Scholar] [CrossRef]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, e93751. [Google Scholar] [CrossRef]
- Maguire, M.G.; Martin, D.F.; Ying, G.S.; Jaffe, G.J.; Daniel, E.; Grunwald, J.E.; Toth, C.A.; Ferris, F.L., III; Fine, S.L.; Comparison of Age-related Macular Degeneration Treatments Trials (CATT) Research Group. Five-Year Outcomes with Anti-Vascular Endothelial Growth Factor Treatment of Neovascular Age-Related Macular De-generation: The Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2016, 123, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Viñals, F.; Pouysségur, J. Confluence of Vascular Endothelial Cells Induces Cell Cycle Exit by Inhibiting P42/P44 Mi-togen-Activated Protein Kinase Activity. Mol. Cell Biol. 1999, 19, 2763–2772. [Google Scholar] [CrossRef]
- Miloudi, K.; Oubaha, M.; Ménard, C.; Dejda, A.; Guber, V.; Cagnone, G.; Wilson, A.M.; Tétreault, N.; Mawambo, G.; Binet, F.; et al. NOTCH1 signaling induces pathological vascular permeability in diabetic retinopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 4538–4547. [Google Scholar] [CrossRef]
- Hasan, S.S.; Fischer, A. Notch Signaling in the Vasculature: Angiogenesis and Angiocrine Functions. Cold Spring Harb. Perspect. Med. 2022, 13, a041166. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Huang, J.; Wang, D.; Chen, L.; Sun, D.; Zhao, C. Endothelium-Specific Cyp2j2 Overexpression Improves Cardiac Dysfunction by Promoting Angiogenesis Via Jagged1/Notch1 Signaling. J. Mol. Cell Cardiol. 2018, 123, 118–127. [Google Scholar] [CrossRef]
- Kong, D.-H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.; Kazlauskas, A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004, 15, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Aziz, A.A.; Shafi, N.A.; Abbas, T.; Khanani, A.M. Targeting Angiopoietin in Retinal Vascular Diseases: A Literature Review and Summary of Clinical Trials Involving Faricimab. Cells 2020, 9, 1869. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.M.; Neiweem, A.E.; Kansara, V.; Harris, A.; Ciulla, T.A. Tie-2/Angiopoietin Pathway Modulation as a Therapeutic Strategy for Retinal Disease. Expert Opin. Investig. Drugs 2019, 28, 861–869. [Google Scholar] [CrossRef]
- Gurnik, S.; Devraj, K.; Macas, J.; Yamaji, M.; Starke, J.; Scholz, A.; Sommer, K.; Di Tacchio, M.; Vutukuri, R.; Beck, H.; et al. Angiopoietin-2-induced blood–brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol. 2016, 131, 753–773. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; E Ryan, T.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of Angiopoietin-1, a Ligand for the TIE2 Receptor, by Secretion-Trap Expression Cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; Suri, C.; Smith, K.; McClain, J.; Sato, T.N.; Yancopoulos, G.D.; McDonald, D.M. Leakage-Resistant Blood Vessels in Mice Transgenically Overexpressing Angiopoietin-1. Science 1999, 286, 2511–2514. [Google Scholar] [CrossRef] [PubMed]
- Benest, A.V.; Kruse, K.; Savant, S.; Thomas, M.; Laib, A.M.; Loos, E.K.; Fiedler, U.; Augustin, H.G. Angiopoietin-2 Is Critical for Cytokine-Induced Vascular Leakage. PLoS ONE 2013, 8, e70459. [Google Scholar] [CrossRef]
- Kim, M.; Allen, B.; Korhonen, E.A.; Nitschké, M.; Yang, H.W.; Baluk, P.; Saharinen, P.; Alitalo, K.; Daly, C.; Thurston, G.; et al. Opposing Actions of Angiopoietin-2 on Tie2 Signaling and Foxo1 Activation. J. Clin. Investig. 2016, 126, 3511–3525. [Google Scholar] [CrossRef]
- Shirakura, K.; Baluk, P.; Nottebaum, A.F.; Ipe, U.; Peters, K.G.; McDonald, D.M.; Vestweber, D. Shear stress control of vascular leaks and atheromas through Tie2 activation by VE-PTP sequestration. EMBO Mol. Med. 2023, e16128. [Google Scholar] [CrossRef] [PubMed]
- Felcht, M.; Luck, R.; Schering, A.; Seidel, P.; Srivastava, K.; Hu, J.; Bartol, A.; Kienast, Y.; Vettel, C.; Loos, E.K.; et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J. Clin. Investig. 2012, 122, 1991–2005. [Google Scholar] [CrossRef]
- Aman, J.; Margadant, C. Integrin-Dependent Cell–Matrix Adhesion in Endothelial Health and Disease. Circ. Res. 2023, 132, 355–378. [Google Scholar] [CrossRef]
- Roviezzo, F.; Tsigkos, S.; Kotanidou, A.; Bucci, M.; Brancaleone, V.; Cirino, G.; Papapetropoulos, A. Angiopoietin-2 Causes Inflammation in Vivo by Promoting Vascular Leakage. Experiment 2005, 314, 738–744. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 August 2021).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ul-trafast Universal Rna-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic Rna-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Mul-tiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCann, M.; Li, Y.; Baccouche, B.; Kazlauskas, A. VEGF Induces Expression of Genes That Either Promote or Limit Relaxation of the Retinal Endothelial Barrier. Int. J. Mol. Sci. 2023, 24, 6402. https://doi.org/10.3390/ijms24076402
McCann M, Li Y, Baccouche B, Kazlauskas A. VEGF Induces Expression of Genes That Either Promote or Limit Relaxation of the Retinal Endothelial Barrier. International Journal of Molecular Sciences. 2023; 24(7):6402. https://doi.org/10.3390/ijms24076402
Chicago/Turabian StyleMcCann, Maximilian, Yueru Li, Basma Baccouche, and Andrius Kazlauskas. 2023. "VEGF Induces Expression of Genes That Either Promote or Limit Relaxation of the Retinal Endothelial Barrier" International Journal of Molecular Sciences 24, no. 7: 6402. https://doi.org/10.3390/ijms24076402