

Role of Macrophages in Sickle Cell Disease Erythrophagocytosis and Erythropoiesis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sickle Cell Disease Pathophysiology
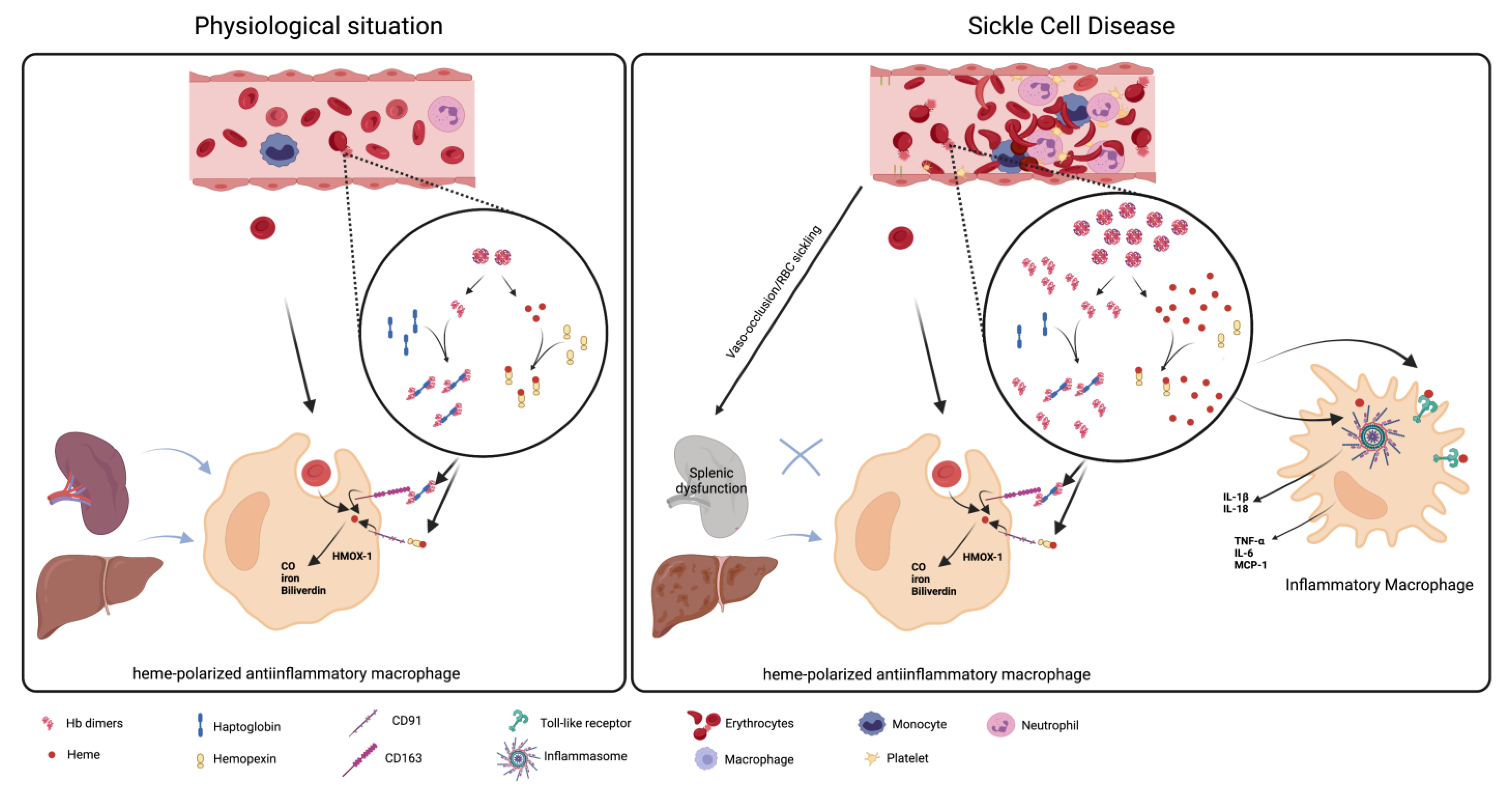
3. The Role of Macrophages in Hemolysis Product Clearance
4. Leukocytosis and Sickle Cell Disease
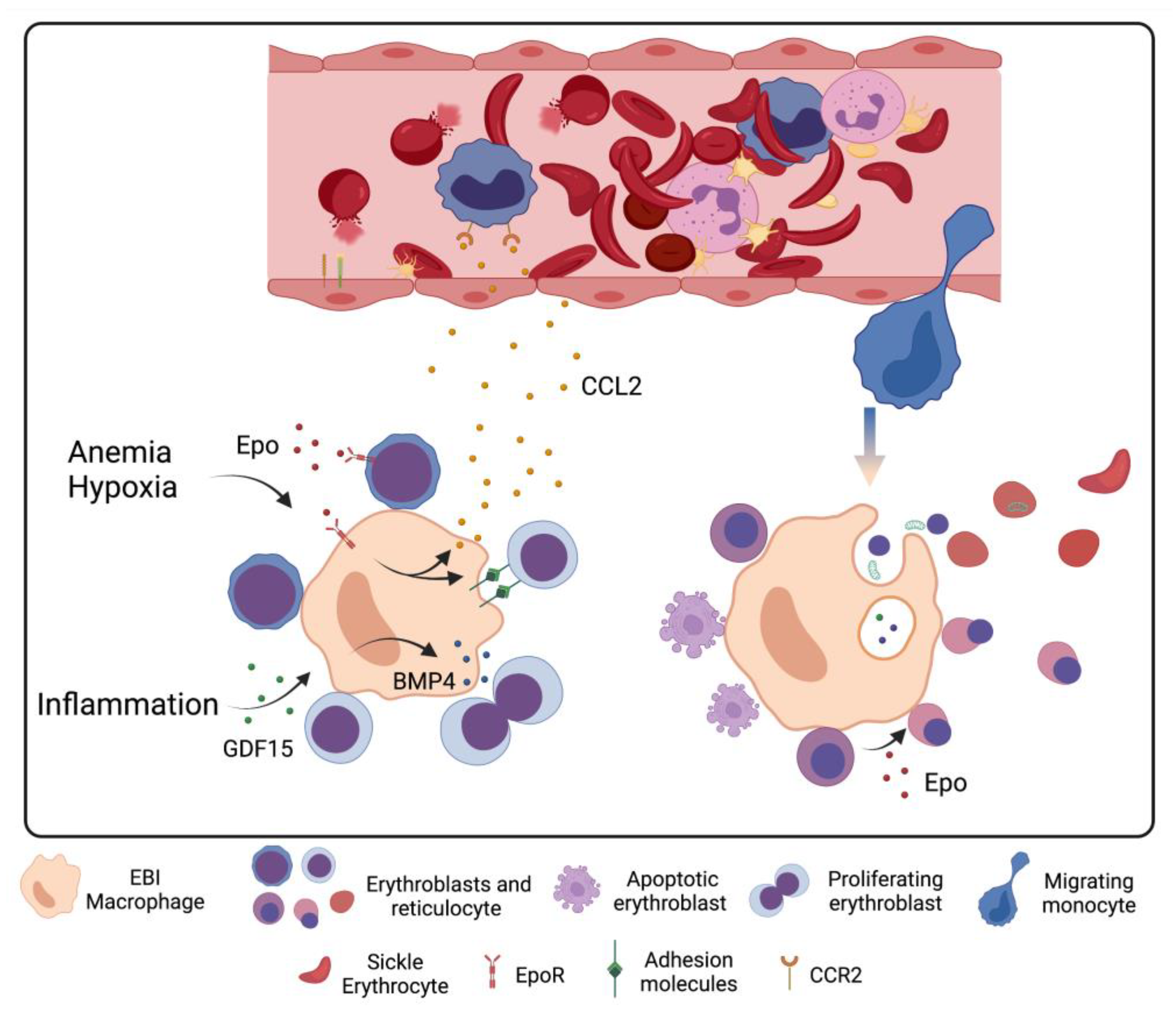
5. Stress Erythropoiesis in Sickle Cell Disease
6. Macrophage–Erythrocyte Interactions in Sickle Cell Disease Erythropoiesis
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Prim. 2018, 4, 18010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, M.H. Fetal Hemoglobin in Sickle Hemoglobinopathies: High HbF Genotypes and Phenotypes. J. Clin. Med. 2020, 9, 3782. [Google Scholar] [CrossRef] [PubMed]
- Hofrichter, J.; Ross, P.D.; Eaton, W.A. Kinetics and mechanism of deoxyhemoglobin S gelation: A new approach to understanding sickle cell disease. Proc. Natl. Acad. Sci. USA 1974, 71, 4864–4868. [Google Scholar] [CrossRef] [Green Version]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Onyekwere, O.C.; Gladwin, M.T. Pulmonary hypertension in sickle cell disease: Relevance to children. Pediatr. Hematol. Oncol. 2007, 24, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Nader, E.; Romana, M.; Connes, P. The Red Blood Cell-Inflammation Vicious Circle in Sickle Cell Disease. Front. Immunol. 2020, 11, 454. [Google Scholar] [CrossRef] [Green Version]
- Conran, N.; Embury, S.H. Sickle cell vaso-occlusion: The dialectic between red cells and white cells. Exp. Biol. Med. 2021, 246, 1458–1472. [Google Scholar] [CrossRef]
- Conran, N.; Belcher, J.D. Inflammation in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 263–299. [Google Scholar] [CrossRef]
- Hebbel, R.P.; Belcher, J.D.; Vercellotti, G.M. The multifaceted role of ischemia/reperfusion in sickle cell anemia. J. Clin. Investig. 2020, 130, 1062–1072. [Google Scholar] [CrossRef]
- Mendonca, R.; Silveira, A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016, 65, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.S.; Hidalgo, A. Neutrophils as drivers of vascular injury in sickle cell disease. Immunol. Rev. 2022. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, A.; Ihunnah, C.A.; Walker, A.L.; Ofori-Acquah, S.F. Inflammatory targets of therapy in sickle cell disease. Transl. Res. 2016, 167, 281–297. [Google Scholar] [CrossRef] [Green Version]
- De Montalembert, M.; Wang, W. Cerebrovascular complications in children with sickle cell disease. Handb. Clin. Neurol. 2013, 113, 1937–1943. [Google Scholar]
- De, A.; Manwani, D.; Rastogi, D. Airway inflammation in sickle cell disease-A translational perspective. Pediatr. Pulmonol. 2018, 53, 400–411. [Google Scholar] [CrossRef]
- Humar, R.; Schaer, D.J.; Vallelian, F. Erythrophagocytes in hemolytic anemia, wound healing, and cancer. Trends Mol. Med. 2022, 28, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Klei, T.R.L.; Dalimot, J.; Nota, B.; Veldthuis, M.; Mul, F.P.J.; Rademakers, T.; Hoogenboezem, M.; Nagelkerke, S.Q.; van IJcken, W.F.J.; Oole, E.; et al. Hemolysis in the spleen drives erythrocyte turnover. Blood 2020, 136, 1579–1589. [Google Scholar] [CrossRef]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Vallelian, F.; Buehler, P.W.; Schaer, D.J. Hemolysis, free hemoglobin toxicity, and scavenger protein therapeutics. Blood 2022, 140, 1837–1844. [Google Scholar] [CrossRef]
- Pfefferle, M.; Ingoglia, G.; Schaer, C.A.; Yalamanoglu, A.; Buzzi, R.; Dubach, I.L.; Tan, G.; Lopez-Cano, E.Y.; Schulthess, N.; Hansen, K.; et al. Hemolysis transforms liver macrophages into antiinflammatory erythrophagocytes. J. Clin. Investig. 2020, 130, 5576–5590. [Google Scholar] [CrossRef]
- Bories, G.F.P.; Yeudall, S.; Serbulea, V.; Fox, T.E.; Isakson, B.E.; Leitinger, N. Macrophage metabolic adaptation to heme detoxification involves CO-dependent activation of the pentose phosphate pathway. Blood 2020, 136, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallelian, F.; Buzzi, R.M.; Pfefferle, M.; Yalamanoglu, A.; Dubach, I.L.; Wassmer, A.; Gentinetta, T.; Hansen, K.; Humar, R.; Schulthess, N.; et al. Heme-stress activated NRF2 skews fate trajectories of bone marrow cells from dendritic cells towards red pulp-like macrophages in hemolytic anemia. Cell Death Differ. 2022, 29, 1450–1465. [Google Scholar] [CrossRef] [PubMed]
- El Hoss, S.; Brousse, V. Considering the spleen in sickle cell disease. Expert Rev. Hematol. 2019, 12, 563–573. [Google Scholar] [CrossRef]
- Nardo-Marino, A.; Glenthoj, A.; Brewin, J.N.; Petersen, J.; Braunstein, T.H.; Kurtzhals, J.A.L.; Williams, T.N.; Rees, D.C. The significance of spleen size in children with sickle cell anemia. Am. J. Hematol. 2022, 97, 1520–1528. [Google Scholar] [CrossRef]
- Quaye, I.K. Haptoglobin, inflammation and disease. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Vinchi, F.; Ingoglia, G.; Tolosano, E.; Buehler, P.W. Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front. Physiol. 2014, 5, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, N.U.; Reddy, S.T. Role of hemoglobin/heme scavenger protein hemopexin in atherosclerosis and inflammatory diseases. Curr. Opin. Lipidol. 2015, 26, 384–387. [Google Scholar] [CrossRef] [Green Version]
- Ragab, S.M.; Safan, M.A.; Badr, E.A. Study of serum haptoglobin level and its relation to erythropoietic activity in Beta thalassemia children. Mediterr. J. Hematol. Infect. Dis. 2015, 7, e2015019. [Google Scholar] [CrossRef] [Green Version]
- Muller-Eberhard, U.; Javid, J.; Liem, H.H.; Hanstein, A.; Hanna, M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 1968, 32, 811–815. [Google Scholar] [CrossRef] [Green Version]
- Bourantas, K.L.; Dalekos, G.N.; Makis, A.; Chaidos, A.; Tsiara, S.; Mavridis, A. Acute phase proteins and interleukins in steady state sickle cell disease. Eur. J. Haematol. 1998, 61, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Foidart, M.; Liem, H.H.; Adornato, B.T.; Engel, W.K.; Muller-Eberhard, U. Hemopexin metabolism in patients with altered serum levels. J. Lab. Clin. Med. 1983, 102, 838–846. [Google Scholar]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [Green Version]
- Gladwin, M.T.; Crawford, J.H.; Patel, R.P. The biochemistry of nitric oxide, nitrite, and hemoglobin: Role in blood flow regulation. Free Radic. Biol. Med. 2004, 36, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Helms, C.C.; Marvel, M.; Zhao, W.; Stahle, M.; Vest, R.; Kato, G.J.; Lee, J.S.; Christ, G.; Gladwin, M.T.; Hantgan, R.R.; et al. Mechanisms of hemolysis-associated platelet activation. J. Thromb. Haemost. 2013, 11, 2148–2154. [Google Scholar] [CrossRef] [Green Version]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The Worst Things in Life are Free: The Role of Free Heme in Sickle Cell Disease. Front. Immunol. 2020, 11, 561917. [Google Scholar] [CrossRef]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Hojrup, P.; Moller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Visca, P.; Altruda, F.; Tolosano, E.; Beringhelli, T.; Fasano, M. Hemoglobin and heme scavenging. IUBMB Life 2005, 57, 749–759. [Google Scholar] [CrossRef]
- Miguel, L.I.; Leonardo, F.C.; Torres, L.S.; Garcia, F.; Mendonca, R.; Ferreira, W.A., Jr.; Gotardo, E.M.F.; Fabris, F.C.Z.; Brito, P.L.; Costa, F.F.; et al. Heme induces significant neutrophil adhesion in vitro via an NFkappaB and reactive oxygen species-dependent pathway. Mol. Cell. Biochem. 2021, 476, 3963–3974. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, B.N.; Alves, L.S.; Fernandez, P.L.; Dutra, T.P.; Figueiredo, R.T.; Graca-Souza, A.V.; Bozza, M.T. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem. 2007, 282, 24430–24436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [Green Version]
- Abboud, M.; Laver, J.; Blau, C.A. Granulocytosis causing sickle-cell crisis. Lancet 1998, 351, 959. [Google Scholar] [CrossRef]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef]
- Anyaegbu, C.C.; Okpala, I.E.; Akren’Ova, Y.A.; Salimonu, L.S. Peripheral blood neutrophil count and candidacidal activity correlate with the clinical severity of sickle cell anaemia (SCA). Eur. J. Haematol. 1998, 60, 267–268. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Turhan, A.; Weiss, L.A.; Mohandas, N.; Coller, B.S.; Frenette, P.S. Primary role for adherent leukocytes in sickle cell vascular occlusion: A new paradigm. Proc. Natl. Acad. Sci. USA 2002, 99, 3047–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenette, P.S. Sickle cell vasoocclusion: Heterotypic, multicellular aggregations driven by leukocyte adhesion. Microcirculation 2004, 11, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Manwani, D.; Frenette, P.S. Vaso-occlusion in sickle cell disease: Pathophysiology and novel targeted therapies. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 362–369. [Google Scholar] [CrossRef]
- Conran, N.; Saad, S.T.; Costa, F.F.; Ikuta, T. Leukocyte numbers correlate with plasma levels of granulocyte-macrophage colony-stimulating factor in sickle cell disease. Ann. Hematol. 2007, 86, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Methe, B.; Amar, S.; Morris, A.; Lim, S.H. Intestinal injury and gut permeability in sickle cell disease. J. Transl. Med. 2019, 17, 183. [Google Scholar] [CrossRef] [Green Version]
- Mtatiro, S.N.; Makani, J.; Mmbando, B.; Thein, S.L.; Menzel, S.; Cox, S.E. Genetic variants at HbF-modifier loci moderate anemia and leukocytosis in sickle cell disease in Tanzania. Am. J. Hematol. 2015, 90, E1–E4. [Google Scholar] [CrossRef] [Green Version]
- Charache, S.; Barton, F.B.; Moore, R.D.; Terrin, M.L.; Steinberg, M.H.; Dover, G.J.; Ballas, S.K.; McMahon, R.P.; Castro, O.; Orringer, E.P. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive "switching" agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine 1996, 75, 300–326. [Google Scholar] [CrossRef]
- Singhal, R.; Rathore, D.K.; Bhakuni, T.; Seth, T.; Guchhait, P. Absence of Nonclassical Monocytes in Hemolytic Patients: Free Hb and NO-Mediated Mechanism. J. Immunol. Res. 2019, 2019, 1409383. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, H.; Vinchi, F.; Mendelson, A.; Yazdanbakhsh, K. Patrolling monocytes in sickle cell hemolytic conditions. Transfus. Clin. Biol. 2019, 26, 128–129. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Kapetanaki, M.G.; Ross, M.; Ghosh, S.; Weidert, F.; Bullock, G.C.; Watkins, S.; Ofori-Acquah, S.F.; Kato, G.J. Nrf2 deficiency in mice attenuates erythropoietic stress-related macrophage hypercellularity. Exp. Hematol. 2020, 84, 19–28.e4. [Google Scholar] [CrossRef]
- Paulson, R.F.; Hariharan, S.; Little, J.A. Stress erythropoiesis: Definitions and models for its study. Exp. Hematol. 2020, 89, 43–54.e2. [Google Scholar] [CrossRef]
- Bessis, M. Erythroblastic island, functional unity of bone marrow. Rev. Hematol. 1958, 13, 8–11. [Google Scholar] [PubMed]
- Li, W.; Guo, R.; Song, Y.; Jiang, Z. Erythroblastic Island Macrophages Shape Normal Erythropoiesis and Drive Associated Disorders in Erythroid Hematopoietic Diseases. Front. Cell Dev. Biol. 2020, 8, 613885. [Google Scholar] [CrossRef]
- Paulson, R.F.; Shi, L.; Wu, D.C. Stress erythropoiesis: New signals and new stress progenitor cells. Curr. Opin. Hematol. 2011, 18, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Xiang, J.; Qian, F.; Diwakar, B.T.; Ruan, B.; Hao, S.; Prabhu, K.S.; Paulson, R.F. Epo receptor signaling in macrophages alters the splenic niche to promote erythroid differentiation. Blood 2020, 136, 235–246. [Google Scholar] [CrossRef]
- Liao, C.; Prabhu, K.S.; Paulson, R.F. Monocyte-derived macrophages expand the murine stress erythropoietic niche during the recovery from anemia. Blood 2018, 132, 2580–2593. [Google Scholar] [CrossRef] [Green Version]
- Gilboa, D.; Haim-Ohana, Y.; Deshet-Unger, N.; Ben-Califa, N.; Hiram-Bab, S.; Reuveni, D.; Zigmond, E.; Gassmann, M.; Gabet, Y.; Varol, C.; et al. Erythropoietin enhances Kupffer cell number and activity in the challenged liver. Sci. Rep. 2017, 7, 10379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef]
- Papadaki, H.A.; Kritikos, H.D.; Valatas, V.; Boumpas, D.T.; Eliopoulos, G.D. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: Improvement following anti-tumor necrosis factor-alpha antibody therapy. Blood 2002, 100, 474–482. [Google Scholar] [CrossRef]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Libregts, S.F.; Gutierrez, L.; de Bruin, A.M.; Wensveen, F.M.; Papadopoulos, P.; van Ijcken, W.; Ozgur, Z.; Philipsen, S.; Nolte, M.A. Chronic IFN-gamma production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood 2011, 118, 2578–2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, R.F.; Ruan, B.; Hao, S.; Chen, Y. Stress Erythropoiesis is a Key Inflammatory Response. Cells 2020, 9, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenox, L.E.; Shi, L.; Hegde, S.; Paulson, R.F. Extramedullary erythropoiesis in the adult liver requires BMP-4/Smad5-dependent signaling. Exp. Hematol. 2009, 37, 549–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harandi, O.F.; Hedge, S.; Wu, D.C.; McKeone, D.; Paulson, R.F. Murine erythroid short-term radioprotection requires a BMP4-dependent, self-renewing population of stress erythroid progenitors. J. Clin. Investig. 2010, 120, 4507–4519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, J.; Wu, D.C.; Chen, Y.; Paulson, R.F. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood 2015, 125, 1803–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, S.; Xiang, J.; Wu, D.C.; Fraser, J.W.; Ruan, B.; Cai, J.; Patterson, A.D.; Lai, Z.C.; Paulson, R.F. Gdf15 regulates murine stress erythroid progenitor proliferation and the development of the stress erythropoiesis niche. Blood Adv. 2019, 3, 2205–2217. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Rosenberg, P.S.; Day, T.; Menzel, S.; Giri, N.; Savage, S.A.; Thein, S.L. Genetic regulation of fetal haemoglobin in inherited bone marrow failure syndromes. Br. J. Haematol. 2013, 162, 542–546. [Google Scholar] [CrossRef]
- Heshusius, S.; Heideveld, E.; von Lindern, M.; van den Akker, E. CD14+ monocytes repress gamma globin expression at early stages of erythropoiesis. Sci. Rep. 2021, 11, 1507. [Google Scholar] [CrossRef]
- El Hoss, S.; Cochet, S.; Godard, A.; Yan, H.; Dussiot, M.; Frati, G.; Boutonnat-Faucher, B.; Laurance, S.; Renaud, O.; Joseph, L.; et al. Fetal hemoglobin rescues ineffective erythropoiesis in sickle cell disease. Haematologica 2021, 106, 2707–2719. [Google Scholar] [CrossRef]
- Hasegawa, S.; Rodgers, G.P.; Dwyer, N.; Noguchi, C.T.; Blanchette-Mackie, E.J.; Uyesaka, N.; Schechter, A.N.; Fibach, E. Sickling of nucleated erythroid precursors from patients with sickle cell anemia. Exp. Hematol. 1998, 26, 314–319. [Google Scholar]
- El Nemer, W.; Godard, A.; El Hoss, S. Ineffective erythropoiesis in sickle cell disease: New insights and future implications. Curr. Opin. Hematol. 2021, 28, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Ballantine, J.D.; Kwon, S.; Liem, R.I. Nucleated Red Blood Cells in Children With Sickle Cell Disease Hospitalized for Pain. J. Pediatr. Hematol. Oncol. 2019, 41, e487–e492. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; van Rooijen, N.; et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Kawane, K.; Koike, M.; Mori, Y.; Uchiyama, Y.; Nagata, S. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 2005, 437, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, M.M.; Kopsombut, P.; Bondurant, M.C.; Price, J.O.; Koury, M.J. Adherence to macrophages in erythroblastic islands enhances erythroblast proliferation and increases erythrocyte production by a different mechanism than erythropoietin. Blood 2008, 111, 1700–1708. [Google Scholar] [CrossRef] [Green Version]
- Heideveld, E.; Hampton-O’Neil, L.A.; Cross, S.J.; van Alphen, F.P.J.; van den Biggelaar, M.; Toye, A.M.; van den Akker, E. Glucocorticoids induce differentiation of monocytes towards macrophages that share functional and phenotypical aspects with erythroblastic island macrophages. Haematologica 2018, 103, 395–405. [Google Scholar] [CrossRef]
- Fabriek, B.O.; Polfliet, M.M.; Vloet, R.P.; van der Schors, R.C.; Ligtenberg, A.J.; Weaver, L.K.; Geest, C.; Matsuno, K.; Moestrup, S.K.; Dijkstra, C.D.; et al. The macrophage CD163 surface glycoprotein is an erythroblast adhesion receptor. Blood 2007, 109, 5223–5229. [Google Scholar] [CrossRef] [Green Version]
- Sadahira, Y.; Yoshino, T.; Monobe, Y. Very late activation antigen 4-vascular cell adhesion molecule 1 interaction is involved in the formation of erythroblastic islands. J. Exp. Med. 1995, 181, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Lo, A.; Short, S.A.; Mankelow, T.J.; Spring, F.; Parsons, S.F.; Yazdanbakhsh, K.; Mohandas, N.; Anstee, D.J.; Chasis, J.A. Targeted gene deletion demonstrates that the cell adhesion molecule ICAM-4 is critical for erythroblastic island formation. Blood 2006, 108, 2064–2071. [Google Scholar] [CrossRef] [Green Version]
- Hampton-O’Neil, L.A.; Severn, C.E.; Cross, S.J.; Gurung, S.; Nobes, C.D.; Toye, A.M. Ephrin/Eph receptor interaction facilitates macrophage recognition of differentiating human erythroblasts. Haematologica 2020, 105, 914–924. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Boulais, P.E.; Zhang, D.; Pinho, S.; Tanaka, M.; Frenette, P.S. Maea expressed by macrophages, but not erythroblasts, maintains postnatal murine bone marrow erythroblastic islands. Blood 2019, 133, 1222–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Spring, F.A.; Parsons, S.F.; Mankelow, T.J.; Peters, L.L.; Koury, M.J.; Mohandas, N.; Anstee, D.J.; Chasis, J.A. Novel secreted isoform of adhesion molecule ICAM-4: Potential regulator of membrane-associated ICAM-4 interactions. Blood 2003, 101, 1790–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seu, K.G.; Papoin, J.; Fessler, R.; Hom, J.; Huang, G.; Mohandas, N.; Blanc, L.; Kalfa, T.A. Unraveling Macrophage Heterogeneity in Erythroblastic Islands. Front. Immunol. 2017, 8, 1140. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.H.; McAllan, B.M.; Fraser, S.T. Scanning Electron Microscopy Reveals Two Distinct Classes of Erythroblastic Island Isolated from Adult Mammalian Bone Marrow. Microsc. Microanal. 2016, 22, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Ulyanova, T.; Jiang, Y.; Padilla, S.; Nakamoto, B.; Papayannopoulou, T. Combinatorial and distinct roles of alpha(5) and alpha(4) integrins in stress erythropoiesis in mice. Blood 2011, 117, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Tolu, S.S.; Wang, K.; Yan, Z.; Zhang, S.; Roberts, K.; Crouch, A.S.; Sebastian, G.; Chaitowitz, M.; Fornari, E.D.; Schwechter, E.M.; et al. Characterization of Hematopoiesis in Sickle Cell Disease by Prospective Isolation of Stem and Progenitor Cells. Cells 2020, 9, 2159. [Google Scholar] [CrossRef]
- Heideveld, E.; Masiello, F.; Marra, M.; Esteghamat, F.; Yagci, N.; von Lindern, M.; Migliaccio, A.R.; van den Akker, E. CD14+ cells from peripheral blood positively regulate hematopoietic stem and progenitor cell survival resulting in increased erythroid yield. Haematologica 2015, 100, 1396–1406. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhong, H.; Bao, W.; Mendelson, A.; An, X.; Shi, P.; Chou, S.T.; Manwani, D.; Yazdanbakhsh, K. Patrolling monocytes scavenge endothelial-adherent sickle RBCs: A novel mechanism of inhibition of vaso-occlusion in SCD. Blood 2019, 134, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Endoh, M.; Tan, D.Q.; Nakamura-Ishizu, A.; Takihara, Y.; Matsumura, T.; Suda, T. Mitochondria transfer from early stages of erythroblasts to their macrophage niche via tunnelling nanotubes. Br. J. Haematol. 2021, 193, 1260–1274. [Google Scholar] [CrossRef]
- Jagadeeswaran, R.; Vazquez, B.A.; Thiruppathi, M.; Ganesh, B.B.; Ibanez, V.; Cui, S.; Engel, J.D.; Diamond, A.M.; Molokie, R.E.; DeSimone, J.; et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp. Hematol. 2017, 50, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Moriconi, C.; Dzieciatkowska, M.; Roy, M.; D’Alessandro, A.; Roingeard, P.; Lee, J.Y.; Gibb, D.R.; Tredicine, M.; McGill, M.A.; Qiu, A.; et al. Retention of functional mitochondria in mature red blood cells from patients with sickle cell disease. Br. J. Haematol. 2022, 198, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Kawane, K.; Fukuyama, H.; Kondoh, G.; Takeda, J.; Ohsawa, Y.; Uchiyama, Y.; Nagata, S. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 2001, 292, 1546–1549. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.A.; Kusy, S.; Luong, R.; Wong, R.J.; Stevenson, D.K.; Contag, C.H. Heme oxygenase-1 deletion affects stress erythropoiesis. PLoS ONE 2011, 6, e20634. [Google Scholar] [CrossRef] [PubMed]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toobiak, S.; Shaklai, M.; Shaklai, N. Carbon monoxide induced erythroid differentiation of K562 cells mimics the central macrophage milieu in erythroblastic islands. PLoS ONE 2012, 7, e33940. [Google Scholar] [CrossRef] [Green Version]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [Green Version]
- Leimberg, M.J.; Prus, E.; Konijn, A.M.; Fibach, E. Macrophages function as a ferritin iron source for cultured human erythroid precursors. J. Cell. Biochem. 2008, 103, 1211–1218. [Google Scholar] [CrossRef]
- Nyffenegger, N.; Zennadi, R.; Kalleda, N.; Flace, A.; Ingoglia, G.; Buzzi, R.M.; Doucerain, C.; Buehler, P.W.; Schaer, D.J.; Durrenberger, F.; et al. The oral ferroportin inhibitor vamifeport improves hemodynamics in a mouse model of sickle cell disease. Blood 2022, 140, 769–781. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Zhao, H.; Zhang, H.; Xu, Y.; Wang, S.; Guo, X.; Huang, Y.; Zhang, S.; Han, Y.; et al. Identification and transcriptome analysis of erythroblastic island macrophages. Blood 2019, 134, 480–491. [Google Scholar] [CrossRef]
- Mukherjee, K.; Bieker, J.J. Transcriptional Control of Gene Expression and the Heterogeneous Cellular Identity of Erythroblastic Island Macrophages. Front. Genet. 2021, 12, 756028. [Google Scholar] [CrossRef]
- Lopez-Yrigoyen, M.; Yang, C.T.; Fidanza, A.; Cassetta, L.; Taylor, A.H.; McCahill, A.; Sellink, E.; von Lindern, M.; van den Akker, E.; Mountford, J.C.; et al. Genetic programming of macrophages generates an in vitro model for the human erythroid island niche. Nat. Commun. 2019, 10, 881. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, K.; Xue, L.; Planutis, A.; Gnanapragasam, M.N.; Chess, A.; Bieker, J.J. EKLF/KLF1 expression defines a unique macrophage subset during mouse erythropoiesis. eLife 2021, 10, e61070. [Google Scholar] [CrossRef]
- Porcu, S.; Manchinu, M.F.; Marongiu, M.F.; Sogos, V.; Poddie, D.; Asunis, I.; Porcu, L.; Marini, M.G.; Moi, P.; Cao, A.; et al. Klf1 affects DNase II-alpha expression in the central macrophage of a fetal liver erythroblastic island: A non-cell-autonomous role in definitive erythropoiesis. Mol. Cell. Biol. 2011, 31, 4144–4154. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Basatemur, G.; Scott, I.C.; Chiarugi, D.; Clement, M.; Harrison, J.; Jugdaohsingh, R.; Yu, X.; Newland, S.A.; Jolin, H.E.; et al. Interleukin-33 Signaling Controls the Development of Iron-Recycling Macrophages. Immunity 2020, 52, 782–793.e5. [Google Scholar] [CrossRef]
- Wei, J.; Zhao, J.; Schrott, V.; Zhang, Y.; Gladwin, M.; Bullock, G.; Zhao, Y. Red Blood Cells Store and Release Interleukin-33. J. Investig. Med. 2015, 63, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Miyagawa, S.; Kobayashi, M.; Konishi, N.; Sato, T.; Ueda, K. Insulin and insulin-like growth factor I support the proliferation of erythroid progenitor cells in bone marrow through the sharing of receptors. Br. J. Haematol. 2000, 109, 555–562. [Google Scholar] [CrossRef]
- Salgar, S.; Bolivar, B.E.; Flanagan, J.M.; Anum, S.J.; Bouchier-Hayes, L. The NLRP3 inflammasome fires up heme-induced inflammation in hemolytic conditions. Transl. Res. 2023, 252, 34–44. [Google Scholar] [CrossRef]
- Ramos, P.; Casu, C.; Gardenghi, S.; Breda, L.; Crielaard, B.J.; Guy, E.; Marongiu, M.F.; Gupta, R.; Levine, R.L.; Abdel-Wahab, O.; et al. Macrophages support pathological erythropoiesis in polycythemia vera and beta-thalassemia. Nat. Med. 2013, 19, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Romano, L.; Seu, K.G.; Papoin, J.; Muench, D.E.; Konstantinidis, D.; Olsson, A.; Schlum, K.; Chetal, K.; Chasis, J.A.; Mohandas, N.; et al. Erythroblastic islands foster granulopoiesis in parallel to terminal erythropoiesis. Blood 2022, 140, 1621–1634. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sesti-Costa, R.; Costa, F.F.; Conran, N. Role of Macrophages in Sickle Cell Disease Erythrophagocytosis and Erythropoiesis. Int. J. Mol. Sci. 2023, 24, 6333. https://doi.org/10.3390/ijms24076333
Sesti-Costa R, Costa FF, Conran N. Role of Macrophages in Sickle Cell Disease Erythrophagocytosis and Erythropoiesis. International Journal of Molecular Sciences. 2023; 24(7):6333. https://doi.org/10.3390/ijms24076333
Chicago/Turabian StyleSesti-Costa, Renata, Fernando F. Costa, and Nicola Conran. 2023. "Role of Macrophages in Sickle Cell Disease Erythrophagocytosis and Erythropoiesis" International Journal of Molecular Sciences 24, no. 7: 6333. https://doi.org/10.3390/ijms24076333