
Novel Insights in the Potential of Halogenated Polyketide–Peptide Molecules as Lead Compounds in Cancer Drug Discovery

 , ,
, ,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
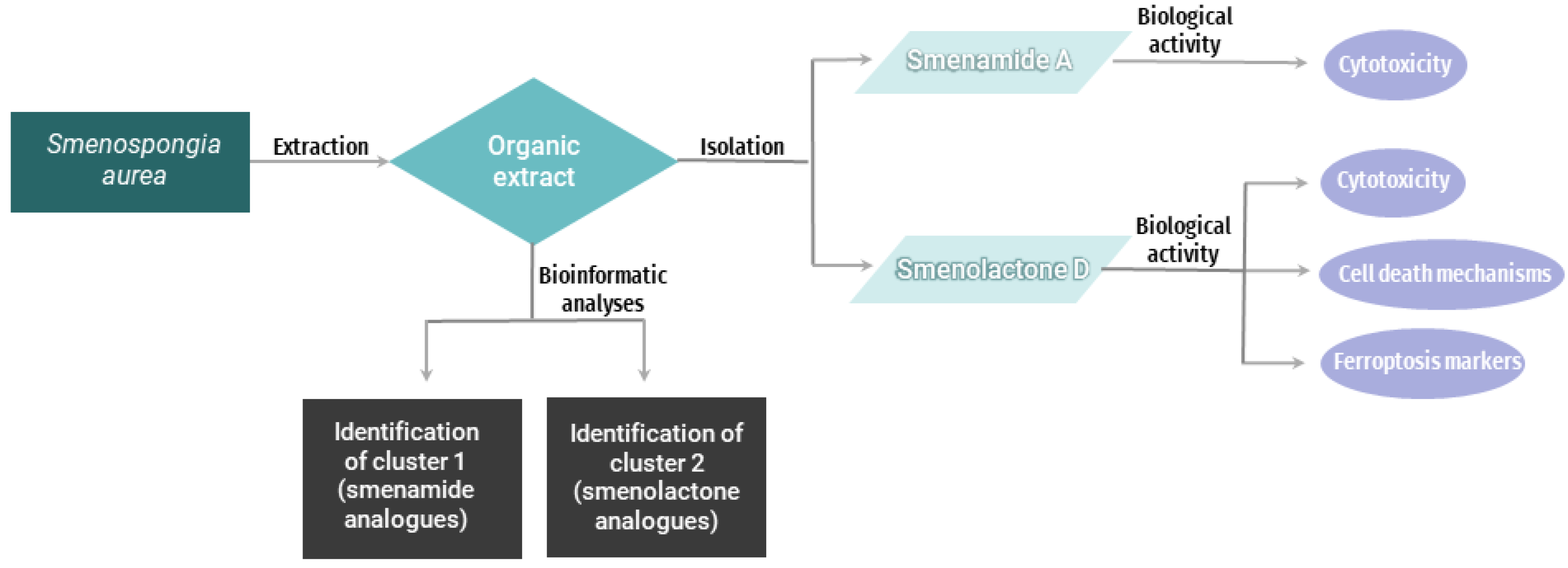
2. Results and Discussion
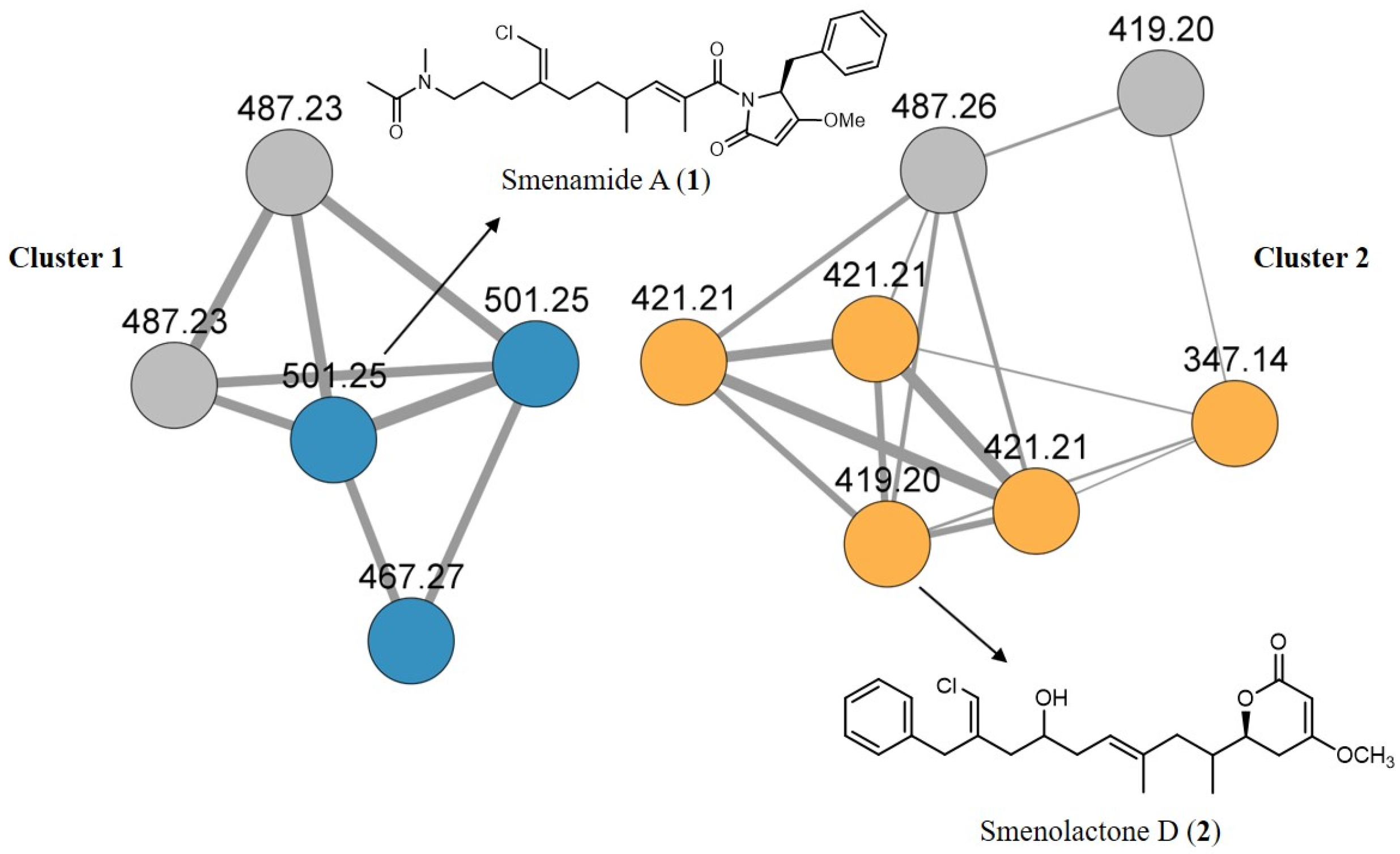
2.1. The Bioinformatic Analyses Highlight the Presence of Two Clusters of Novel Chlorinated Molecules in S. aurea Extract
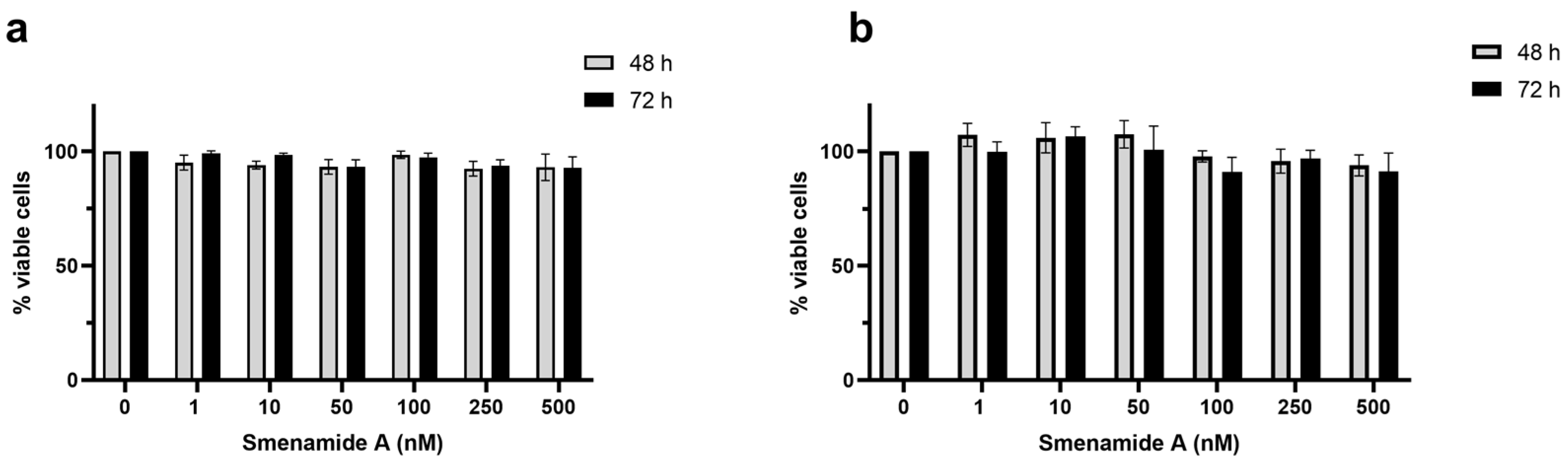
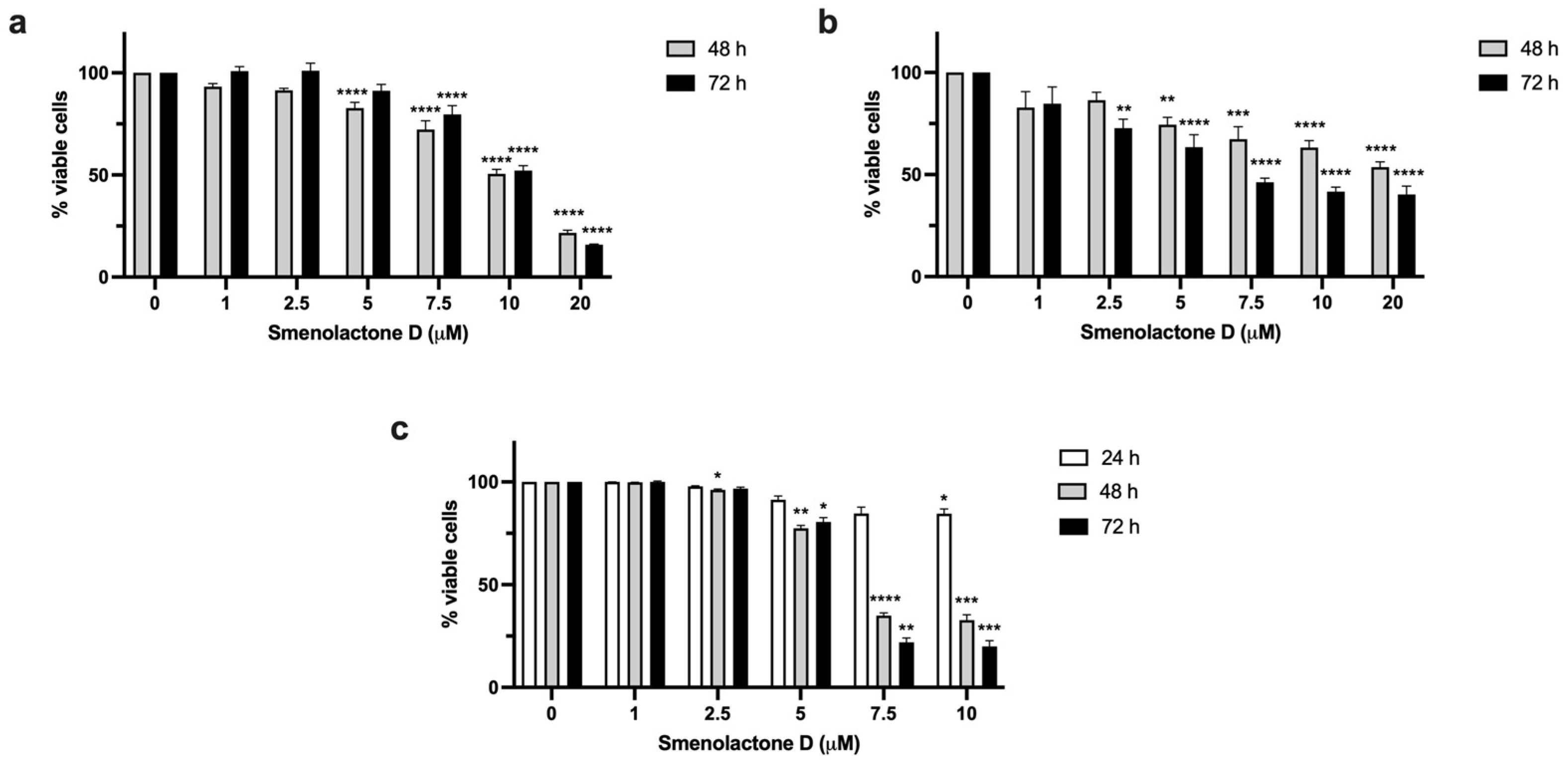
2.2. Smenolactone D Is Cytotoxic in a Panel of Human Cancer Cell Lines
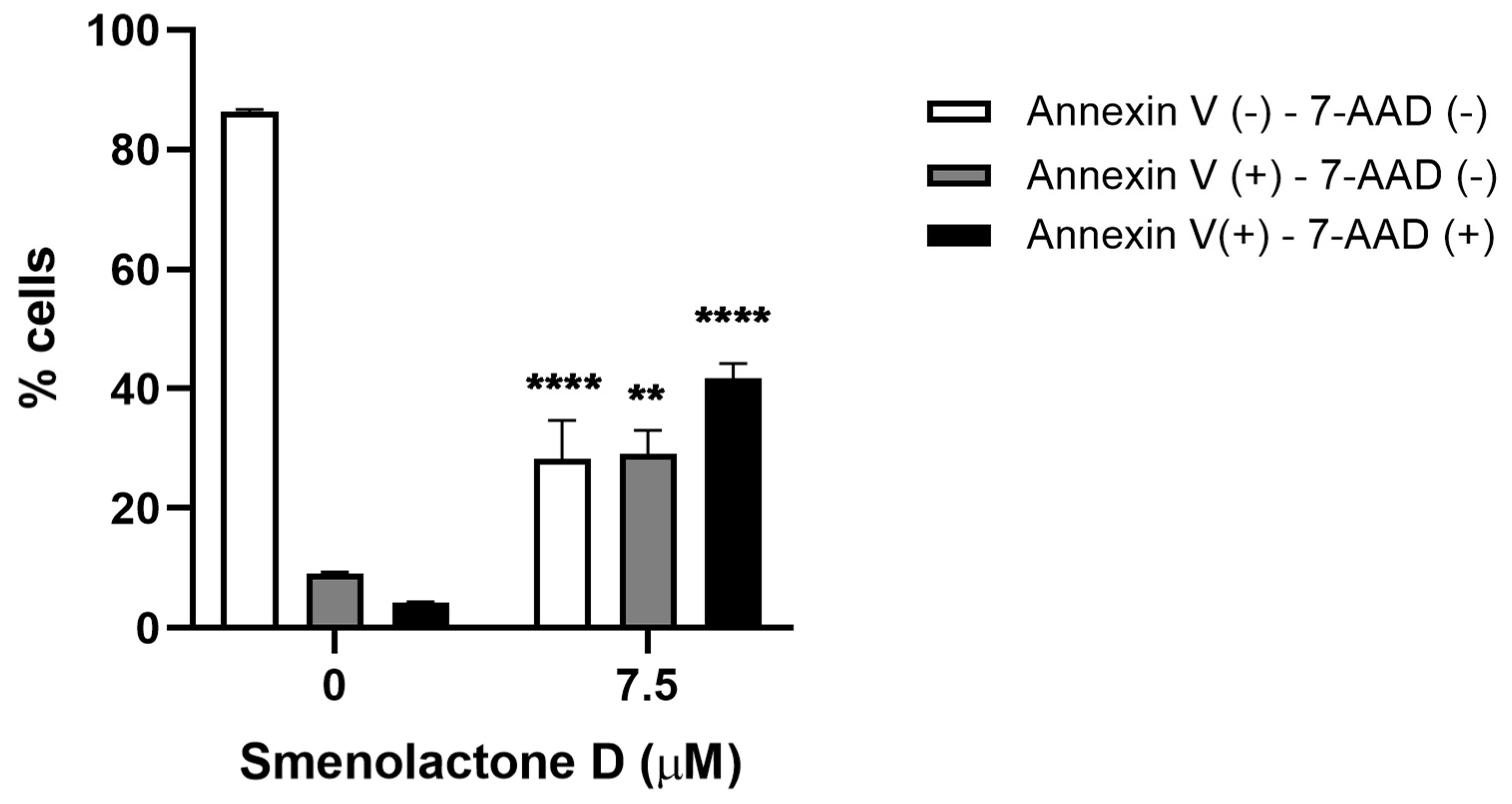
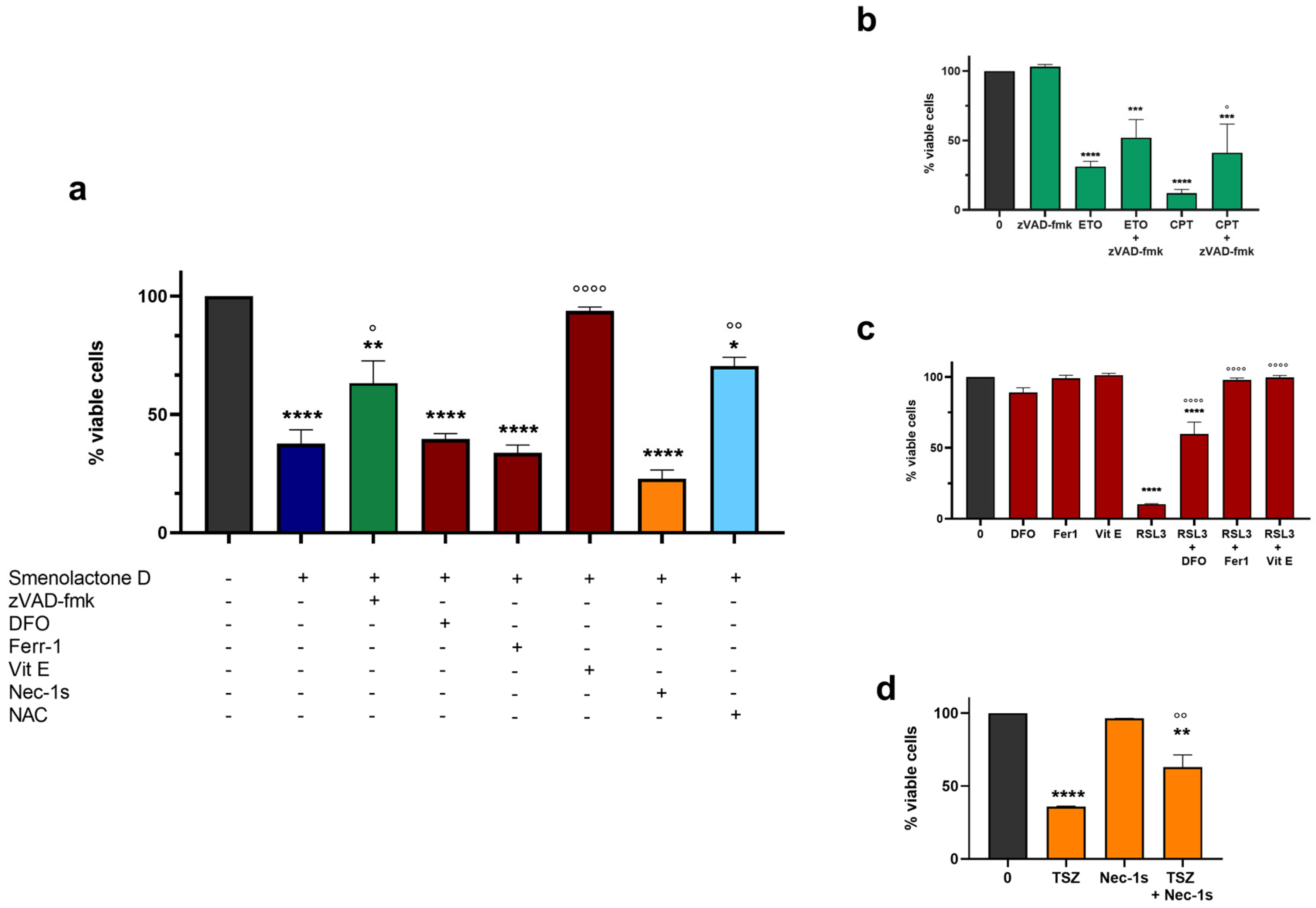
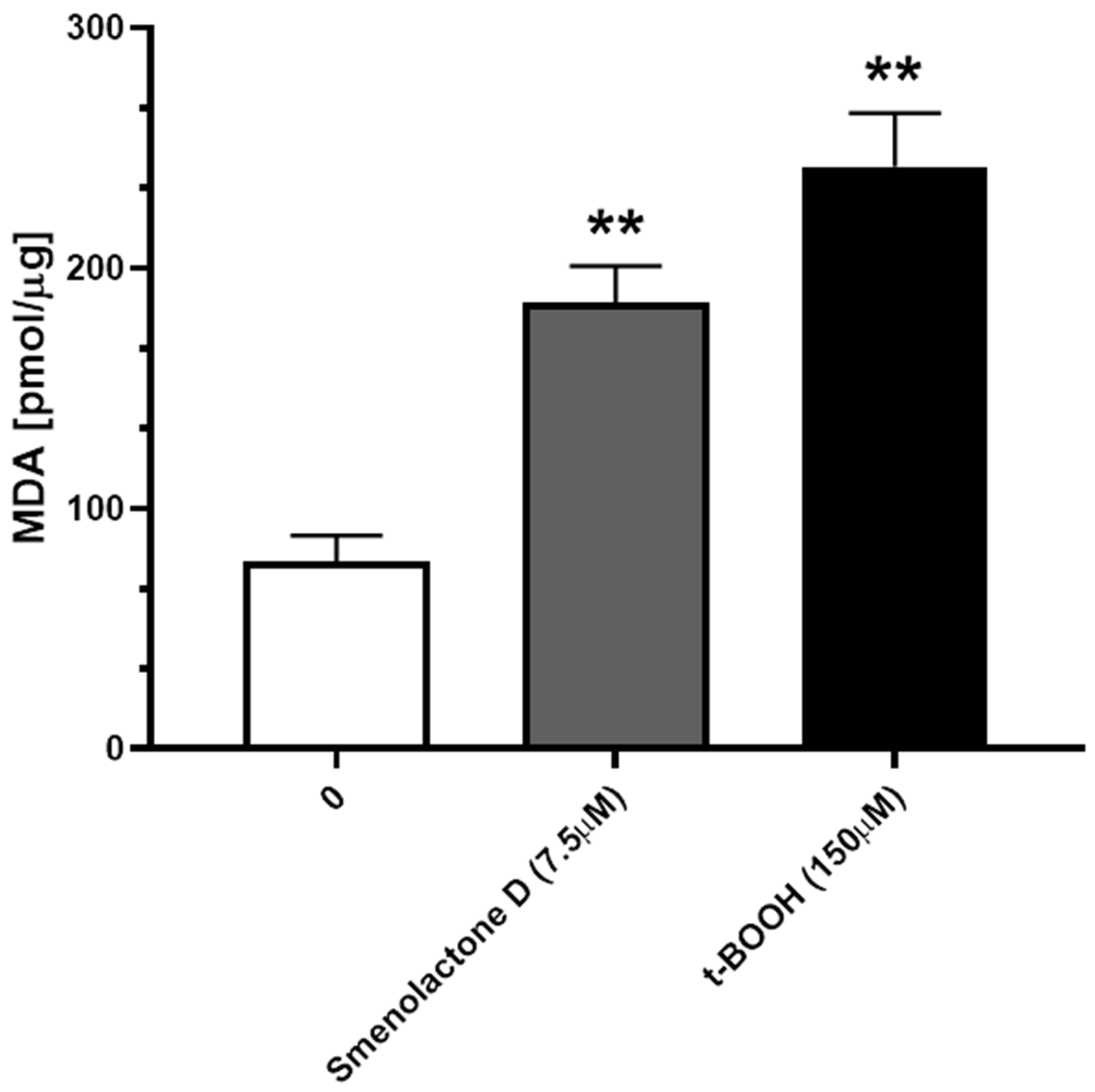
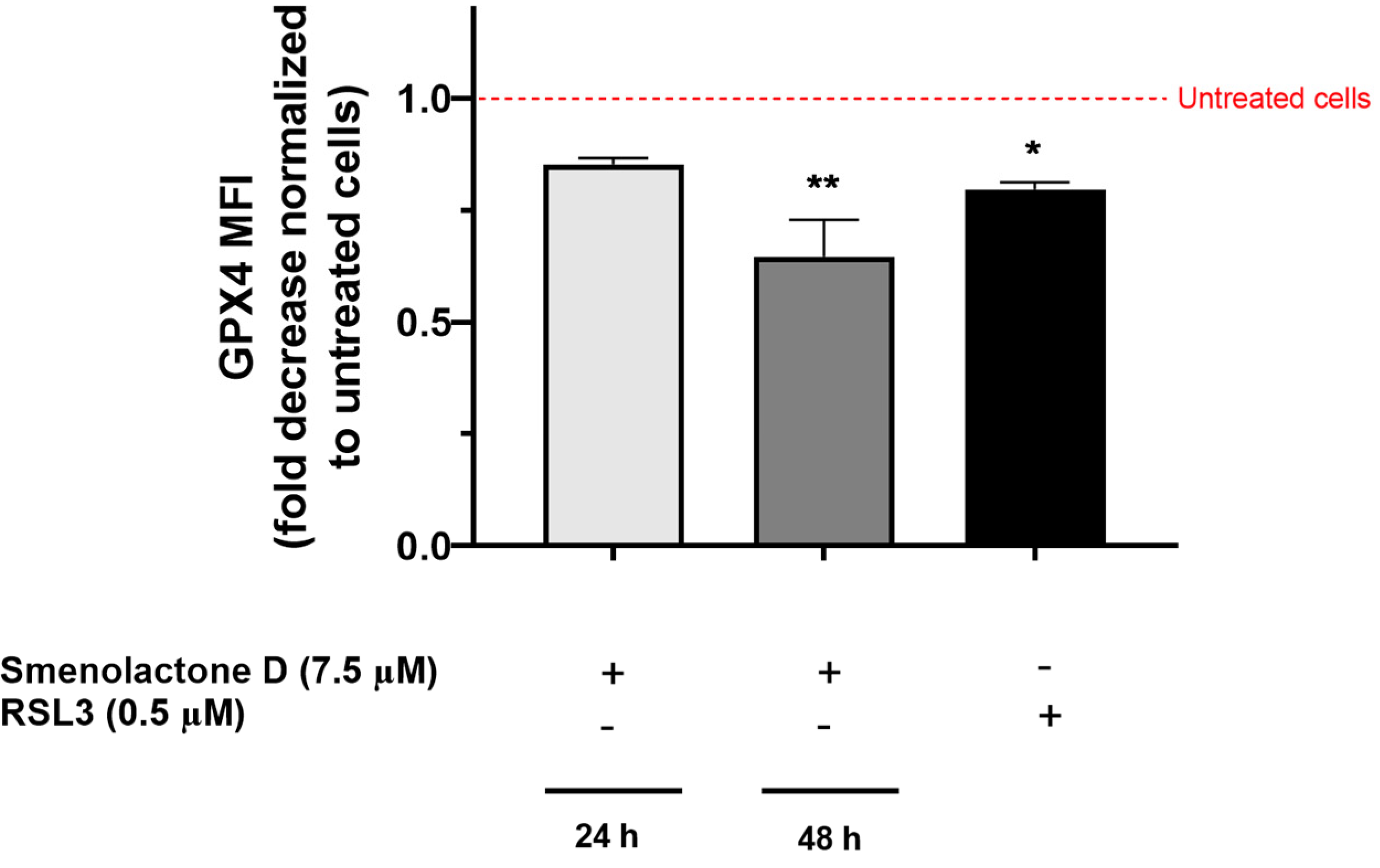
2.3. Smenolactone D Induces Apoptosis and Ferroptosis in Jurkat Cells
3. Materials and Methods
3.1. Isolation and Structural Elucidation
3.2. MZmine and Molecular Networking
3.3. Cell Culture and Treatment
3.4. Cell Viability Assays
3.5. Annexin-V Assay
3.6. Determination of Lipid Peroxidation
3.7. Evaluation of GPX4 Expression
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine Sponge Natural Products with Anticancer Po-tential: An Updated Review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef] [Green Version]
- Sutton, T.T.; Clark, M.R.; Dunn, D.C.; Halpin, P.N.; Rogers, A.D.; Guinotte, J.; Bograd, S.J.; Angel, M.V.; Perez, J.A.A.; Wishner, K.; et al. A global biogeographic classification of the mesopelagic zone. Deep. Sea Res. Part I: Oceanogr. Res. Pap. 2017, 126, 85–102. [Google Scholar] [CrossRef]
- Barzkar, N.; Jahromi, S.T.; Poorsaheli, H.B.; Vianello, F. Metabolites from Marine Microorganisms, Micro, and Macroalgae: Immense Scope for Pharmacology. Mar. Drugs 2019, 17, 464. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2023, 40, 275–325. [Google Scholar] [CrossRef] [PubMed]
- Varijakzhan, D.; Loh, J.-Y.; Yap, W.-S.; Yusoff, K.; Seboussi, R.; Lim, S.-H.; Lai, K.-S.; Chong, C.-M. Bioactive Compounds from Marine Sponges: Fundamentals and Applications. Mar. Drugs 2021, 19, 246. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.N.A.; Van Soest, R.W.M. (Eds.) Systema Porifera: A Guide to the Classification of Sponges; Springer: Boston, MA, USA, 2002; pp. 1–7. ISBN 978-0-306-47260-2. [Google Scholar]
- Priyanto, J.A.; Purwaningtyas, W.E.; Ferantika, D.; Tohir, D.; Astuti, R.I.; Wahyudi, A.T. Natural extracts derived from marine sponge-associated bacteria with diverse nonribosomal peptide synthetase genes have anticancer activities. J. Appl. Pharm. Sci. 2022, 12, 54–63. [Google Scholar] [CrossRef]
- Chu, M.-J.; Li, M.; Ma, H.; Li, P.-L.; Li, G.-Q. Secondary metabolites from marine sponges of the genus Agelas: A comprehensive update insight on structural diversity and bioactivity. RSC Adv. 2022, 12, 7789–7820. [Google Scholar] [CrossRef]
- Ercolano, G.; De Cicco, P.; Ianaro, A. New Drugs from the Sea: Pro-Apoptotic Activity of Sponges and Algae Derived Compounds. Mar. Drugs 2019, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- Mioso, R.; Marante, F.J.T.; Bezerra, R.D.S.; Borges, F.V.P.; Santos, B.V.D.O.; De Laguna, I.H.B. Cytotoxic Compounds Derived from Marine Sponges. A Review (2010–2012). Molecules 2017, 22, 208. [Google Scholar] [CrossRef] [Green Version]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine Sponge Derived Natural Products between 2001 and 2010: Trends and Opportunities for Discovery of Bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [Green Version]
- Costantino, V.; Fattorusso, E.; Mangoni, A.; Di Rosa, M.; Ianaro, A. Glycolipids from Sponges. 6.1 Plakoside A and B, Two Unique Prenylated Glycosphingolipids with Immunosuppressive Activity from the Marine Sponge Plakortis simplex. J. Am. Chem. Soc. 1997, 119, 12465–12470. [Google Scholar] [CrossRef]
- Esposito, G.; Bourguet-Kondracki, M.-L.; Mai, L.H.; Longeon, A.; Teta, R.; Meijer, L.; Van Soest, R.; Mangoni, A.; Costantino, V. Chloromethylhalicyclamine B, a Marine-Derived Protein Kinase CK1δ/ε Inhibitor. J. Nat. Prod. 2016, 79, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Della Sala, G.; Teta, R.; Caso, A.; Bourguet-Kondracki, M.-L.; Pawlik, J.R.; Mangoni, A.; Costantino, V. Chlorinated Thiazole-Containing Polyketide-Peptides from the Caribbean Sponge Smenospongia conulosa: Structure Elucidation on Microgram Scale. Eur. J. Org. Chem. 2016, 2016, 2871–2875. [Google Scholar] [CrossRef]
- Karanam, G.; Arumugam, M.K.; Natesh, N.S. Anticancer Effect of Marine Sponge-Associated Bacillus pumilus AMK1 Derived Dipeptide Cyclo (-Pro-Tyr) in Human Liver Cancer Cell Line Through Apoptosis and G2/M Phase Arrest. Int. J. Pept. Res. Ther. 2019, 26, 445–457. [Google Scholar] [CrossRef]
- Natesh, N.S.; Arumugam, M.; Karanam, G. Apoptotic role of marine sponge symbiont Bacillus subtilis NMK17 through the activation of caspase-3 in human breast cancer cell line. Mol. Biol. Rep. 2018, 45, 2641–2651. [Google Scholar] [CrossRef]
- Dhaneesha, M.; Hasin, O.; Sivakumar, K.C.; Ravinesh, R.; Naman, C.B.; Carmeli, S.; Sajeevan, T.P. DNA Binding and Molecular Dynamic Studies of Polycyclic Tetramate Macrolactams (PTM) with Potential Anticancer Activity Isolated from a Sponge-Associated Streptomyces zhaozhouensis subsp. mycale subsp. nov. Mar. Biotechnol. 2018, 21, 124–137. [Google Scholar] [CrossRef]
- Chang, W.-T.; Bow, Y.-D.; Fu, P.-J.; Li, C.-Y.; Wu, C.-Y.; Chang, Y.-H.; Teng, Y.-N.; Li, R.-N.; Lu, M.-C.; Liu, Y.-C.; et al. A Marine Terpenoid, Heteronemin, Induces Both the Apoptosis and Ferroptosis of Hepatocellular Carcinoma Cells and Involves the ROS and MAPK Pathways. Oxidative Med. Cell. Longev. 2021, 2021, 7689045. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Xu, G. Fascaplysin Induces Apoptosis and Ferroptosis, and Enhances Anti-PD-1 Immunotherapy in Non-Small Cell Lung Cancer (NSCLC) by Promoting PD-L1 Expression. Int. J. Mol. Sci. 2022, 23, 13774. [Google Scholar] [CrossRef]
- Riccio, G.; Nuzzo, G.; Zazo, G.; Coppola, D.; Senese, G.; Romano, L.; Costantini, M.; Ruocco, N.; Bertolino, M.; Fontana, A.; et al. Bioactivity Screening of Antarctic Sponges Reveals Anticancer Activity and Potential Cell Death via Ferroptosis by Mycalols. Mar. Drugs 2021, 19, 459. [Google Scholar] [CrossRef]
- Teta, R.; Irollo, E.; Della Sala, G.; Pirozzi, G.; Mangoni, A.; Costantino, V. Smenamides A and B, Chlorinated Peptide/Polyketide Hybrids Containing a Dolapyrrolidinone Unit from the Caribbean Sponge Smenospongia aurea. Evaluation of Their Role as Leads in Antitumor Drug Research. Mar. Drugs 2013, 11, 4451–4463. [Google Scholar] [CrossRef]
- Esposito, G.; Teta, R.; Miceli, R.; Ceccarelli, L.S.; Della Sala, G.; Camerlingo, R.; Irollo, E.; Mangoni, A.; Pirozzi, G.; Costantino, V. Isolation and Assessment of the in Vitro Anti-Tumor Activity of Smenothiazole A and B, Chlorinated Thiazole-Containing Peptide/Polyketides from the Caribbean Sponge, Smenospongia aurea. Mar. Drugs 2015, 13, 444–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teta, R.; Della Sala, G.; Esposito, G.; Via, C.W.; Mazzoccoli, C.; Piccoli, C.; Bertin, M.J.; Costantino, V.; Mangoni, A. A joint molecular networking study of a Smenospongia sponge and a cyanobacterial bloom revealed new antiproliferative chlorinated polyketides. Org. Chem. Front. 2019, 6, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Caso, A.; Esposito, G.; Della Sala, G.; Pawlik, J.R.; Teta, R.; Mangoni, A.; Costantino, V. Fast Detection of Two Smenamide Family Members Using Molecular Networking. Mar. Drugs 2019, 17, 618. [Google Scholar] [CrossRef] [Green Version]
- Caso, A.; Mangoni, A.; Piccialli, G.; Costantino, V.; Piccialli, V. Studies toward the Synthesis of Smenamide A, an Antiproliferative Metabolite from Smenospongia aurea: Total Synthesis of ent-Smenamide A and 16-epi-Smenamide A. ACS Omega 2017, 2, 1477–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caso, A.; Laurenzana, I.; Lamorte, D.; Trino, S.; Esposito, G.; Piccialli, V.; Costantino, V. Smenamide A Analogues. Synthesis and Biological Activity on Multiple Myeloma Cells. Mar. Drugs 2018, 16, 206. [Google Scholar] [CrossRef] [Green Version]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Demchak, B.; Ideker, T. Cytoscape Tools for the Web Age: D3.Js and Cytoscape.Js Exporters . F1000Research 2014, 3, 143. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, T.P.; Freeman, C.J.; Paul, V.J.; Agarwal, V.; Garg, N. Mass Spectrometry-Based Integration and Expansion of the Chemical Diversity Harbored Within a Marine Sponge. J. Am. Soc. Mass Spectrom. 2019, 30, 1373–1384. [Google Scholar] [CrossRef]
- Shlomovitz, I.; Speir, M.; Gerlic, M. Flipping the dogma—Phosphatidylserine in non-apoptotic cell death. Cell Commun. Signal. 2019, 17, 139. [Google Scholar] [CrossRef] [Green Version]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.; Catanzaro, E.; Fimognari, C. Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer. Cancers 2021, 13, 304. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Central Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Hinman, A.; Holst, C.R.; Latham, J.C.; Bruegger, J.J.; Ulas, G.; McCusker, K.P.; Amagata, A.; Davis, D.; Hoff, K.G.; Kahn-Kirby, A.H.; et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS ONE 2018, 13, e0201369. [Google Scholar] [CrossRef] [Green Version]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Central Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2018, 133, 144–152. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Yu, X.; Long, Y.C. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci. Rep. 2016, 6, 30033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horchani, M.; Della Sala, G.; Caso, A.; D’Aria, F.; Esposito, G.; Laurenzana, I.; Giancola, C.; Costantino, V.; Jannet, H.; Romdhane, A. Molecular Docking and Biophysical Studies for Antiproliferative Assessment of Synthetic Pyrazolo-Pyrimidinones Tethered with Hydrazide-Hydrazones. Int. J. Mol. Sci. 2021, 22, 2742. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, M.; Cocchi, V.; Novaković, A.; Karaman, M.; Sakač, M.; Mandić, A.; Pojić, M.; Barbalace, M.C.; Angeloni, C.; Hrelia, P.; et al. Meripilus giganteus ethanolic extract exhibits pro-apoptotic and anti-proliferative effects in leukemic cell lines. BMC Complement. Altern. Med. 2018, 18, 300. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Lee, D.H.; Lee, Y.S.; Jo, M.J.; Jeong, Y.A.; Kwon, W.T.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Molecular crosstalk between ferroptosis and apoptosis: Emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 2017, 8, 115164–115178. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (µM) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Jurkat | 37. 43 | 7.08 | 6.39 |
| A-431 | n.c. | 10.68 | 10.76 |
| MCF-7 | n.c. | 26.58 | 8.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellicioni, V.; Esposito, G.; Greco, G.; Cruz-Chamorro, I.; Ferrini, F.; Sestili, P.; Teta, R.; Fimognari, C.; Costantino, V. Novel Insights in the Potential of Halogenated Polyketide–Peptide Molecules as Lead Compounds in Cancer Drug Discovery. Int. J. Mol. Sci. 2023, 24, 6208. https://doi.org/10.3390/ijms24076208
Pellicioni V, Esposito G, Greco G, Cruz-Chamorro I, Ferrini F, Sestili P, Teta R, Fimognari C, Costantino V. Novel Insights in the Potential of Halogenated Polyketide–Peptide Molecules as Lead Compounds in Cancer Drug Discovery. International Journal of Molecular Sciences. 2023; 24(7):6208. https://doi.org/10.3390/ijms24076208
Chicago/Turabian StylePellicioni, Valentina, Germana Esposito, Giulia Greco, Ivan Cruz-Chamorro, Fabio Ferrini, Piero Sestili, Roberta Teta, Carmela Fimognari, and Valeria Costantino. 2023. "Novel Insights in the Potential of Halogenated Polyketide–Peptide Molecules as Lead Compounds in Cancer Drug Discovery" International Journal of Molecular Sciences 24, no. 7: 6208. https://doi.org/10.3390/ijms24076208