
Linking Benzene, in Utero Carcinogenicity and Fetal Hematopoietic Stem Cell Niches: A Mechanistic Review


Abstract
:1. Introduction
2. Survey Methodology
3. Hematopoiesis
3.1. Hematopoiesis in the Adult Phase
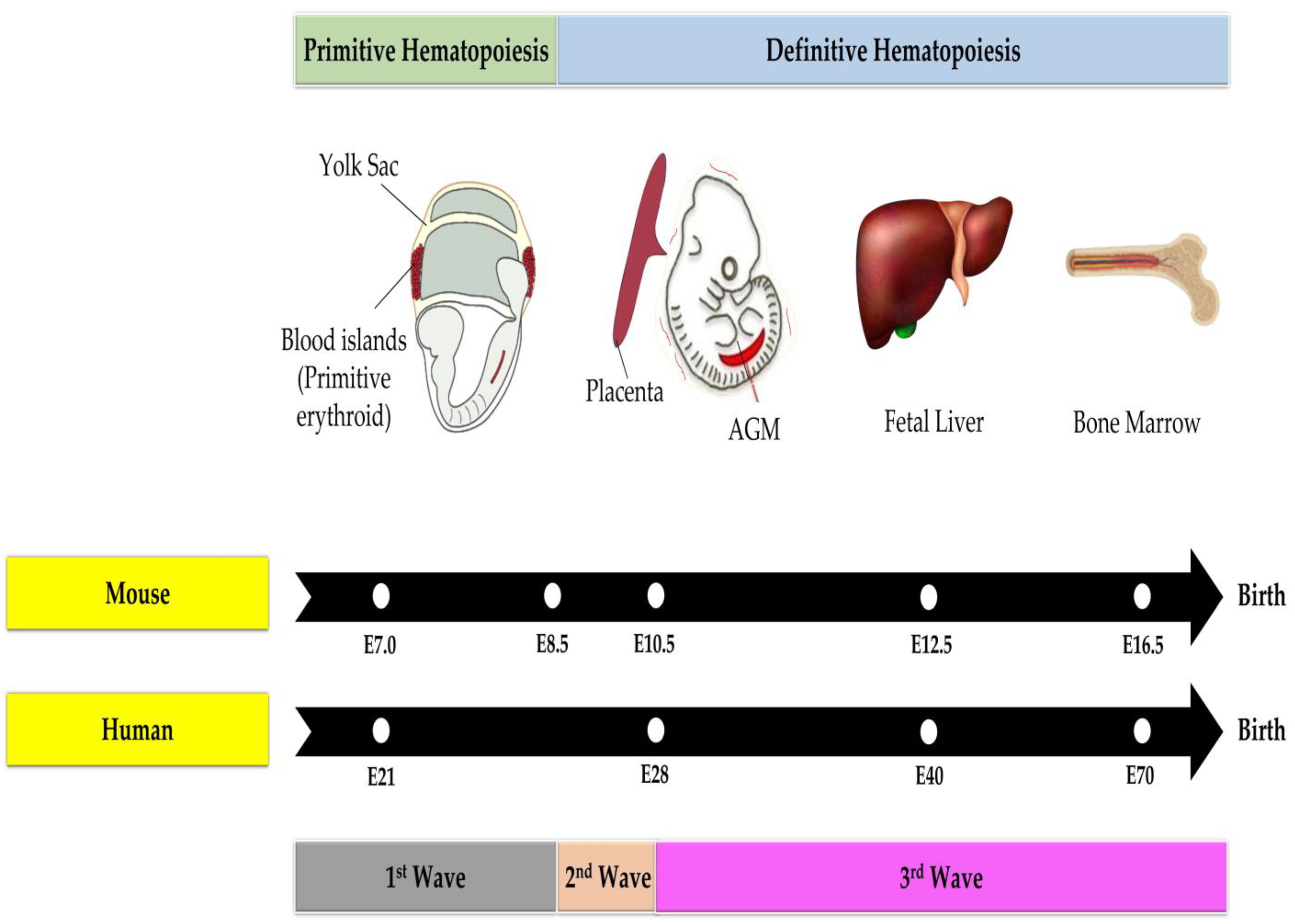
3.2. Hematopoiesis in the Fetal Phase
4. Benzene Metabolism Linked to Fetal Toxicity via Maternal and Paternal Exposure
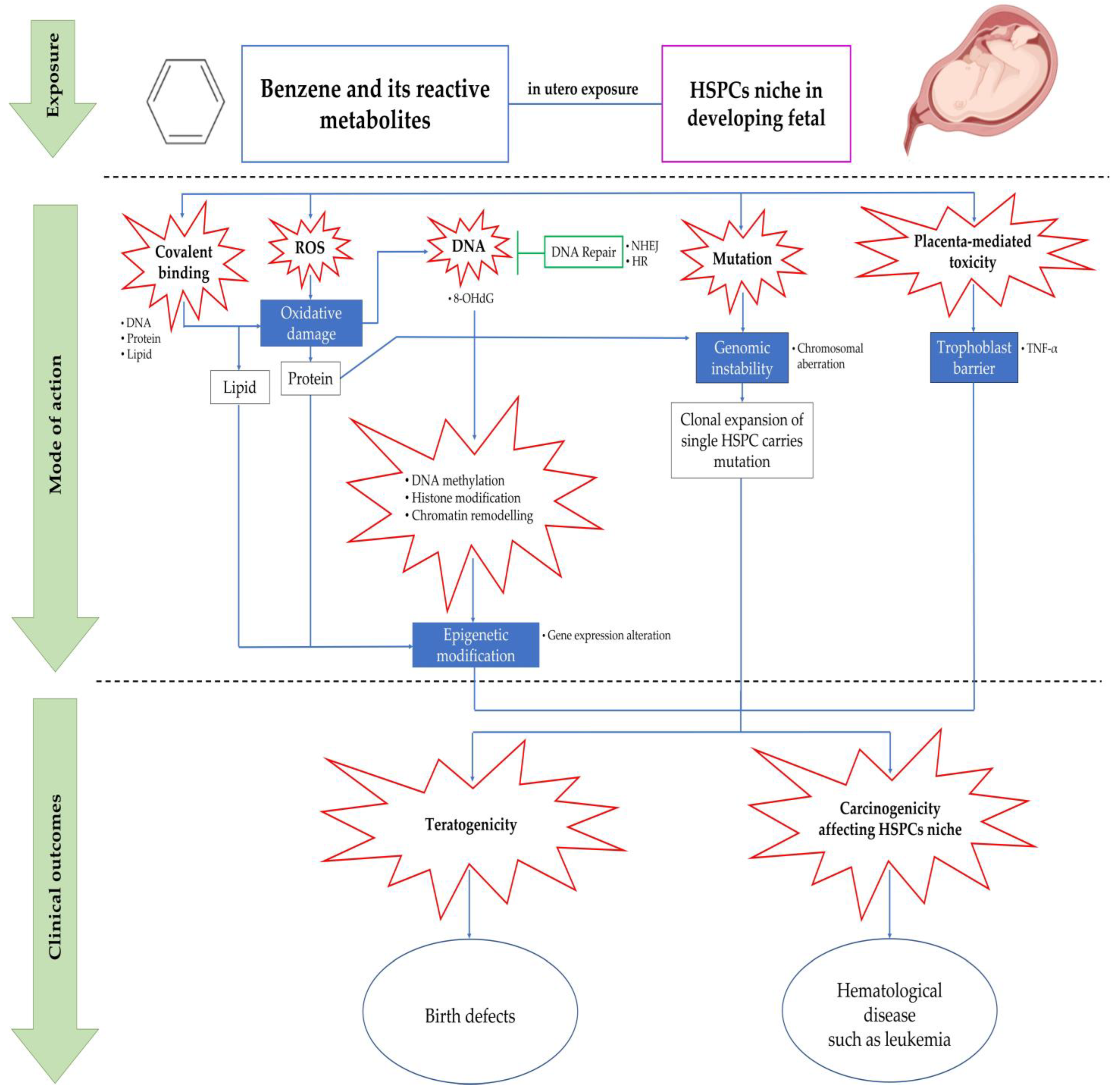
5. Mechanisms of Benzene-Induced in Utero Carcinogenicity Involving Hematopoietic Stem Cells and Multilineage Progenitors
5.1. Covalent Binding
5.2. Oxidative Stress
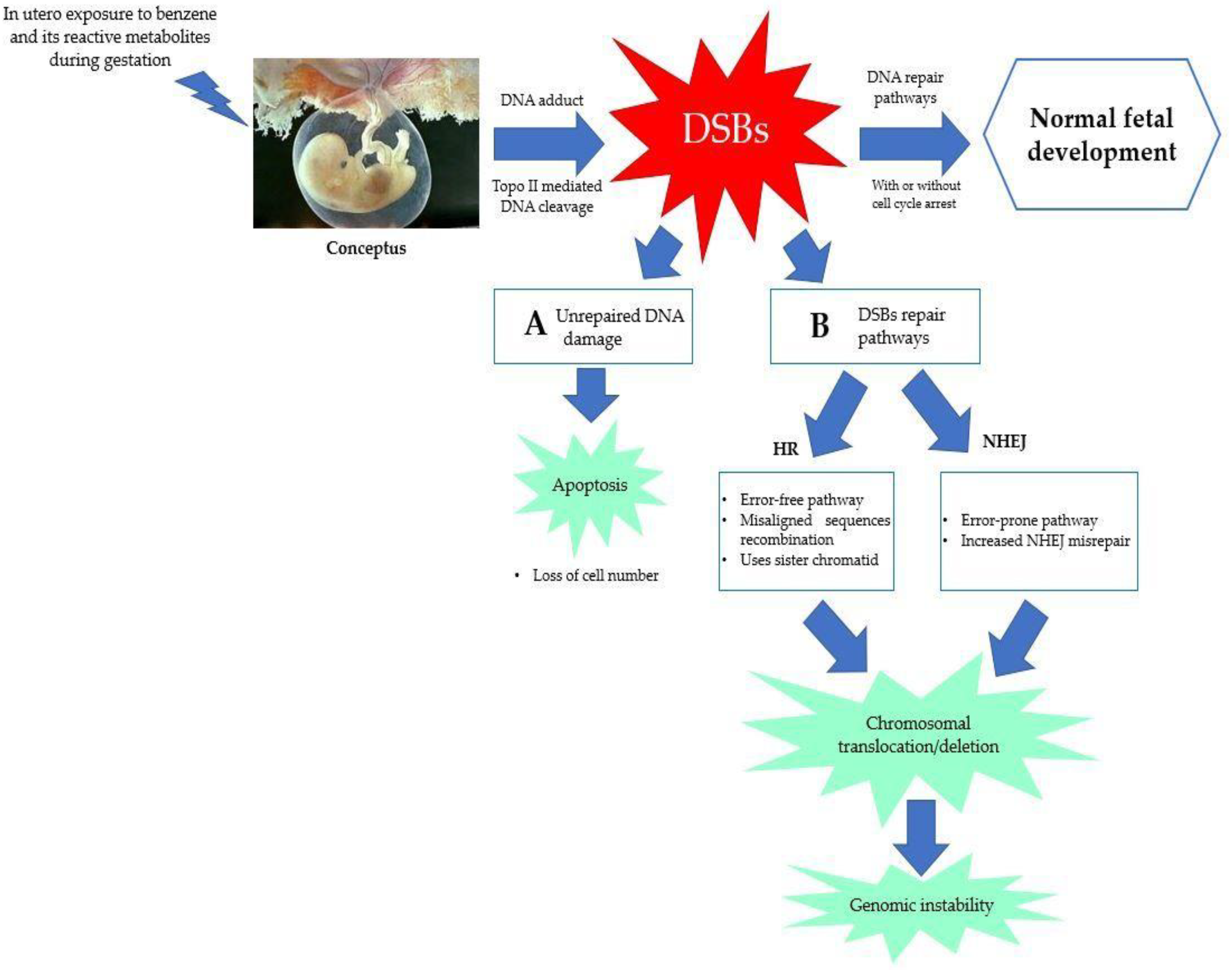
5.3. Error in DNA Repair Pathways
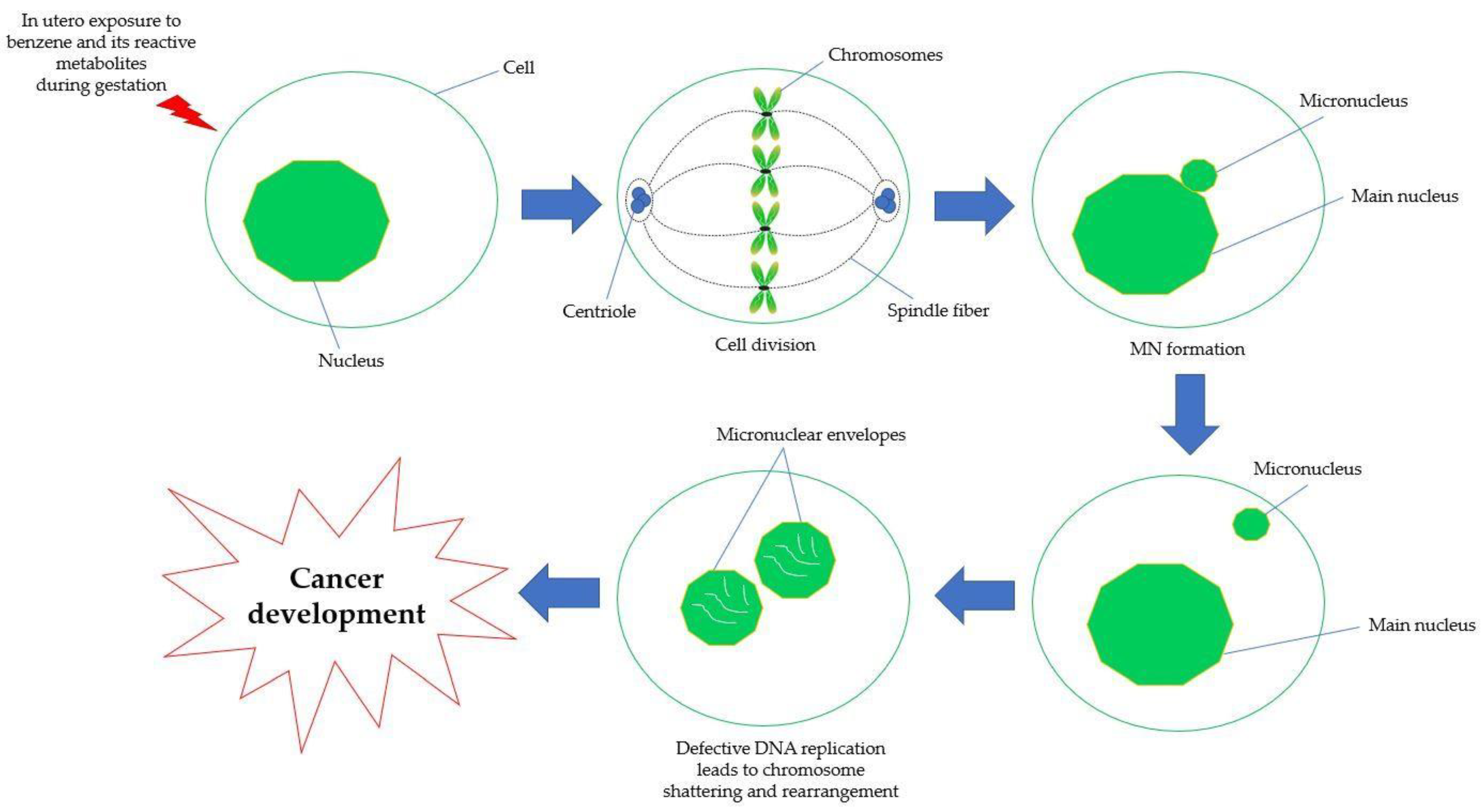
5.4. Chromosomal Aberration and Genetic Damage
5.5. Epigenetic Modification
5.5.1. DNA Methylation
5.5.2. Histone Modification and Chromatin Remodeling
5.6. Placenta-Mediated Toxicity
6. The Origin of Hematological Diseases from In-Utero Benzene Exposure
7. Conclusions and Future Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACGIH | The American Conference of Governmental Industrial Hygienists |
| AGM | Aorta-gonad-mesonephros |
| ALL | Acute lymphocytic leukemia |
| AML | Acute myeloid leukemia |
| ATSDR | Agency for Toxic Substances and Disease Registry |
| BER | Base excision repair |
| BFU-E | Burst-forming unit erythroid |
| BM | Bone marrow |
| BM-YSCs | Bone marrow hematopoietic stem cells |
| BQ | Benzoquinone |
| 1,4-BQ | 1,4-benzoquinone |
| C6H6 | Benzene |
| CFU-E | Colony-forming unit erythroid |
| CFU-G | Colony-forming unit granulocyte |
| CFU-GM | Colony-forming unit granulocyte-macrophage |
| CLL | Chronic lymphoid leukemia |
| CLP | Common lymphoid progenitor |
| CMP | Common myeloid progenitor |
| CpGs | Cytosine-phosphate-guanine dinucleotides |
| CYP | Cytochrome |
| DCFDA | 2′,7′-dichlorodihydrofluorescein diacetate |
| DDR | Damage response and repair |
| DNA | Deoxyribonucleic acid |
| DNA-PKcs | DNA-dependent protein kinase |
| DNMT | DNA methyltransferases |
| DR | Direct reversal |
| DSB | Double-strand break |
| E | Embryonic day |
| Fe2+ | Iron |
| GD | Gestation day |
| GSH | Glutathione |
| GST | Glutathione S-transferase |
| H2O2 | Hydrogen peroxide |
| H3K4me3 | Histone H3 trimethylated at lysine 4 |
| HPCs | Hematopoietic progenitor cells |
| Hb | Hemoglobin |
| HQ | Hydroquinone |
| HR | Homologous recombination |
| HSCs | Hematopoietic stem cells |
| HSPCs | Hematopoietic stem/progenitor cells |
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| IARC | International Agency for Research on Cancer |
| ICR | Imprinting control region |
| i.p | Intraperitoneal |
| LSC | Leukemic stem cell |
| MDA | Malondialdehyde |
| MAPK | Mitogen-activated protein kinases |
| MDR | Multidrug resistance |
| MDS | Myelodysplastic syndromes |
| mEH | Microsomal epoxide hydrolase |
| MMR | Mismatch repair |
| MN | Micronuclei |
| MNPCE | Micronucleated bone marrow polychromatic erythrocytes |
| MPO | Myeloperoxidase |
| mtDNA | Mitochondria DNA |
| MSC | Mesenchymal stem cell |
| MSPC | Human mesenchymal stem and progenitor cell |
| NER | Nucleotide excision repair |
| NF-κB | Nuclear factor kappa B |
| NHEJ | Non-homologous end-joining |
| NIOSH | National Institute of Occupational Safety and Health |
| NK | Natural killer |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| O2•− | Anion radical |
| o-BQ | o-BQ |
| OH | Hydroxide |
| OSHA | Occupational Safety and Health Administration |
| p-BQ | p-benzoquinone |
| ppm | Parts per million |
| PC | Protein carbonyl |
| PTMs | Post-translationally modified |
| RNA | Ribonucleic acid |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SCE | Sister chromatid exchange |
| SQ•− | Semiquinone |
| SOD | Superoxide dismutase |
| SPF | Specific pathogen-free |
| TNF-α | Tumor Necrosis Factor alpha |
| Topo II | Topoisomerase II |
| TWA | Time-weighted average |
| UGTs | Uridine 5′-diphospho-glucuronosyltransferase |
| µM | Micromole |
| YS-HSCs | Yolk sac hematopoietic stem cells |
References
- International Agency for Research on Cancer. Monographs on the Evaluation of Carcinogenic Hazards to Humans; World Health Organization: Lyon, France, 2020. [Google Scholar]
- Wang, L.; He, X.; Bi, Y.; Ma, Q. Stem Cell and Benzene-Induced Malignancy and Hematotoxicity. Chem. Res. Toxicol. 2012, 25, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Nithyanandam, R.; Mun, Y.K.; Fong, T.S.; Siew, T.C.; Yee, O.S.; Ismail, N. Review on Production of Benzene from Petroleum Associated Gas by Dehydroaromatization, Partial Oxidation of Methane and Methanol-to-Aromatics Processes. J. Eng. Sci. Technol. 2018, 13, 4290–4309. [Google Scholar]
- Dewi, R.; Abd Hamid, Z.; Ng, Q.M.; Rajab, N.F.; Shuib, S.; Abdul Razak, S.R. Bone Marrow Oxidative Stress and Acquired Lineage-Specific Genotoxicity in Hematopoietic Stem/Progenitor Cells Exposed to 1,4-Benzoquinone. Int. J. Environ. Res. Public Health 2020, 17, 5865. [Google Scholar]
- Warden, H.; Richardson, H.; Richardson, L.; Siemiatycki, J.; Ho, V. Associations between Occupational Exposure to Benzene, Toluene and Xylene and Risk of Lung Cancer in Montréal. Occup. Environ. Med. 2018, 75, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Mchale, C.M.; Zhang, L.; Smith, M.T. Current Understanding of the Mechanism of Benzene-induced Leukemia in Humans: Implications for Risk Assessment. Carcinogenesis 2012, 33, 240–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, D.; Zhou, H. Relationships between Metabolic and Non-metabolic Susceptibility Factors in Benzene Toxicity. Chem. Biol. Interact. 2010, 184, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passegué, E.; Jamieson, C.H.; Ailles, L.E.; Weissman, I.L. Normal and Leukemic Hematopoiesis: Are Leukemias A Stem Cell Disorder or A Reacquisition of Stem Cell Characteristics? Proc. Natl. Acad. Sci. USA 2003, 100, 11842–11849. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Cho, S.; Spangrude, G.J. Hematopoietic Stem Cells: Generation and Self-renewal. Cell Death Differ. 2007, 11, 1851–1859. [Google Scholar] [CrossRef]
- Warr, M.R.; Pietras, E.M.; Passegué, E. Mechanisms Controlling Hematopoietic Stem Cell Functions during Normal Hematopoiesis and Hematological Malignancies. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 681–701. [Google Scholar] [CrossRef]
- Abernethy, D.J.; Kleymenova, E.V.; Rose, J.; Recio, L.; Faiola, B. Human CD34+ Hematopoietic Progenitor Cells are Sensitive Targets for Toxicity Induced by 1, 4-Benzoquinone. Toxicol. Sci. 2004, 79, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Chow, P.W.; Rajab, N.F.; Chua, K.H.; Chan, K.M.; Abd Hamid, Z. Differential Responses of Lineage-committed Hematopoietic Progenitors and Altered Expression of Self-renewal and Differentiation-related Genes in 1,4-benzoquinone (1,4-BQ) Exposure. Toxicol. Vitr. 2018, 46, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Dewi, R.; Abd Hamid, Z.; Ng, Q.M.; Rajab, N.F.; Shuib, S.; Abdul Razak, S.R. Penilaian Profil Genotoksisiti, Epigenetik dan Mekanisme Detoksifikasi Sel Stem dan Sel Progenitor Hematopoietik Mencit Berlainan Keturunan Aruhan 1,4-Benzokuinon. Ph.D. Thesis, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia, 21 April 2022. [Google Scholar]
- Chow, P.W.; Abdul Hamid, Z.; Chan, K.M.; Inayat-Hussain, S.H.; Rajab, N.F. Lineage-related Cytotoxicity and Clonogenic Profile of 1,4-benzoquinone-exposed Hematopoietic Stem and Progenitor Cells. Toxicol. Appl. Pharmacol. 2015, 284, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Greim, H.; Kaden, D.A.; Larson, R.A.; Palermo, C.M.; Rice, J.M.; Ross, D.; Snyder, R. The Bone Marrow Niche, Stem Cells, and Leukemia: Impact of Drugs, Chemicals, and the Environment. Ann. N. Y. Acad. Sci. 2014, 1310, 7–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; Rusyn, I.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.F. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Pyatt, D.; Hays, S. A Review of the Potential Association between Childhood Leukemia and Benzene. Chem. Biol. Interact. 2010, 184, 151–164. [Google Scholar] [CrossRef]
- Freedman, D.M.; Stewart, P.; Kleinerman, R.A.; Wacholder, S.; Hatch, E.E.; Tarone, R.E.; Robison, L.L.; Linet, M.S. Household Solvent Exposures and Childhood Acute Lymphoblastic Leukemia. Am. J. Public Health 2001, 91, 564–567. [Google Scholar] [PubMed] [Green Version]
- Woods, L.; Perez-Garcia, V.; Hemberger, M. Regulation of Placental Development and Its Impact on Fetal Growth-New Insights from Mouse Models. Front. Endocrinol. 2018, 9, 570. [Google Scholar] [CrossRef] [Green Version]
- Howard, S.C.; Metzger, M.L.; Wilimas, J.A.; Quintana, Y.; Pui, C.H.; Robison, L.L.; Ribeiro, R.C. Childhood Cancer Epidemiology in Low-Income Countries. Cancer 2008, 112, 461–472. [Google Scholar] [CrossRef]
- Pietras, E.M.; Warr, M.R.; Passegué, E. Cell Cycle Regulation in Hematopoietic Stem Cells. J. Cell Biol. 2011, 1, 34–48. [Google Scholar]
- Pietras, E.M.; Lakshminarasimhan, R.; Techner, J.M.; Fong, S.; Flach, J.; Binnewies, M.; Passegué, E. Re-entry into Quiescence Protects Hematopoietic Stem Cells from the Killing Effect of Chronic Exposure to Type I Interferons. J. Exp. Med. 2014, 211, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Jassinskaja, M.; Johansson, E.; Kristiansen, T.A.; Åkerstrand, H.; Sjöholm, K.; Hauri, S.; Hansson, J. Comprehensive Proteomic Characterization of Ontogenic Changes in Hematopoietic Stem and Progenitor Cells. Cell Rep. 2017, 21, 3285–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, R.; Witz, G.; Goldstein, B.D. The Toxicology of Benzene. Environ. Health Perspect. 1993, 100, 293–306. [Google Scholar] [CrossRef]
- Nandakumar, S.K.; Ulirsch, J.C.; Sankaran, V.G. Advances in Understanding Erythropoiesis: Evolving Perspectives. Br. J. Haematol. 2016, 173, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hendriks, M.; Chatzis, A.; Ramasamy, S.K.; Kusumbe, A.P. Bone Vasculature and Bone Marrow Vascular Niches in Health and Disease. J. Bone Miner. Res. 2020, 35, 2103–2120. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, A.; Bhartiy, D. Pluripotent Stem Cells in Bone Marrow and Cord Blood. In Blood Cell—An Overview of Studies in Hematology; IntechOpen: London, UK, 2012; pp. 60–88. [Google Scholar] [CrossRef]
- Carrelha, J.; Meng, Y.; Kettyle, L.M.; Luis, T.C.; Norfo, R.; Alcolea, V.; Boukarabila, H.; Grasso, F.; Gambardella, A.; Grover, A.; et al. Hierarchically Related Lineage-Restricted Fates of Multipotent Haematopoietic Stem Cells. Nature 2018, 554, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fraticelli, A.; Wolock, S.; Weinreb, C.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal Analysis of Lineage Fate in Native Haematopoiesis. Nature 2018, 553, 212–216. [Google Scholar] [CrossRef]
- Sanjuan-Pla, A.; Macaulay, I.; Jensen, C.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carella, C.; Matsuoka, S.; Jones, T.B.; et al. Platelet-Biased Stem Cells Reside at the Apex of the Haematopoietic Stem–Cell Hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Pucella, J.N.; Upadhaya, S.; Reizis, B. The Source and Dynamics of Adult Hematopoiesis: Insights from Lineage Tracing. Annu. Rev. Cell Dev. Biol. 2020, 36, 529–550. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Kaushansky, K. Lineage-specific Hematopoietic Growth Factors. N. Engl. J. Med. 2006, 354, 2034–2045. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Trumpp, A. Bone-marrow Haematopoietic-stem-cell Niches. Nat. Rev. Immunol. 2006, 6, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Li, L. The Stem Cell Niches in Bone. J. Clin. Investig. 2006, 116, 1195–1201. [Google Scholar] [CrossRef]
- Heo, J.S.; Choi, Y.; Kim, H.S.; Kim, H.O. Comparison of Molecular Profiles of Human Mesenchymal Stem Cells Derived from Bone Marrow, Umbilical Cord Blood, Placenta and Adipose Tissue. Int. J. Mol. Med. 2015, 37, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and Haematopoietic Stem Cells Form A Unique Bone Marrow Niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Villar, O.; Garcia, J.L.; Sanchez-Guijo, F.M.; Robledo, C.; Villaron, E.M.; Hernandez-Campo, P.; Lopez-Holgado, N.; Diez-Campelo, M.; Barbado, M.V.; Perez-Simon, J.A.; et al. Both Expanded and Uncultured Mesenchymal Stem Cells from MDS Patients are Genomically Abnormal, Showing A Specific Genetic Profile for the 5q- Syndrome. Leukemia 2009, 23, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Geyh, S.; Oz, S.; Cadeddu, R.P.; Frobel, J.; Bruckner, B.; Kundgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient Stromal Support in MDS Results from Molecular and Functional Deficits of Mesenchymal Stromal Cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhao, M.; Li, X.; Ma, L.; Zhang, J.; Shi, J.; Zhou, Y. The Cytotoxic Effect of the Benzene Metabolite Hydroquinone is Mediated by the Modulation of MDR1 Expression via the NF-κB Signaling Pathway. Cell Physiol. Biochem. 2015, 37, 592–602. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Kondo, M. Developmental Switch of Mouse Hematopoietic Stem Cells from Fetal to Adult Type Occurs in Bone Marrow after Birth. Proc. Natl. Acad. Sci. USA 2006, 103, 17852–17857. [Google Scholar] [CrossRef] [Green Version]
- Dzierzak, E.; Speck, N.A. Of lineage and Legacy: The Development of Mammalian Hematopoietic Stem Cells. Nat. Immunol. 2008, 9, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Ciriza, J.; Thompson, H.; Petrosian, R.; Manilay, J.O.; García-Ojeda, M.E. The Migration of Hematopoietic Progenitors from the Fetal Liver to the Fetal Bone Marrow: Lessons Learned and Possible Clinical Applications. Exp. Hematol. 2013, 41, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Chen, C.; Cheng, T. Cell Cycle Regulation of Hematopoietic Stem or Progenitor Cells. Int. J. Hematol. 2016, 103, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, O.C.; Kang, Y.A.; Passegué, E. Normal Hematopoiesis is a Balancing Act of Self-Renewal and Regeneration. Cold Spring Harb. Perspect. Med. 2020, 10, a035519. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.; Yoshimoto, M.; Takebe, T. Fetal Liver Hematopoiesis: From Development to Delivery. Stem Cell Res. Ther. 2021, 12, 139. [Google Scholar] [CrossRef]
- Bertrand, J.Y.; Chi, N.C.; Santoso, B.; Teng, S.; Stainier, D.Y.R.; Traver, D. Haematopoietic Stem Cells Derive Directly from Aortic Endothelium during Development. Nature 2010, 464, 108–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palis, J. Primitive and Definitive Erythropoiesis in Mammals. Front. Physiol. 2014, 28, 3. [Google Scholar] [CrossRef] [Green Version]
- Yamane, T. Mouse Yolk Sac Hematopoiesis. Front. Cell Dev. Biol. 2018, 6, 80. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, J.Y.; Jalil, A.; Klaine, M.; Jung, S.; Cumano, A.; Godin, I. Three Pathways to Mature Macrophages in the Early Mouse Yolk Sac. Blood 2005, 106, 3004–3011. [Google Scholar] [CrossRef]
- Palis, J.; Malik, J.; McGrath, K.E.; Kingsley, P.D. Primitive Erythropoiesis in the Mammalian Embryo. Int. J. Dev. Biol. 2010, 54, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Stefanska, M.; Batta, K.; Patel, R.; Florkowska, M.; Kouskoff, V.; Lacaud, G. Primitive Erythrocytes are Generated from Hemogenic Endothelial Cells. Sci Rep. 2017, 25, 6401. [Google Scholar] [CrossRef] [Green Version]
- Potts, K.S.; Sargeant, T.J.; Markham, J.F.; Shi, W.; Biben, C.; Josefsson, E.C.; Whitehead, L.W.; Rogers, K.L.; Liakhovitskaia, A.; Smyth, G.K.; et al. A Lineage of Diploid Platelet-Forming Cells Precedes Polyploid Megakaryocyte Formation in the Mouse Embryo. Blood 2014, 124, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tober, J.; Koniski, A.; McGrath, K.E.; Vemishetti, R.; Emerson, R.; de Mesy-Bentley, K.K.; Waugh, R.; Palis, J. The Megakaryocyte Lineage Originates from Hemangioblast Precursors and is An Integral Component Both of Primitive and of Definitive Hematopoiesis. Blood 2007, 109, 1433–1441. [Google Scholar] [CrossRef] [Green Version]
- Boiers, C.; Carrelha, J.; Lutteropp, M.; Luc, S.; Green, J.C.; Azzoni, E.; Woll, P.S.; Mead, A.J.; Hultquist, A.; Swiers, G. Lymphomyeloid Contribution of an Immune-restricted Progenitor Emerging prior to Definitive Hematopoietic Stem Cells. Cell Stem Cell 2013, 13, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, K.E.; Frame, J.M.; Fegan, K.H.; Bowen, J.R.; Conway, S.J.; Catherman, S.C.; Kingsley, P.D.; Koniski, A.D.; Palis, J. Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep. 2015, 11, 1892–1904. [Google Scholar] [CrossRef] [Green Version]
- Ivanovs, A.; Rybtsov, S.; Welch, L.; Anderson, R.A.; Turner, M.L.; Medvinsky, A. Highly Potent Human Hematopoietic Stem Cells First Emerge in the Intraembryonic Aorta–Gonad–Mesonephros Region. J. Exp. Med. 2011, 208, 2417–2427. [Google Scholar] [CrossRef] [Green Version]
- Medvinsky, A.; Rybtsov, S.; Taoudi, S. Embryonic Origin of the Adult Hematopoietic System: Advances and Questions. Development 2011, 138, 1017–1031. [Google Scholar] [CrossRef] [Green Version]
- Boisset, J.C.; van Cappellen, W.; Andrieu–Soler, C.; Galjart, N.; Dzierzak, E.; Robin, C. In vivo Imaging of Haematopoietic Cells Emerging from the Mouse Aortic Endothelium. Nature 2010, 464, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Dzierzak, E.; Robin, C. Placenta as A Source of Hematopoietic Stem Cells. Trends Mol. Med. 2010, 16, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Lobo, S.E.; Leonel, L.C.P.C.; Miranda, C.M.F.C.; Coelho, T.M.; Ferreira, G.A.S.; Mess, A.; Miglino, M.A. The Placenta as an Organ and a Source of Stem Cells and Extracellular Matrix: A Review. Cells Tissues Organs 2016, 201, 239–252. [Google Scholar] [CrossRef]
- Portilho, N.A.; Pelajo-Machado, M. Mechanism of Hematopoiesis and Vasculogenesis in Mouse Placenta. Placenta 2018, 69, 140–145. [Google Scholar] [CrossRef]
- Ottersbach, K.; Dzierzak, E. The Murine Placenta Contains Hematopoietic Stem Cells within the Vascular Labyrinth Region. Dev. Cell 2005, 8, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gekas, C.; Dieterlen-Lièvre, F.; Orkin, S.H.; Mikkola, H.K.A. The Placenta is a Niche for Hematopoietic Stem Cells. Dev. Cell 2005, 8, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Holmes, T.H.; Winn, L.M. DNA Damage and Perturbed Topoisomerase IIα as a Target of 1,4-Benzoquinone Toxicity in Murine Fetal Liver Cells. Toxicol. Sci. 2019, 171, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Badham, H.J.; Winn, L.M. In Utero Exposure to Benzene Disrupts Fetal Hematopoietic Progenitor Cell Growth via Reactive Oxygen Species. Toxicol. Sci. 2010, 113, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbel, C.; Salaun, J.; Belo-Diabangouaya, P.; Dieterlen-Lievre, F. Hematopoietic Potential of the Pre-Fusion Allantois. Dev Biol. 2007, 301, 478–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisan, M.; Dzierzak, E. The Many Faces of Hematopoietic Stem Cell Heterogeneity. Development 2016, 143, 4571–4581. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Soman–Faulkner, K.; Sugumar, K. Embryology, Hematopoiesis; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK544245/ (accessed on 1 October 2022).
- Cao, H.; Oteiza, A.; Nilsson, S.K. Understanding the Role of the Microenvironment during Definitive Hemopoietic Development. Exp. Hematol. 2013, 41, 761–768. [Google Scholar] [CrossRef]
- Dzierzak, E. The Emergence of Definitive Hematopoietic Stem Cells in the Mammal. Curr. Opin. Hematol. 2005, 12, 197–202. [Google Scholar] [CrossRef]
- Smith, M.T. Advances in Understanding Benzene Health Effects and Susceptibility. Annu. Rev. Public Health 2010, 31, 133–482. [Google Scholar] [CrossRef] [Green Version]
- American Conference of Governmental Industrial Hygienists. TLVs and BEIs: Based on the Documentation of the Threshold Limit Values for Chemicals and Physical Agents & Biological Exposure Indices; American Conference of Governmental Industrial Hygienists: Cincinnati, OH, USA, 2004. [Google Scholar]
- National Institute for Occupational Health and Safety. NIOSH Pocket Guide to Chemical Hazards; Department of Health and Human Service. Centers for Diseases Control and Prevention: Cincinnati, OH, USA, 2005. [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile For Toluene, Georgia. 2000. Available online: http://www.atsdr.cdc.gov/toxprofiles (accessed on 10 March 2023).
- Aksoy, M.; Dinçol, K.; Erdem, S.; Akgün, T.; Dinçol, G. Details of Blood Changes in 32 Patients with Pancytopenia Associated with Long-Term Exposure to Benzene. Br. J. Ind. Med. 1972, 29, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.K.; Bajpayee, M.; Parmar, D.; Kumar, R.; Rastogi, S.K.; Mathur, N.; Thorning, P.; de Matas, M.; Shao, Q.; Anderson, D.; et al. Multipronged Evaluation of Genotoxicity in Indian Petrol-Pump Workers. Environ. Mol. Mutagen. 2008, 49, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.A.; Sellakumar, A.R.; James, D.J.; Albert, R.E. The Carcinogenicity of Discontinuous Inhaled Benzene Exposures in CD-1 and C57Bl/6 Mice. Arch. Toxicol. 1988, 62, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Linnea-Niemi, J.V.; Kudłak, B.; Williams, M.J.; Jönsson, J.; Schiöth, H.B. Role of the Synergistic Interactions of Environmental Pollutants in the Development of Cancer. Geohealth 2022, 6, e2021GH000552. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Yuan, L.; Wei, C.; Zhao, Y.; Qian, Y.; Ma, P.; Ding, S.; Yang, X.; Wang, X. Effects of Combined Exposure to Formaldehyde and Benzene on Immune Cells in the Blood and Spleen in Balb/c Mice. Environ. Toxicol. Pharmacol. 2016, 45, 265–273. [Google Scholar] [CrossRef]
- Huang, L.J.; Fang, S.B.; Chen, C.D. Effect of Benzene, Toluene, Xylene Occupational Exposure on DNA Damage of Peripheral Blood Cells in Female Jewel Processing Workers. Mod. Prev. Med. 2010, 13, 006. [Google Scholar]
- McCollough, C.H.; Schueler, B.A.; Atwell, T.D.; Braun, N.N.; Regner, D.M.; Brown, D.L. Radiation Exposure and Pregnancy: When Should We Be Concerned? Radiographics 2007, 27, 909–918. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Monographs Program. Benzene; World Health Organization Press: Lyon, France, 2018. [Google Scholar]
- Myren, M.; Mose, T.; Mathiesen, L.; Knudsen, L.E. The Human Placenta—An Alternative for Studying Foetal Exposure. Toxicol. Vitr. 2007, 21, 1332–1340. [Google Scholar] [CrossRef]
- McKinney, P.A.; Raji, O.Y.; van Tongeren, M.; Feltbower, R.G. The UK Childhood Cancer Study: Maternal Occupational Exposures and Childhood Leukaemia and Lymphoma. Radiat. Prot. Dosim. 2008, 132, 232–240. [Google Scholar] [CrossRef]
- Snyder, R.; Hedli, C.C. An Overview of Benzene Metabolism. Environ. Health Perspect. 1996, 104, 1165–1171. [Google Scholar]
- Synder, R. Benzene’s Toxicity: A Consolidated Short Review of Human and Animal Studies. Hum. Exp. Toxicol. 2007, 26, 687–696. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450s and Other Enzymes in Drug Metabolism and Toxicity. AAPS J. 2006, 8, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.M.; El-Bayoumy, K.; Hosey, J.; Cunningham, J.; Aliaga, C.; Melikian, A.A. Benzene Increases Protein-Bound 3-Nitrotyrosine in Bone Marrow of B6C3F1 Mice. Chem. Biol. Interact. 2005, 156, 81–91. [Google Scholar] [CrossRef]
- Corti, M.; Snyder, C.A. Influences of Gender, Development, Pregnancy and Ethanol Consumption on the Hematotoxicity of Inhaled 10 Ppm Benzene. Arch. Toxicol. 1996, 70, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.A.; Snyder, C.A. Mice Exposed in Utero to 20 ppm Benzene Exhibit Altered Numbers of Recognizable Hematopoietic Cells up to Seven Weeks after Exposure. Fundam. Appl. Toxicol. 1988, 10, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, H.; Danielsson, B.R. Placental Transfer and Distribution of Toluene, Xylene and Benzene, and Their Metabolites during Gestation in Mice. Biol. Res. Pregnancy Perinatol. 1986, 7, 98–105. [Google Scholar]
- Dowty, B.J.; Laseter, J.L.; Storer, J. The Transplacental Migration and Accumulation in Blood of Volatile Organic Constituents. Pediatr. Res. 1976, 10, 696–701. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.A.; Waidyanatha, S.; Rappaport, S.M. Production of Benzoquinone Adducts with Hemoglobin and Bone-Marrow Proteins following Administration of Benzene to Rats. Carcinogenesis 1994, 14, 1921–1925. [Google Scholar] [CrossRef] [PubMed]
- Yeowell-O’Connell, K.; Rothman, N.; Waidyanatha, S.; Smith, M.T.; Hayes, R.B.; Li, G.; Bechtold, W.E.; Dosemeci, M.; Zhang, L.; Yin, S.; et al. Protein Adducts of 1,4-Benzoquinone and Benzene Oxide among Smokers and Nonsmokers Exposed to Benzene in China. Cancer Epidemiol. Biomark. Prev. 2001, 10, 831–838. [Google Scholar]
- Badham, H.J.; Renaud, S.J.; Wan, J.; Winn, L.M. Benzene-initiated Oxidative Stress: Effects on Embryonic Signaling Pathways. Chem. Biol. Interact. 2010, 184, 218–221. [Google Scholar] [CrossRef]
- Stillman, W. The Benzene Metabolite, Hydroquinone, Selectively Induces 5q31− and −7 in Human CD34+CD19− Bone Marrow Cells. Exp. Hematol. 2000, 28, 169–176. [Google Scholar] [CrossRef]
- Ning, H.; Kado, N.Y.; Kuzmicky, P.A.; Hsieh, D.P. Benzene-induced Micronuclei Formation in Mouse Fetal Liver Blood, Peripheral Blood, and Maternal Bone Marrow Cells. Environ. Mol. Mutagen 1991, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ma, H.; Zhang, W.; Yu, Z.; Sheng, G.; Fu, J. Effects of Benzene and Its Metabolites on Global DNA Methylation in Human Normal Hepatic L02 Cells. Environ. Toxicol. 2014, 29, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Carver, T.A.; Miranda, J.L.; Kautiainen, A.; Vogel, J.S.; Dingley, K.; Burlingame, A.L. Attomole Detection of in Vivo Protein Targets of Benzene in Mice. Mol. Cell Proteom. 2002, 1, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Xing, X.; Zhang, X.; Liang, B.; He, Z.; Gao, C.; Li, D. Enhanced H3k4me3 Modifications are Involved in Transactivation of DNA Damage Responsive Genes in Workers Exposed to Low-Level Benzene. Environ. Pollut. 2018, 234, 127–135. [Google Scholar] [CrossRef]
- Apidi, E.; Wan Taib, W.R.; Hassan, R.; Ab Mutalib, N.; Ismail, I. A Review on Effect of Genetic Features on Treatment Responses in Acute Myeloid Leukemia. Meta. Gene. 2018, 18, 31–38. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Yang, S.; Guo, L.; Li, Z.; Wang, W.; Wang, S.; Huang, W.; Wang, L.; Yang, T.; et al. Comparison of Toxicity of Benzene Metabolite Hydroquinone in Hematopoietic Stem Cells Derived from Murine Embryonic Yolk Sac and Adult Bone Marrow. PLoS ONE 2013, 8, e71153. [Google Scholar] [CrossRef] [Green Version]
- Snyder, R. Overview of the Toxicology of Benzene. J. Toxicol. Environ. Health A 2000, 61, 339–346. [Google Scholar] [CrossRef]
- Whysner, J.; Reddy, M.V.; Ross, P.M.; Mohan, M.; Lax, E.A. Genotoxicity of Benzene and Its Metabolites. Mutat. Res. 2004, 566, 99–130. [Google Scholar] [CrossRef]
- Pandey, A.K.; Gurbani, D.; Bajpayee, M.; Parmar, D.; Ajmani, S.; Dhawan, A. In Silico Studies with Human DNA Topoisomerase-II Alpha to Unravel The Mechanism of in Vitro Genotoxicity of Benzene and Its Metabolites. Mutat. Res. 2009, 661, 57–70. [Google Scholar] [CrossRef]
- Ejiri, N.; Katayama, K.; Kiyosawa, N.; Baba, Y.; Doi, K. Microarray Analysis on Phase II Drug Metabolizing Enzymes Expression in Pregnant Rats after Treatment with Pregnenolone-16alpha-Carbonitrile or Phenobarbital. Exp. Mol. Pathol. 2005, 79, 272–277. [Google Scholar] [CrossRef]
- Lin, Y.; Lu, P.; Tang, C.; Mei, Q.; Sandig, G.; Rodrigues, A.D.; Rushmore, T.H.; Shou, M. Substrate Inhibition Kinetics for Cytochrome P450-Catalyzed Reactions. Drug Metab. Dispos. 2001, 29, 368–374. [Google Scholar] [PubMed]
- Collom, S.L.; Laddusaw, R.M.; Burch, A.M.; Kuzmic, P.; Perry, M.D.; Miller, G.P. CYP2E1 Substrate Inhibition. Mechanistic Interpretation through An Effector Site for Monocyclic Compounds. J. Biol. Chem. 2008, 283, 3487–3496. [Google Scholar] [CrossRef] [Green Version]
- Ross, D. Metabolic Basis of Benzene Toxicity. Eur. J. Haematol. 2009, 57, 111–118. [Google Scholar] [CrossRef]
- Ross, D.; Zhou, H.; Siegel, D. Benzene Toxicity: The Role of the Susceptibility Factor NQO1 in Bone Marrow Endothelial Cell Signaling and Function. Chem. Biol. Interact. 2011, 192, 145–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philbrook, N.A.; Winn, L.M. Investigating the Effects of in utero Benzene Exposure on Epigenetic Modifications in Maternal and Fetal CD-1 Mice. Toxicol. Appl. Pharmacol. 2015, 289, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.A.; Snyder, C.A. Mice Exposed in Utero to Low Concentrations of Benzene Exhibit Enduring Changes in Their Colony Forming Hematopoietic Cells. Toxicology 1986, 42, 171–181. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, S.; Li, Z.; Zhu, J.; Bi, Y.; Bai, Y.; Wang, H. Maternal Benzene Exposure during Pregnancy and Risk of Childhood Acute Lymphoblastic Leukemia: A Meta-analysis of Epidemiologic Studies. PLoS ONE 2014, 15, e110466. [Google Scholar] [CrossRef]
- Scelo, G.; Metayer, C.; Zhang, L.; Wiemels, J.L.; Aldrich, M.C. Household Exposure to Paint and Petroleum Solvents, Chromosomal Translocations, and the Risk of Childhood Leukemia. Environ. Health Perspect. 2009, 117, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, A.; Glass, D.C.; Bailey, H.D.; Milne, E.; Armstrong, B.K.; Alvaro, F.; Fritschi, L. Parental Occupational Exposure to Exhausts, Solvents, Glues and Paints, and Risk of Childhood Leukemia. Cancer Causes Control 2011, 22, 1575–1585. [Google Scholar] [CrossRef]
- Swaen, G.M.; Slangen, J.J. Gasoline Consumption and Leukemia Mortality and Morbidity in 19 European Countries: An Ecological Study. Int. Arch. Occup. Environ. Health 1995, 67, 85–93. [Google Scholar]
- Bechtold, W.E.; Sun, J.D.; Birnbaum, L.S.; Yin, S.N.; Li, G.L.; Kasicki, S.; Henderson, R.F. S-Phenylcysteine Formation in Hemoglobin as A Biological Exposure Index to Benzene. Arch. Toxicol. 1992, 66, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Lutz, W.K.; Schlatter, C. Mechanism of the Carcinogenic Action of Benzene: Irreversible Binding to Rat Liver DNA. Chem. Biol. Interact. 1977, 18, 241–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenna, A.; Hang, B.; Rydberg, B.; Kim, E.; Pongracz, K.; Bodel, W.J.; Singer, B. The Benzene Metabolite p-Benzoquinone Forms Adducts with DNA Bases that are Excised by a Repair Activity from Human Cells that Differs from an Ethenoadenine Glycosylase. Proc. Natl. Acad. Sci. USA 1995, 92, 5890–5894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, R. Benzene and Leukemia. Crit. Rev. Toxicol. 2002, 32, 155–210. [Google Scholar] [CrossRef] [PubMed]
- Kolachana, P.; Subrahmanyam, V.V.; Meyer, K.B.; Zhang, L.; Smith, M.T. Benzene and Its Phenolic Metabolites Produce Oxidative DNA Damage in HL60 Cells in Vitro and in the Bone Marrow in Vivo. Cancer Res. 1993, 53, 1023–1026. [Google Scholar]
- Wiemels, J.L.; Cazzaniga, G.; Daniotti, M.; Eden, O.B.; Addison, G.M.; Masera, G.; Saha, V.; Biondi, A.; Greaves, M.F. Prenatal Origin of Acute Lymphoblastic Leukaemia in Children. Lancet 1999, 354, 1499–1503. [Google Scholar] [CrossRef]
- Shen, M.; Zhang, L.; Bonner, M.R.; Liu, C.S.; Li, G.; Vermeulen, R.; Dosemeci, M.; Yin, S.; Lan, Q. Association between Mitochondrial DNA Copy Number, Blood Cell Counts and Occupational Benzene Exposure. Environ. Mol. Mutagen. 2008, 49, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Ness, S.A. Myb Binding Proteins: Regulators and Cohorts in Transformation. Oncogene 1999, 18, 3039–3046. [Google Scholar] [CrossRef] [Green Version]
- Mucenski, M.L.; McLain, K.; Kier, A.B.; Swerdlow, S.H.; Schreiner, C.M.; Miller, T.A.; Pietryga, D.W.; Scott, W.J.; Potter, S.S. A Functional C-Myb Gene is Required for Normal Murine Fetal Hepatic Hematopoiesis. Cell 1991, 65, 677–689. [Google Scholar] [CrossRef]
- Amson, R.; Sigaux, F.; Przedborski, S.; Flandrin, G.; Givol, D.; Telerman, A. The Human Protooncogene Product P33pim is Expressed during Fetal Hematopoiesis and in Diverse Leukemias. Proc. Natl. Acad. Sci. USA 1989, 86, 8857–8861. [Google Scholar] [CrossRef] [Green Version]
- Perera, F.; Hemminki, K.; Jedrychowski, R.; Whyatt, W.; Campbell, U.; Hsu, Y.; Santella, R.; Albertini, R.; O’Neill, J.P. In Utero DNA Damage from Environmental Pollution is Associated with Somatic Gene Mutation in Newborns. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1134–1137. [Google Scholar]
- Boysen, G.; Pachkowski, B.F.; Nakamura, J.J.; Swenberg, A. The Formation and Biological Significance of N7-Guanine Adducts. Mutat. Res. 2009, 678, 76–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.Y.; Deng, C.X.; Hoeijmarkers, J.H.; Rebel, V.I.; Hasty, P. A Mechanism for 1,4-Benzoquinone-induced Genotoxicity. Oncotarget 2016, 7, 46433–46447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, K.H.; Sobol, R.W. A Unified View of Base Excision Repair: Lesion-Dependent Protein Complexes Regulated by Post-Translational Modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Eker, A.P.; Quayle, C.; Chaves, I.; van der Horst, G.T. DNA Repair in Mammalian Cells: Direct DNA Damage Reversal: Elegant Solutions for Nasty Problems. Cell Mol. Life Sci. 2009, 66, 968–980. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Nouspikel, T. DNA Repair in Mammalian Cells: Nucleotide Excision Repair: Variations on Versatility. Cell Mol. Life Sci. 2009, 66, 994–1009. [Google Scholar] [CrossRef]
- Pardo, B.; Gomez-Gonzalez, B.; Aguilera, A. DNA Repair in Mammalian Cells: DNA Double-Strand Break Repair: How to Fix A Broken Relationship. Cell Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA Double-strand Break Repair Pathway Choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Hooker, A.M.; Morley, A.A.; Tilley, W.D.; Sykes, P.J. Cancer-associated Genes Can Affect Somatic Intrachromosomal Recombination Early in Carcinogenesis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2004, 550, 1–10. [Google Scholar] [CrossRef]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The Life and Death of DNA-PK. Oncogene 2005, 24, 949–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiruvella, K.K.; Sebastian, R.; Sharma, S.; Karande, A.A.; Choudhary, B.; Raghavan, S.C. Time-dependent Predominance of Nonhomologous DNA End-Joining Pathways during Embryonic Development in Mice. J. Mol. Biol. 2012, 417, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Pachkowski, B.F.; Guyton, K.Z.; Sonawane, B. DNA Repair during in Utero Development: A Review of the Current State of Knowledge, Research Needs, and Potential Application in Risk Assessment. Mutat. Res. 2011, 728, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Li, Y.; Kong, M.; Xiao, X.; Zhao, Z.; He, X.; Ma, Q. Gene Expression in Benzene-Exposed Workers by Microarray Analysis of Peripheral Mononuclear Blood Cells: Induction and Silencing of CYP4F3A and Regulation of DNA-Dependent Protein Kinase Catalytic Subunit in DNA Double Strand Break Repair. Chem. Biol. Interact. 2010, 184, 207–211. [Google Scholar] [CrossRef]
- Xiao, X.; Song, W.; Bi, Y. Role of DNA-PKcs in the Biological Effect of A Benzene Metabolite: Phenol Toxicity to Human K562 Cells in Vitro. Chem. Biol. Interact. 2010, 184, 302–305. [Google Scholar] [CrossRef]
- Ishihama, M.; Toyooka, T.; Ibuki, Y. Generation of Phosphorylated Histone H2AX by Benzene Metabolites. Toxicol. Vitr. 2008, 22, 1861–1868. [Google Scholar] [CrossRef]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegué, E. Hematopoietic Stem Cell Quiescence Promotes Error-Prone DNA Repair and Mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Mondrala, S.; Eastmond, D.A. Topoisomerase II Inhibition by the Bioactivated Benzene Metabolite Hydroquinone Involves Multiple Mechanisms. Chem. Biol. Interact. 2010, 184, 259–268. [Google Scholar] [CrossRef]
- Lau, A.; Belanger, C.L.; Winn, L.M. In Utero and Acute Exposure to Benzene: Investigation of DNA Double-Strand Breaks and DNA Recombination in Mice. Mutat. Res. 2009, 676, 74–82. [Google Scholar] [CrossRef]
- Kakehashi, A.; Wei, M.; Fukushima, S.; Wanibuchi, H. Oxidative Stress in the Carcinogenicity of Chemical Carcinogens. Cancers 2013, 5, 1332–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wang, K.; Wang, B.; Pu, Y.; Zhang, J. Occupational Benzene Exposure and the Risk of Genetic Damage: A Systematic Review and Meta-Analysis. BMC Public Health 2020, 20, 1113. [Google Scholar] [CrossRef] [PubMed]
- Joo, W.A.; Kang, M.J.; Son, W.K.; Lee, D.Y.; Lee, E.; Kim, C.W. Monitoring Protein Expression by Proteomics: Human Plasma Exposed to Benzene. Proteomics 2003, 3, 2402–2411. [Google Scholar] [CrossRef]
- Zhang, L.; Venkatesh, P.; Creek, M.L.; Smith, M.T. Detection of 1,2,4-Benzenetriol Induced Aneuploidy and Microtubule Disruption by Fluorescence in Situ Hybridization and Immunocytochemistry. Mutat. Res. 1994, 320, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Nakamura, A.; Kovalchuk, O.; Koturbash, I.; Mitchell, S.A.; Marino, S.A. DNA Double-Strand Breaks Form in Bystander Cells after Microbeam Irradiation of Three-Dimensional Human Tissue Models. Cancer Res. 2007, 67, 4295–4302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J. Molecular Mechanisms of Micronucleus, Nucleoplasmic Bridge and Nuclear Bud Formation in Mammalian and Human Cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Mateuca, R.; Lombaert, N.; Aka, P.V.; Decordier, I.; Kirsch-Volders, M. Chromosomal Changes: Induction, Detection Methods and Applicability in Human Biomonitoring. Biochimie 2006, 88, 1515–1531. [Google Scholar] [CrossRef]
- Ciranni, R.; Barale, R.; Marrazzini, A.; Loprieno, N. Benzene and the Genotoxicity of Its Metabolites. Transplacental Activity in Mouse Fetuses and in Their Dams. Mutat. Res. 1988, 208, 61–67. [Google Scholar] [CrossRef]
- Cooke, G.M. Biomonitoring of Human Fetal Exposure to Environmental Chemicals in Early Pregnancy. J. Toxicol. Environ. Health Part B 2014, 17, 205–224. [Google Scholar] [CrossRef]
- Shames, D.S.; Minna, J.D.; Gazdar, A.F. DNA Methylation in Health, Disease, and Cancer. Curr. Mol. Med. 2007, 7, 85–102. [Google Scholar] [CrossRef]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-Throughput DNA Methylation Profiling using Universal Bead Arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Daxinger, L.; Whitelaw, E. Understanding Transgenerational Epigenetic Inheritance via the Gametes in Mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Aaron, D.; Goldberg, C.; Allis, D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 28, 635–638. [Google Scholar]
- Lestari, S.W.; Rizki, M.D. Epigenetic: A New Approach to Etiology of Infertility. Med. J. Indones. 2016, 24, 4. [Google Scholar] [CrossRef] [Green Version]
- Rivera, R.M.; Ross, J.W. Epigenetics in Fertilization and Preimplantation Embryo Development. Prog. Biophys. Mol. 2013, 113, 423–432. [Google Scholar] [CrossRef]
- Jones, P.A.; Liang, G. Rethinking How DNA Methylation Patterns are Maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential Role for De Novo DNA Methyltransferase Dnmt3a in Paternal and Maternal Imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic Modifications and Human Disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Christensen, B.C.; Marsit, C.J. Epigenomics in Environmental Health. Front. Genet. 2011, 2, 84. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.A.; Stempor, P.; Down, T.A.; Zeiser, E.; Feuer, S.K.; Ahringer, J. Extreme HOT Regions are CpG–dense Promoters in C. elegans and Humans. Genome Res. 2014, 24, 1138–1146. [Google Scholar] [CrossRef] [Green Version]
- Bollati, V.; Baccarelli, A.; Hou, L.; Bonzini, M.; Fustinoni, S.; Cavallo, D.; Byun, H.M.; Jiang, J.; Marinelli, B.; Pesatori, A.C.; et al. Changes in DNA Methylation Patterns in Subjects Exposed to Low-Dose Benzene. Cancer Res. 2007, 67, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; McHale, C.M.; Rothman, N.; Li, G.; Ji, Z.; Vermeulen, R.; Hubbard, A.E.; Ren, X.; Shen, M.; Rappaport, S.M.; et al. Systems Biology of Human Benzene Exposure. Chem. Biol. Interact. 2010, 184, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fustinoni, S.; Rossella, F.; Polledri, E.; Bollati, V.; Campo, L.; Byun, H.M.; Agnello, L.; Consonni, D.; Pesatori, A.C.; Baccarelli, A.; et al. Global DNA Methylation and Low-Level Exposure to Benzene. La Med. Del Lav. 2012, 103, 84–95. [Google Scholar]
- Chopra, M.; Bohlander, S.K. Disturbing the Histone Code in Leukemia: Translocations and Mutations Affecting Histone Methyl Transferases. Cancer Genet. 2015, 208, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Husmann, D.; Gozani, O. Histone Lysine Methyltransferases in Biology and Disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Davie, J.R.; Xu, W.; Delcuve, G.P. Histone H3K4 Trimethylation: Dynamic Interplay with Pre-mRNA Splicing. Biochem. Cell Biol. 2015, 94, 1–11. [Google Scholar] [CrossRef]
- Minciullo, P.L.; Navarra, M.; Calapai, G.; Gangemi, S. Cytokine Network Involvement in Subjects Exposed to Benzene. J. Immunol. Res. 2014, 2014, 937987. [Google Scholar] [CrossRef] [Green Version]
- Myöhänen, K.; Vähäkangas, K. Foetal Exposure to Food and Environmental Carcinogens in Human Beings. Basic Clin. Pharmacol. Toxicol. 2011, 110, 101–112. [Google Scholar]
- Prouillac, C.; Lecoeur, S. The Role of the Placenta in Fetal Exposure to Xenobiotics: Importance of Membrane Transporters and Human Models for Transfer Studies. Drug Metab. Dispos. 2010, 38, 1623–1635. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Fowden, A.L. The Placenta: A Multifaceted, Transient Organ. Philos. Trans. R. Soc. B 2015, 5, 20140066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myllynen, P.; Karttunen, V.; Sieppi, E.; Kummu, M.; Rautio, A.; Vähäkangas, K. Fetal Exposure to Chemical Carcinogens and Environmental Contaminants. In Proceedings of the SkyPro Conference, Oulu, Finland, 3 June 2010; Pongrácz, E., Hyvärinen, M., Pitkäaho, S., Keiski, R.L., Eds.; Clean Air Research at the University of Oulu: Oulu, Finland; pp. 28–29. [Google Scholar]
- Karttunen, V.; Myllynen, P.; Prochazka, G.; Pelkonen, O.; Segerbäck, D.; Vähäkangas, K. Placental Transfer and DNA Binding of Benzo(A)Pyrene in Human Placental Perfusion. Toxicol. Lett. 2010, 197, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Cesario, A.; Granone, P.; Dominioni, L.; Puntoni, R.; D’Angelillo, R.M.; Russo, P. Prognostic Role Of K-Ras Mutations in Non–Small Cell Lung Cancer: Still An Issue for Open Debate. Lung Cancer 2006, 53, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M. The Role of the Placenta in Fetal Programming—A Review. Placenta 2002, 23, 20–27. [Google Scholar] [CrossRef] [PubMed]
- McKay, R. Developmental Biology: Remarkable Role for the Placenta. Nature 2011, 472, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B. The Anatomy of the Normal Placenta. J. Clin. Pathol. 2008, 61, 1296–1302. [Google Scholar] [CrossRef]
- Jones, A.; Gokhale, P.; Allison, T.; Sampson, B.; Athwal, S.; Grant, S.; Andrews, P.W.; Allen, N.D.; Patrick, C. Case Evidence for Bystander Signalling between Human Trophoblast Cells and Human Embryonic Stem Cells. Sci. Rep. 2015, 5, 11694. [Google Scholar] [CrossRef] [Green Version]
- Cogan, N.; Baird, D.M.; Phillips, R.; Crompton, L.A.; Caldwell, M.A.; Rubio, M.A.; Newson, R.; Lyng, F.; Case, C.P. DNA Damaging Bystander Signalling from Stem Cells, Cancer Cells and Fibroblasts after Cr(VI) Exposure and Its Dependence on Telomerase. Mutat. Res. 2010, 683, 1–8. [Google Scholar] [CrossRef]
- Bhabra, G.; Sood, A.; Fisher, B.; Cartwright, L.; Saunders, M.; Evans, W.H.; Case, C.P. Nanoparticles Can Cause DNA Damage Across A Cellular Barrier. Nat. Nanotechnol. 2009, 4, 876–883. [Google Scholar] [CrossRef]
- Sood, A.; Salih, S.; Roh, D.; Lacharme-Lora, L.; Parry, M.; Hardiman, B.; Case, C.P. Signalling of DNA Damage and Cytokines Across Cell Barriers Exposed to Nanoparticles Depends on Barrier Thickness. Nat. Nanotechnol. 2011, 6, 824–833. [Google Scholar] [CrossRef]
- Curtis, D.J.; Sood, A.; Phillips, T.J.; Leinster, V.H.; Nishiguchi, A.; Coyle, C.; Lacharme-Lora, L.; Beaumont, O.; Kemp, H.; Goodall, R.; et al. Secretions from Placenta, after Hypoxia/Reoxygenation, Can Damage Developing Neurones of Brain Under Experimental Conditions. Exp. Neurol. 2014, 261, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Kalnas, J.; Teitelbaum, D.T. Dermal Absorption of Benzene: Implications for Work Practices and Regulations. Int. J. Occup. Med. Environ. Health 2000, 6, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Chiu, B.C.; Hou, N. Epidemiology and Etiology of Non-Hodgkin Lymphoma. Cancer Treat. Res. 2015, 165, 1–25. [Google Scholar]
- Vlaanderen, J.; Lan, Q.; Kromhout, H.; Rothman, N.; Vermeulen, R. Occupational Benzene Exposure and the Risk of Chronic Myeloid Leukemia: A Meta-Analysis of Cohort Studies Incorporating Study Quality Dimensions. Am. J. Ind. Med. 2012, 55, 779–785. [Google Scholar] [CrossRef]
- Carlos-Wallace, F.M.; Zhang, L.; Smith, M.T.; Rader, G.; Steinmaus, C. Parental, In Utero, and Early-Life Exposure to Benzene and the Risk of Childhood Leukemia: A Meta-Analysis. Am. J. Epidemiol. 2016, 183, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Wiemels, J. Origins of Chromosome Translocations in Childhood Leukaemia. Nat. Rev. Cancer 2003, 3, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Caron-Beaudoin, É.; Valter, N.; Chevrier, J.; Ayotte, P.; Frohlich, K.; Verner, M.A. Gestational Exposure to Volatile Organic Compounds (VOCs) in Northeastern British Columbia, Canada: A Pilot Study. Environ. Int. 2018, 110, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T. The Leukemic Stem Cell. Best Pract. Res. Clin. Haematol. 2007, 20, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.C.; Armstrong, S.A. Therapeutic Implications of Leukemia Stem Cell Development. Clin. Cancer. Res. 2007, 15, 3439–3442. [Google Scholar] [CrossRef] [Green Version]
- Eden, T. Aetiology of Childhood Leukemia. Cancer Trea. Rev. 2010, 3, 286–297. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. Is electrophile or can be metabolically activated to electrophiles. 2. Is genotoxic. 3. Alters DNA repair and causes genomic instability. 4. Induces epigenetic alterations. 5. Induces oxidative stress. 6. Induces chronic inflammation. 7. Is immunosuppressive. 8. Modulates receptor-mediated effects. 9. Causes immortalization. 10. Alters cell proliferation, cell death or nutrient supply. |
| No | Category | Description |
|---|---|---|
| 1 | Journal Databases |
|
| 2 | Inclusion Criteria |
|
| 3 | Exclusion Criteria | None |
| 4 | Types of articles |
|
| Exposure Time Windows | Periconception (Pre- and Around Conception) | Intrauterine (Gestational) | Postnatal |
| Maternal |
|
|
|
| Paternal |
|
|
|
| Benzene or Metabolites | Study Design | Dose or Concentration | Experimental Model | Toxicity Effects | References |
|---|---|---|---|---|---|
| 1,4-BQ | In vitro | 0, 1.25, 2.5, 5 μM | ICR mice | Induce concentration-dependent cytotoxicity & apoptosis in BM cells, ↓total counts of Sca-1+, CD11b+, Gr-1+ & CD45+ cells, ↓clonogenicity in 1,4-BQ-treated cells | [12] |
| 1,4-BQ | In vitro | 0, 5, 7, 12 μM | ICR mice | ↓colony-forming capacity of the myeloid progenitor at 1.25 & 2.5 μM, ↑expression HoxB4 level at all concentrations & ↑Bmi-1 expression level at 5 μM, ↑GATA3 expression level at 2.5 µM | [14] |
| Benzene | Epidemiology | – | Pregnant women | ↑risk of childhood ALL | [18] |
| BQ | In vitro | 6.25, 9.375 12.5, 15.625 μM | Murine CD-1 | ↓c-kit+Lin-Sca-1-Il7rα-cell population in BQ-treated, ↓Topo IIα activity in concentration-dependent, ↑γH2AX levels at 12.5 µM BQ exposure | [66] |
| Benzene | In vivo (i.p.) | 200, 400 mg/kg (GD8, 10, 12, 14, 16) | Pregnant C57Bl/6N mice | ↑numbers of CFU-E, BFU-E, CFU-GM & CFU-G colonies at 200 mg/kg, ↓numbers of CFU-M colonies in hematopoietic tissue of GD16 C57Bl/6N fetuses at 200 mg/kg, ↑numbers of CFU-E & CFU-G colonies at 400 mg/kg benzene on GD8, 10, 12, 14, ↑ROS production in fetal liver | [67] |
| Benzene | In vivo (i.p.) | 50, 100, 200, 400 mg/kg | B6C3F1 mice | ↑nitration of tyrosine residues in bone marrow proteins from 50 to 200 mg/kg | [90] |
| Benzene | In vivo (inhalation) | 10 ppm (GD6–15) | Pregnant Swiss Webster mice | ↑alteration in cell numbers for progenitor & hematopoietic precursor | [91] |
| Benzene | In vivo (inhalation) | 5, 10, 20 ppm (GD6–15) | Pregnant Swiss Webster mice | ↓numbers of circulating erythroid precursor cells, ↑numbers of hepatic hematopoietic blast cells & granulopoietic precursor cells, ↑granulopoiesis. | [92] |
| Radio- labelled benzene | In vivo (inhalation) | – | Pregnant C57BL mice | Radioactivity detected in the embryonic hematopoietic tissue | [93] |
| Benzene | Epidemiology | – | Pregnant women | Benzene detected in fetal umbilical cord blood at the same or greater level than in the mother’s blood, ↑benzene accumulation in the fetal-placenta unit | [94] |
| Benzene | In vivo (oral) | 50, 100, 200, 400 mg/kg | Fischer (F344) rats | ↑BQ adducts of Hb and bone-marrow proteins with dose-dependent manner | [95] |
| Benzene | Epidemiology | ≤31, >31 ppm | Workers | ↑levels of BO & HQ adducts of Hb and albumin | [96] |
| 1,4-BQ | In vitro | 0, 1.25, 2.5, 5, 7, 12 μM | ICR mice | ↓GSH level, ↓SOD activity, ↑MDA level, ↑PC level, ↑DNA damage in BM cells (↑DNA in tail % at 7 & 12 μM as well as ↑tail moment at 12 μM), ↑DNA damage in myeloid & pre-B lymphoid progenitors at 2.5 µM, ↑DNA damage in the erythroid progenitor at 5 µM 1,4-BQ, ↑in tail moment at 7 µM & 12 µM 1,4-BQ exposure for all progenitors | [4] |
| Benzene | In vivo (i.p.) | 200 mg/kg (GD8, 10, 12, 14) | Pregnant CD mice | ↑oxidative stress in fetal tissue from embryos, ↑ROS sensitive fluorescent probe DCFDA, ↑expression of fetal Pim-1, ↑Pim-1 phosphorylation, ↑c-Myb, ↑phosphorylated p38-MAPK, ↓protein levels of Iҡßα | [97] |
| HQ | In vitro | 0, 5, 10, 15, 20 μM | Human | ↑frequency of the specific chromosomal aberrations in CD34+ bone marrow cells | [98] |
| Benzene | In vivo (i.p.) | 0, 109, 219, 437, 874 mg/kg (GD14) | Pregnant Swiss Webster mice | ↑frequency of MNPCE in fetal liver & fetal peripheral blood cells at 219 to 874 mg/kg, ↑frequency of MNPCE in maternal bone marrow cells at 437 & 874 mg/kg | [99] |
| HQ & 1,4-BQ | In vitro | 5, 10, 25, 50 μM | L02 cell line | ↑global DNA methylation, ↓DNMT activity | [100] |
| Benzene | In vivo (i.p.) | 155, 800 µg/kg | B6C3F1 mice | ↑protein adducts in bone marrow & liver | [101] |
| Hydroquinone | In vitro | 0, 0.1, 1.0, 10.0 µM | Worker’s blood | ↑global H3K4me3 modification & hematotoxicity, ↓white blood cells count, ↓neutrophils count, ↓lymphocytes count, ↓monocytes count | [102] |
| Muconic acid | Epidemiology | – | Pregnant women | ↑muconic acid in urine of pregnant women living close to natural-gas hydraulic fracturing sites | [103] |
| Hydroquinone | In vitro | 0, 1.25, 2.5, 5.0 µM | SPF Kunming mice | ↓proliferation & differentiation as well as ↓colony formation of both YS-HSCs & BM-HSCs, ↑apoptosis of both YS-HSCs & BM-HSCs | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yusoff, N.A.; Abd Hamid, Z.; Budin, S.B.; Taib, I.S. Linking Benzene, in Utero Carcinogenicity and Fetal Hematopoietic Stem Cell Niches: A Mechanistic Review. Int. J. Mol. Sci. 2023, 24, 6335. https://doi.org/10.3390/ijms24076335
Yusoff NA, Abd Hamid Z, Budin SB, Taib IS. Linking Benzene, in Utero Carcinogenicity and Fetal Hematopoietic Stem Cell Niches: A Mechanistic Review. International Journal of Molecular Sciences. 2023; 24(7):6335. https://doi.org/10.3390/ijms24076335
Chicago/Turabian StyleYusoff, Nur Afizah, Zariyantey Abd Hamid, Siti Balkis Budin, and Izatus Shima Taib. 2023. "Linking Benzene, in Utero Carcinogenicity and Fetal Hematopoietic Stem Cell Niches: A Mechanistic Review" International Journal of Molecular Sciences 24, no. 7: 6335. https://doi.org/10.3390/ijms24076335