
The Role of Hydrogen Sulfide in Regulation of Cell Death following Neurotrauma and Related Neurodegenerative and Psychiatric Diseases
 , ,
, ,
Abstract
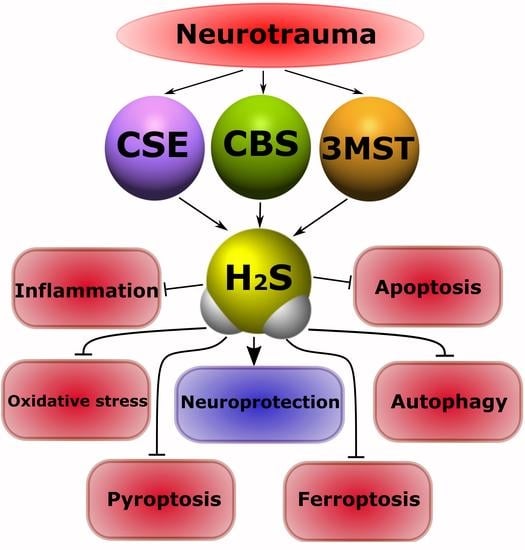
:
1. Introduction
2. Materials and Methods
2.1. Finding Sources
2.2. Study Quality Assessment
2.3. Conceptualization and Synthesis of the Received Data
3. Classification and Molecular Mechanisms of Neurotrauma
4. The Role of Neurotrauma in the Pathogenesis of Mental Disorders
4.1. Traumatic Brain Injury
4.2. Spinal Cord Injury
4.3. Trauma of the Peripheral Nervous System
5. Metabolism and Functions of H2S
5.1. Biosynthesis of H2S and Its Deposition
5.2. Catabolism of H2S
5.3. Various Biological Effects of Endogenous H2S
6. Endogenous and Exogenous H2S in Neurotrauma
6.1. Endogenous H2S Levels in Neurotrauma
6.2. Exogenous H2S: Between Neuroprotection and Neurodegeneration
7. The Role of H2S in Cell Death in Neurotrauma
7.1. Participation of H2S in Oxidative Stress
7.2. Modulation of the H2S Activity of NMDARs and Intracellular Ca2+ Homeostasis
7.3. Anti- and Pro-Inflammatory Effects of H2S
7.4. The Effect of H2S on the Level of Neurotrophic Factors
7.5. Effects of H2S on the Blood-Brain Barrier and Cerebral Edema
7.6. The Role of H2S in Remyelination Processes
7.7. H2S-Associated Anti- and Pro-Apoptotic Signaling Mechanisms
7.8. H2S-Associated Mechanisms of Autophagy
7.9. H2S-Associated Mechanisms of Ferroptosis
7.10. H2S-Associated Mechanisms of Pyroptosis
8. The Role of H2S in Mental Disorders and Neurodegenerative Diseases
8.1. Cognitive Impairment
8.2. Encephalopathy
8.3. Depression and Anxiety Disorders
8.4. Epilepsy
8.5. Chronic Pain
9. Neurodegenerative Diseases
9.1. Alzheimer’s Disease
9.2. Parkinson’s Disease
10. Therapeutic Approaches Using H2S as a Neuroprotector
11. Conclusions
12. Main Conclusions
12.1. Effects of H2S in Neurons and Glial Cells in Injuries of the Central and Peripheral Nervous System
- H2S reduces oxidative stress via uptake of ROS and increased levels of GSH, Trx-1, COD, CAT and GPx, and p66Shcis. However, high levels of H2S can induce oxidative stress;
- H2S can modulate NMDAR activity through cAMP\PKA, and H2S can directly interact with cysteine residues of NMDAR subunits, modifying them by S-sulfhydration. H2S can activate slow L-type Ca2+ channels, as well as fast T-type Ca-type CaV 3.2 channels;
- H2S can inhibit inflammation by reducing ROS, NF-κB, leukocyte adhesion to the endothelium, TNF, IL-1β, and NLRP3/caspase-1/GSDMD. However, excessive production of ROS can cause the oxidation of H2S to form sulfite, leading to leukocyte adhesion and neurophilic activation. H2S may increase inflammation through the inhibition of caspase-3 and the activation of p38 protein kinase;
- H2S can increase the level of the neurotrophic factors GDNF, NGF, BDNF, and VEGF;
- H2S is involved in maintaining the integrity of the blood-brain barrier via inhibition of PKC-α, β I, β II and δ, and activation of PKC-ε and increased levels of Claudin-5, Occlusin and ZO-1, as well as in the suppression of the expression of AQP4 on astrocytes and the inhibition of MMP-9 and NOS level modulation;
- H2S can lead to both the destruction of the myelin sheath and the processes of remyelination and the repair of axons by activating the PI3K/AKT/mTOR signaling pathway;
- H2S can regulate apoptosis by acting as an anti- or pro-oxidant, or by interacting with proteins involved in apoptotic signaling. H2S can reduce the expression of p53, caspase-3, Bax, NF-κB p65, NOX4, iNOS, COX-2, and increase Bcl-2, ncRNA CasC7 and the phosphorylation of Akt;
- H2S can reduce autophagy by modeling the PI3K/Akt /Nrf2 and ROS signaling pathways. H2S may enhance autophagy by increasing miR-30c, Beclin 1, and LC3 levels;
- H2S can inhibit ferroptosis by reducing the level of ROS, the LDH accumulation of Fe2+, and by increasing the antioxidant enzyme GSH, as well as through the activation of NRF2/KEAP1 and AMPK to phosphorylate p62;
- H2S can reduce pyroptosis by inhibiting NOD-, LRR-, NLRP 3, GSDMD, caspase-1, and ASC.
12.2. Effects of H2S in Neurons and Glial Cells in Mental Disorders:
- H2S may reduce cognitive impairment through the inhibition of endoplasmic reticulum stress, caspase-12, CHOP, and TLR4 /NF-κB; a decrease in the level of TNF-α, IL-1β and IL-6, Sirt1, ROS, LP, CPR78, CHOP, caspase-12, Bax; and an increase in synapsin-1 and PSD-95, Bcl-2, HO-2, M2-RK, LDHA, and PDK in the hippocampus. H2S modulates the level of catecholamines and reduces the level of apoptosis;
- H2S may reduce apoptosis in encephalopathy through Nrf2/ARE activation. However, a high content of H2S reduces the activity of CS, Aco, and CK, and enhances LP in the brain. H2S reduces neuroinflammation through a decrease in the level of IL-1β, IL-6, TNF-α, and also restores the level of SIRT1 and phosphorylation mTOR and NF-κB p65 for encephalopathy;
- H2S has an antidepressant and anxiolytic effect through an increase in the expression of Sirt1, Sirt6, IL-4, and IL-10, the activation of PI3K/p-Akt, and a decrease in the level of IL-1β, IL-6, TNF-α, Fe2+ deposition, ROS, NOS2, H3K9ac, Notch1, Beclin 1, and GRP78. H2S prevents the loss of dendritic spines and increases the level of mTORC1, TrkB PSD-95, synaptophysin, and the AMPA receptor GluR1/2 subunit in depression and anxiety disorders;
- H2S reduces epileptic seizures. H2S reduces the level of aquaporin 4, 1β, IL-6, TNF-α, and c-fos, and increases the expression of PKC, Kir6.2 and SUR1, GABABR1 and GABABR2. However, H2S can also lead to seizures via activation of NMDARs and AMPARs;
- H2S can reduce neuropathic pain through the inhibition of the expression of microglial activation, a decrease in the level of apoptosis, PI3K, TNF-α, IL-1β, and IL-6, and increased levels of Nrf2, HO-1, NQO1 and GSTM1.
12.3. Effects of H2S in Neurons and Glial Cells in Neurodegenerative Diseases:
- H2S may reduce the progression of Alzheimer’s disease. H2S inhibits hyperphosphorylation Tau; reduces amyloid β-plaques in the hippocampal cortex, neuroinflammation, oxidative stress, JNK expression, p38, TNF-α, IL-6, IL-1β, miR-155, pAkt, Bax, caspase-3, Aβ1-40, Aβ42, the phosphorylation of p38 MAPK, p65 NF-κB, and BACE1; and increases synaptophysin levels, PSD-95, Bcl-2, Sirt1, and Nrf2;
- H2S may reduce the progression of Parkinson’s disease. H2S reduces the death of dopaminergic neurons in the SN; increases the expression of Nrf-2, dopamine, and GSH; activates BDNF/TrkB, and miR-135a-5p; and inhibits ROS/NO, LP, and ROCK2.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furlan, J.C.; Gulasingam, S.; Craven, B.C. Epidemiology of War-Related Spinal Cord Injury Among Combatants: A Systematic Review. Glob. Spine J. 2019, 9, 545–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowitz, D.; Grant, G.E. Translational Research in Traumatic Brain Injury; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Rodkin, S.V.; Dzreyan, V.A.; Demyanenko, S.V.; Uzdensky, A.B. The Role of p53-Dependent Signaling Pathways in Survival and Death of Neurons and Glial Cells after Peripheral Nerve Injury. Biochem. (Moscow) Suppl. Ser. A Membr. Cell Biol. 2021, 15, 334–347. [Google Scholar] [CrossRef]
- Karimi, S.A.; Hosseinmardi, N.; Janahmadi, M.; Sayyah, M.; Hajisoltani, R. The protective effect of hydrogen sulfide (H2S) on traumatic brain injury (TBI) induced memory deficits in rats. Brain Res. Bull. 2017, 134, 177–182. [Google Scholar] [CrossRef]
- Calvillo, M.; Irimia, A. Neuroimaging and Psychometric Assessment of Mild Cognitive Impairment After Traumatic Brain Injury. Front. Psychol. 2020, 11, 1423. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Nightingale, T.E.; Krassioukov, A.V. The Blood Pressure Pendulum following Spinal Cord Injury: Implications for Vascular Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 2464. [Google Scholar] [CrossRef] [Green Version]
- Nightingale, T.E.; Zheng, M.M.Z.; Sachdeva, R.; Phillips, A.A.; Krassioukov, A.V. Diverse cognitive impairment after spinal cord injury is associated with orthostatic hypotension symptom burden. Physiol. Behav. 2020, 213, 112742. [Google Scholar] [CrossRef]
- Chiaravalloti, N.D.; Weber, E.; Wylie, G.; Dyson-Hudson, T.; Wecht, J.M. Patterns of cognitive deficits in persons with spinal cord injury as compared with both age-matched and older individuals without spinal cord injury. J. Spinal Cord Med. 2020, 43, 88–97. [Google Scholar] [CrossRef]
- Gupta, R.; Sen, N. Traumatic brain injury: A risk factor for neurodegenerative diseases. Rev. Neurosci. 2016, 27, 93–100. [Google Scholar] [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef]
- Yeh, T.-S.; Huang, Y.-P.; Wang, H.-I.; Pan, S.-L. Spinal cord injury and Parkinson’s disease: A population-based, propensity score-matched, longitudinal follow-up study. Spinal Cord 2016, 54, 1215–1219. [Google Scholar] [CrossRef]
- Xu, X.-J.; Yang, M.-S.; Zhang, B.; Niu, F.; Dong, J.-Q.; Liu, B.-Y. Glucose metabolism: A link between traumatic brain injury and Alzheimer’s disease. Chin. J. Traumatol. 2021, 24, 5–10. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Kunik, M.E.; Yu, H.; Lin, H.L.; Richardson, P.A.; Moore, S.; Sarwar, A.I.; Marsh, L.; Jorge, R.E. Post-Traumatic Stress Disorder is Associated with further Increased Parkinson’s Disease Risk in Veterans with Traumatic Brain Injury. Ann. Neurol. 2020, 88, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.B.; Fan, L.; Levasseur, R.A.; Faden, A.I. Sustained Sensory/Motor and Cognitive Deficits With Neuronal Apoptosis Following Controlled Cortical Impact Brain Injury in the Mouse. J. Neurotrauma 1998, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-N.; Chen, L.; Luo, B.; Li, X.; Wang, C.-Y.; Zou, W.; Zhang, P.; You, Y.; Tang, X.-Q. Hydrogen sulfide attenuates chronic restrain stress-induced cognitive impairment by upreglulation of Sirt1 in hippocampus. Oncotarget 2017, 8, 100396–100410. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.-H.; Chu, M.; Wang, Y.-P. Up-Regulation of Trem2 Inhibits Hippocampal Neuronal Apoptosis and Alleviates Oxidative Stress in Epilepsy via the PI3K/Akt Pathway in Mice. Neurosci. Bull. 2019, 35, 471–485. [Google Scholar] [CrossRef]
- Saleem, S. Apoptosis, Autophagy, Necrosis and Their Multi Galore Crosstalk in Neurodegeneration. Neuroscience 2021, 469, 162–174. [Google Scholar] [CrossRef]
- Ozdamar Unal, G.; Demirdas, A.; Nazıroglu, M.; Ovey, I.S. Agomelatine attenuates calcium signaling and apoptosis via the inhibition of TRPV1 channel in the hippocampal neurons of rats with chronic mild stress depression model. Behav. Brain Res. 2022, 434, 114033. [Google Scholar] [CrossRef]
- Zhang, H.; Li, N.; Li, Z.; Li, Y.; Yu, Y.; Zhang, L. The Involvement of Caspases in Neuroinflammation and Neuronal Apoptosis in Chronic Pain and Potential Therapeutic Targets. Front. Pharmacol. 2022, 13, 1561. [Google Scholar] [CrossRef]
- Mumtaz, S.; Rana, J.N.; Choi, E.H.; Han, I. Microwave Radiation and the Brain: Mechanisms, Current Status, and Future Prospects. Int. J. Mol. Sci. 2022, 23, 9288. [Google Scholar] [CrossRef]
- Pérez-González, A.; Castañeda-Arriaga, R.; Guzmán-López, E.G.; Hernández-Ayala, L.F.; Galano, A. Chalcone Derivatives with a High Potential as Multifunctional Antioxidant Neuroprotectors. ACS Omega 2022, 7, 38254–38268. [Google Scholar] [CrossRef]
- Ali, A.; Wang, Y.; Wu, L.; Yang, G. Gasotransmitter signaling in energy homeostasis and metabolic disorders. Free Radic. Res. 2021, 55, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, K.D.; Maassen, H.; van Dijk, P.R.; Henning, R.H.; van Goor, H.; Hillebrands, J.-L. Gasotransmitters in health and disease: A mitochondria-centered view. Curr. Opin. Pharmacol. 2019, 45, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sen, N. Functional and Molecular Insights of Hydrogen Sulfide Signaling and Protein Sulfhydration. J. Mol. Biol. 2017, 429, 543–561. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, S.; Shan, H.; Zhang, M. Biologic Effect of Hydrogen Sulfide and Its Role in Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2020, 2020, 7301615. [Google Scholar] [CrossRef] [PubMed]
- Pomierny, B.; Krzyżanowska, W.; Jurczyk, J.; Skórkowska, A.; Strach, B.; Szafarz, M.; Przejczowska-Pomierny, K.; Torregrossa, R.; Whiteman, M.; Marcinkowska, M.; et al. The Slow-Releasing and Mitochondria-Targeted Hydrogen Sulfide (H2S) Delivery Molecule AP39 Induces Brain Tolerance to Ischemia. Int. J. Mol. Sci. 2021, 22, 7816. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Wu, N.; Wang, Q.; Yang, L. Endogenous hydrogen sulfide alleviates methotrexate-induced cognitive impairment by attenuating endoplasmic reticulum stress-induced apoptosis via CHOP and caspase-12. Fundam. Clin. Pharmacol. 2020, 34, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488. [Google Scholar] [CrossRef]
- Sun, J.; Li, X.; Gu, X.; Du, H.; Zhang, G.; Wu, J.; Wang, F. Neuroprotective effect of hydrogen sulfide against glutamate-induced oxidative stress is mediated via the p53/glutaminase 2 pathway after traumatic brain injury. Aging 2021, 13, 7180–7189. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, X.; Zhao, S.; Wei, C.; Yin, Y.; Liu, T.; Jiang, S.; Xie, J.; Wan, X.; Mao, M.; et al. Hydrogen sulfide prevents OGD/R-induced apoptosis via improving mitochondrial dysfunction and suppressing an ROS-mediated caspase-3 pathway in cortical neurons. Neurochem. Int. 2013, 63, 826–831. [Google Scholar] [CrossRef]
- Lu, D.; Wang, L.; Liu, G.; Wang, S.; Wang, Y.; Wu, Y.; Wang, J.; Sun, X. Role of hydrogen sulfide in subarachnoid hemorrhage. CNS Neurosci. Ther. 2022, 28, 805–817. [Google Scholar] [CrossRef]
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majid, A.S.A.; Majid, A.M.S.A.; Yin, Z.Q.; Ji, D. Slow Regulated Release of H2S Inhibits Oxidative Stress Induced Cell Death by Influencing Certain Key Signaling Molecules. Neurochem. Res. 2013, 38, 1375–1393. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Moshal, K.S.; Sen, U.; Vacek, T.P.; Kumar, M.; Hughes, W.M.; Kundu, S.; Tyagi, S.C. H2S Protects Against Methionine–Induced Oxidative Stress in Brain Endothelial Cells. Antioxid. Redox Signal. 2009, 11, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Yu, S.; Yang, K.; Li, C.; Liang, Y. Hydrogen Sulfide Inhibits Autophagic Neuronal Cell Death by Reducing Oxidative Stress in Spinal Cord Ischemia Reperfusion Injury. Oxid. Med. Cell. Longev. 2017, 2017, 8640284. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.; Muqadas, M.; Adlat, S.; Zheng, H.; Li, G.; Zhu, P.; Nasser, M.I. Protective Effect of Hydrogen Sulfide on Cerebral Ischemia–Reperfusion Injury. Cell. Mol. Neurobiol. 2023, 43, 15–25. [Google Scholar] [CrossRef]
- Bhatia, M. Role of Hydrogen Sulfide in the Pathology of Inflammation. Scientifica 2012, 2012, 159680. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Li, Y.; Zhao, Z.; Li, P.; Xie, Y. Hydrogen Sulfide Overproduction Is Involved in Acute Ischemic Cerebral Injury Under Hyperhomocysteinemia. Front. Neurosci. 2020, 14, 582851. [Google Scholar] [CrossRef] [PubMed]
- Calenic, B.; Yaegaki, K.; Ishkitiev, N.; Kumazawa, Y.; Imai, T.; Tanaka, T. p53-Pathway activity and apoptosis in hydrogen sulfide-exposed stem cells separated from human gingival epithelium. J. Periodontal Res. 2013, 48, 322–330. [Google Scholar] [CrossRef]
- Calenic, B.; Yaegaki, K.; Kozhuharova, A.; Imai, T. Oral Malodorous Compound Causes Oxidative Stress and p53-Mediated Programmed Cell Death in Keratinocyte Stem Cells. J. Periodontol. 2010, 81, 1317–1323. [Google Scholar] [CrossRef]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef]
- Rodkin, S.; Nwosu, C.; Sannikov, A.; Tyurin, A.; Chulkov, V.S.; Raevskaya, M.; Ermakov, A.; Kirichenko, E.; Gasanov, M. The Role of Gasotransmitter-Dependent Signaling Mechanisms in Apoptotic Cell Death in Cardiovascular, Rheumatic, Kidney, and Neurodegenerative Diseases and Mental Disorders. Int. J. Mol. Sci. 2023, 24, 6014. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Hussain, Z.; Boyle, J. A beginner’s guide to the literature search in medical education. Scott. Med. J. 2017, 62, 58–62. [Google Scholar] [CrossRef]
- Smith, C. Neurotrauma. Handb. Clin. Neurol. 2018, 145, 115–132. [Google Scholar]
- Chang, W.-T.W.; Badjatia, N. Neurotrauma. Emerg. Med. Clin. N. Am. 2014, 32, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord.–Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Sussman, E.S.; Pendharkar, A.V.; Ho, A.L.; Ghajar, J. Mild Traumatic Brain Injury and Concussion: Terminology and Classification; Elsevier: Amsterdam, The Netherlands, 2018; pp. 21–24. [Google Scholar]
- Robinson, C.P. Moderate and Severe Traumatic Brain Injury. Contin. Lifelong Learn. Neurol. 2021, 27, 1278–1300. [Google Scholar] [CrossRef]
- Zhang, M.; Shan, H.; Chang, P.; Wang, T.; Dong, W.; Chen, X.; Tao, L. Hydrogen Sulfide Offers Neuroprotection on Traumatic Brain Injury in Parallel with Reduced Apoptosis and Autophagy in Mice. PLoS ONE 2014, 9, e87241. [Google Scholar] [CrossRef]
- Wang, K.; Liu, B.; Ma, J. Research progress in traumatic brain penumbra. Chin. Med. J. 2014, 127, 1964–1968. [Google Scholar]
- Sun, G.; Gao, F.; Zhao, Z.; Sun, H.; Xu, W.; Wu, L.; He, Y. Endoplasmic reticulum stress-induced apoptosis in the penumbra aggravates secondary damage in rats with traumatic brain injury. Neural Regen. Res. 2016, 11, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Killen, M.J.; Giorgi-Coll, S.; Helmy, A.; Hutchinson, P.J.; Carpenter, K.L. Metabolism and inflammation: Implications for traumatic brain injury therapeutics. Expert Rev. Neurother. 2019, 19, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Zong, P.; Feng, J.; Yue, Z.; Li, Y.; Wu, G.; Sun, B.; He, Y.; Miller, B.; Yu, A.S.; Su, Z.; et al. Functional coupling of TRPM2 and extrasynaptic NMDARs exacerbates excitotoxicity in ischemic brain injury. Neuron 2022, 110, 1944–1958.e8. [Google Scholar] [CrossRef]
- Moojen, V.K.M.; Damiani-Neves, M.; Bavaresco, D.V.; Pescador, B.B.; Comim, C.M.; Quevedo, J.; Boeck, C.R. NMDA preconditioning prevents object recognition memory impairment and increases brain viability in mice exposed to traumatic brain injury. Brain Res. 2012, 1466, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Sansur, C.; Xu, H.; Jia, X. The Temporal Pattern, Flux, and Function of Autophagy in Spinal Cord Injury. Int. J. Mol. Sci. 2017, 18, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wu, Y.; Han, W.; Li, J.; Xu, K.; Li, Z.; Wang, Q.; Xu, K.; Liu, Y.; Xie, L.; et al. Hydrogen Sulfide Ameliorates Blood-Spinal Cord Barrier Disruption and Improves Functional Recovery by Inhibiting Endoplasmic Reticulum Stress-Dependent Autophagy. Front. Pharmacol. 2018, 9, 858. [Google Scholar] [CrossRef]
- Eckert, M.J.; Martin, M.J. Trauma. Surg. Clin. N. Am. 2017, 97, 1031–1045. [Google Scholar] [CrossRef]
- Anjum, A.; Yazid, M.D.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Athi Kumar, R.K.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533. [Google Scholar] [CrossRef]
- Goulart, C.; Martinez, A.B. Tubular conduits, cell-based therapy and exercise to improve peripheral nerve regeneration. Neural Regen. Res. 2015, 10, 565–567. [Google Scholar] [CrossRef]
- Bhandari, P.S. Management of peripheral nerve injury. J. Clin. Orthop. Trauma 2019, 10, 862–866. [Google Scholar] [CrossRef]
- Renthal, W.; Tochitsky, I.; Yang, L.; Cheng, Y.-C.; Li, E.; Kawaguchi, R.; Geschwind, D.H.; Woolf, C.J. Transcriptional Reprogramming of Distinct Peripheral Sensory Neuron Subtypes after Axonal Injury. Neuron 2020, 108, 128–144.e9. [Google Scholar] [CrossRef]
- Hart, A.M.; Terenghi, G.; Wiberg, M. Neuronal death after peripheral nerve injury and experimental strategies for neuroprotection. Neurol. Res. 2008, 30, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Navarro, X.; Vivó, M.; Valero-Cabré, A. Neural plasticity after peripheral nerve injury and regeneration. Prog. Neurobiol. 2007, 82, 163–201. [Google Scholar] [CrossRef]
- Rishal, I.; Fainzilber, M. Axon–soma communication in neuronal injury. Nat. Rev. Neurosci. 2014, 15, 32–42. [Google Scholar] [CrossRef]
- Patodia, S.; Raivich, G. Role of Transcription Factors in Peripheral Nerve Regeneration. Front. Mol. Neurosci. 2012, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Esposito, E.; Ahmad, A.; Di Paola, R.; Paterniti, I.; Cordaro, M.; Bruschetta, G.; Wallace, J.L.; Cuzzocrea, S. Hydrogen sulfide-releasing cyclooxygenase inhibitor ATB-346 enhances motor function and reduces cortical lesion volume following traumatic brain injury in mice. J. Neuroinflamm. 2014, 11, 196. [Google Scholar] [CrossRef] [Green Version]
- Oh, G.-S.; Pae, H.-O.; Lee, B.-S.; Kim, B.-N.; Kim, J.-M.; Kim, H.-R.; Jeon, S.B.; Jeon, W.K.; Chae, H.-J.; Chung, H.-T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic. Biol. Med. 2006, 41, 106–119. [Google Scholar] [CrossRef]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, G.C.; Twose, C.; Weller, A.; Dougherty, J.W.; Goes, F.S.; Sair, H.I.; Smith, G.S.; Roy, D. Neuroimaging Correlates of Depression after Traumatic Brain Injury: A Systematic Review. J. Neurotrauma 2022, 39, 755–772. [Google Scholar] [CrossRef] [PubMed]
- Delmonico, R.L.; Theodore, B.R.; Sandel, M.E.; Armstrong, M.A.; Camicia, M. Prevalence of depression and anxiety disorders following mild traumatic brain injury. PM R 2022, 14, 753–763. [Google Scholar] [CrossRef]
- Kaur, J.; Ghosh, S.; Singh, P.; Dwivedi, A.K.; Sahani, A.K.; Sinha, J.K. Cervical Spinal Lesion, Completeness of Injury, Stress, and Depression Reduce the Efficiency of Mental Imagery in People With Spinal Cord Injury. Am. J. Phys. Med. Rehabil. 2022, 101, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Mariajoseph, F.P.; Chen, Z.; Sekhar, P.; Rewell, S.S.; O’Brien, T.J.; Antonic-Baker, A.; Semple, B.D. Incidence and risk factors of posttraumatic epilepsy following pediatric traumatic brain injury: A systematic review and meta-analysis. Epilepsia 2022, 63, 2802–2812. [Google Scholar] [CrossRef] [PubMed]
- Pease, M.; Gonzalez-Martinez, J.; Puccio, A.; Nwachuku, E.; Castellano, J.F.; Okonkwo, D.O.; Elmer, J. Risk Factors and Incidence of Epilepsy after Severe Traumatic Brain Injury. Ann. Neurol. 2022, 92, 663–669. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.; Liu, G.; Gao, K.; Zhou, H.; Zhao, Y.; Wang, H.; Zhang, L.; Liu, S. Spinal cord injury and its underlying mechanism in rats with temporal lobe epilepsy. Exp. Ther. Med. 2020, 19, 2103–2112. [Google Scholar] [CrossRef] [Green Version]
- Kruitbosch, J.M.; Schouten, E.J.; Tan, I.Y.; Veendrick-Meekes, M.J.B.M.; de Vocht, J.W.M.M. Cervical spinal cord injuries in patients with refractory epilepsy. Seizure 2006, 15, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.; Johnson, V.E.; Smith, D.H.; Stewart, W. Chronic Traumatic Encephalopathy: The Neuropathological Legacy of Traumatic Brain Injury. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Bailes, J.E.; Huber, J.D.; Rosen, C.L. Linking Traumatic Brain Injury to Chronic Traumatic Encephalopathy: Identification of Potential Mechanisms Leading to Neurofibrillary Tangle Development. J. Neurotrauma 2014, 31, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.; Curran, M.; Lucas, S.; Zumsteg, J. Collaborative Care to Treat Chronic Pain after Traumatic Brain Injury: A Randomized Controlled Trial. Arch. Phys. Med. Rehabil. 2022, 103, e57. [Google Scholar] [CrossRef]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Vergne-Salle, P.; Bertin, P. Chronic pain and neuroinflammation. Jt. Bone Spine 2021, 88, 105222. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Clark, C.M.; Liun, F.; Lee, V.Y.-M.; Trojanowski, J.Q. Increase of Brain Oxidative Stress in Mild Cognitive Impairment. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Penninx, B.W.J.H. Oxidative stress in major depressive and anxiety disorders, and the association with antidepressant use; results from a large adult cohort. Psychol. Med. 2017, 47, 936–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geronzi, U.; Lotti, F.; Grosso, S. Oxidative stress in epilepsy. Expert Rev. Neurother. 2018, 18, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.S.; Strath, L.J.; Sorge, R.E. Dietary Interventions for Treatment of Chronic Pain: Oxidative Stress and Inflammation. Pain Ther. 2020, 9, 487–498. [Google Scholar] [CrossRef]
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Irvine, K.-A.; Clark, J.D. Chronic Pain After Traumatic Brain Injury: Pathophysiology and Pain Mechanisms. Pain Med. 2018, 19, 1315–1333. [Google Scholar] [CrossRef] [Green Version]
- Ghajari, M.; Hellyer, P.J.; Sharp, D.J. Computational modelling of traumatic brain injury predicts the location of chronic traumatic encephalopathy pathology. Brain 2017, 140, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Bjelakovic, B.; Dimitrijevic, L.; Lukic, S.; Golubovic, E. Hypertensive encephalopathy as a late complication of autonomic dysreflexia in a 12-year-old boy with a previous spinal cord injury. Eur. J. Pediatr. 2014, 173, 1683–1684. [Google Scholar] [CrossRef]
- Kennedy, P.; Rogers, B.A. Anxiety and depression after spinal cord injury: A longitudinal analysis. Arch. Phys. Med. Rehabil. 2000, 81, 932–937. [Google Scholar] [CrossRef]
- Hunt, C.; Moman, R.; Peterson, A.; Wilson, R.; Covington, S.; Mustafa, R.; Murad, M.H.; Hooten, W.M. Prevalence of chronic pain after spinal cord injury: A systematic review and meta-analysis. Reg. Anesth. Pain Med. 2021, 46, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Cho, S.S.; Kim, H.Y.; Lee, B.H.; Cho, H.J.; Gwak, Y.S. Regional Hyperexcitability and Chronic Neuropathic Pain Following Spinal Cord Injury. Cell. Mol. Neurobiol. 2020, 40, 861–878. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol. 2012, 124, 643–664. [Google Scholar] [CrossRef] [PubMed]
- Alcántar-Garibay, O.; Incontri-Abraham, D.; Ibarra, A. Spinal cord injury-induced cognitive impairment: A narrative review. Neural Regen. Res. 2022, 17, 2649–2654. [Google Scholar] [CrossRef]
- Li, Y.; Cao, T.; Ritzel, R.M.; He, J.; Faden, A.I.; Wu, J. Dementia, Depression, and Associated Brain Inflammatory Mechanisms after Spinal Cord Injury. Cells 2020, 9, 1420. [Google Scholar] [CrossRef]
- Rekand, T.; Hagen, E.; Grønning, M. Chronic pain following spinal cord injury. Tidsskr. Nor. Legeforening 2012, 132, 974–979. [Google Scholar] [CrossRef] [Green Version]
- Norman, G.J.; Karelina, K.; Zhang, N.; Walton, J.C.; Morris, J.S.; DeVries, A.C. Stress and IL-1β contribute to the development of depressive-like behavior following peripheral nerve injury. Mol. Psychiatry 2010, 15, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Inquimbert, P.; Moll, M.; Latremoliere, A.; Tong, C.-K.; Whang, J.; Sheehan, G.F.; Smith, B.M.; Korb, E.; Athié, M.C.P.; Babaniyi, O.; et al. NMDA Receptor Activation Underlies the Loss of Spinal Dorsal Horn Neurons and the Transition to Persistent Pain after Peripheral Nerve Injury. Cell Rep. 2018, 23, 2678–2689. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Ying, J.; Xiang, L.; Zhang, C. The biologic effect of hydrogen sulfide and its function in various diseases. Medicine 2018, 97, e13065. [Google Scholar] [CrossRef]
- Khattak, S.; Rauf, M.A.; Khan, N.H.; Zhang, Q.-Q.; Chen, H.-J.; Muhammad, P.; Ansari, M.A.; Alomary, M.N.; Jahangir, M.; Zhang, C.-Y.; et al. Hydrogen Sulfide Biology and Its Role in Cancer. Molecules 2022, 27, 3389. [Google Scholar] [CrossRef]
- Kabil, O.; Zhou, Y.; Banerjee, R. Human Cystathionine β-Synthase Is a Target for Sumoylation. Biochemistry 2006, 45, 13528–13536. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine Beta-Synthase (CBS) Contributes to Advanced Ovarian Cancer Progression and Drug Resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Omorou, M.; Liu, N.; Huang, Y.; Al-Ward, H.; Gao, M.; Mu, C.; Zhang, L.; Hui, X. Cystathionine beta-Synthase in hypoxia and ischemia/reperfusion: A current overview. Arch. Biochem. Biophys. 2022, 718, 109149. [Google Scholar] [CrossRef]
- Zhu, H.; Blake, S.; Chan, K.T.; Pearson, R.B.; Kang, J. Cystathionine β-Synthase in Physiology and Cancer. Biomed. Res. Int. 2018, 2018, 3205125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Zhao, T.; Tao, L.; Yu, Q.; Yang, Y.; Cheng, J.; Lu, S.; Ding, Q. Cystathionine β-Synthase-Derived Hydrogen Sulfide Correlates with Successful Aging in Mice. Rejuvenation Res. 2019, 22, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, H.; Kaczor-Kamińska, M.; Bronowicka-Adamska, P.; Wróbel, M. Cystathionine γ-lyase. Postepy Hig. Med. Dosw. 2014, 68, 1–9. [Google Scholar] [CrossRef]
- Zhao, K. Regulation of cystathionine gamma-lyase/H2S system and its pathological implication. Front. Biosci. 2014, 19, 1355–1369. [Google Scholar] [CrossRef]
- Szijártó, I.A.; Markó, L.; Filipovic, M.R.; Miljkovic, J.L.; Tabeling, C.; Tsvetkov, D.; Wang, N.; Rabelo, L.A.; Witzenrath, M.; Diedrich, A.; et al. Cystathionine γ-Lyase–Produced Hydrogen Sulfide Controls Endothelial NO Bioavailability and Blood Pressure. Hypertension 2018, 71, 1210–1217. [Google Scholar] [CrossRef]
- Kabil, O.; Banerjee, R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.Y.; Dugbartey, G.J.; Juriasingani, S.; Sener, A. Hydrogen Sulfide Metabolite, Sodium Thiosulfate: Clinical Applications and Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6452. [Google Scholar] [CrossRef]
- Park, B.S.; Kim, H.-W.; Rhyu, I.J.; Park, C.; Yeo, S.G.; Huh, Y.; Jeong, N.Y.; Jung, J. Hydrogen sulfide is essential for Schwann cell responses to peripheral nerve injury. J. Neurochem. 2015, 132, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Lupoli, R.; Di Minno, A.; Spadarella, G.; Franchini, M.; Sorrentino, R.; Cirino, G.; Di Minno, G. Methylation Reactions, the Redox Balance and Atherothrombosis: The Search for a Link with Hydrogen Sulfide. Semin. Thromb. Hemost. 2015, 41, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Lajin, B.; Francesconi, K.A. The hydrogen sulfide metabolite trimethylsulfonium is found in human urine. Sci. Rep. 2016, 6, 27038. [Google Scholar] [CrossRef] [Green Version]
- Insko, M.A.; Deckwerth, T.L.; Hill, P.; Toombs, C.F.; Szabo, C. Detection of exhaled hydrogen sulphide gas in rats exposed to intravenous sodium sulphide. Br. J. Pharmacol. 2009, 157, 944–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R. Hydrogen Sulfide: The Third Gasotransmitter in Biology and Medicine. Antioxid. Redox Signal. 2010, 12, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, A.V.; Kurmasheva, E.D.; Ishchenko, Y.; Giniatullin, R.; Sitdikova, G.F. Age-Dependent, Subunit Specific Action of Hydrogen Sulfide on GluN1/2A and GluN1/2B NMDA Receptors. Front. Cell. Neurosci. 2017, 11, 375. [Google Scholar] [CrossRef] [Green Version]
- Munaron, L.; Avanzato, D.; Moccia, F.; Mancardi, D. Hydrogen sulfide as a regulator of calcium channels. Cell Calcium 2013, 53, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Disbrow, E.; Stokes, K.Y.; Ledbetter, C.; Patterson, J.; Kelley, R.; Pardue, S.; Reekes, T.; Larmeu, L.; Batra, V.; Yuan, S.; et al. Plasma hydrogen sulfide: A biomarker of Alzheimer’s disease and related dementias. Alzheimer’s Dement. 2021, 17, 1391–1402. [Google Scholar] [CrossRef]
- Liu, L.; Wang, J.; Wang, H. Hydrogen sulfide alleviates oxidative stress injury and reduces apoptosis induced by MPP+ in Parkinson’s disease cell model. Mol. Cell. Biochem. 2020, 472, 231–240. [Google Scholar] [CrossRef]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef] [Green Version]
- Tabassum, R.; Jeong, N.; Jung, J. Therapeutic importance of hydrogen sulfide in age-associated neurodegenerative diseases. Neural Regen. Res. 2020, 15, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Duan, X.; Li, H.; Dang, B.; Yin, J.; Wang, Y.; Gao, A.; Yu, Z.; Chen, G. Hydrogen Sulfide Ameliorates Early Brain Injury Following Subarachnoid Hemorrhage in Rats. Mol. Neurobiol. 2016, 53, 3646–3657. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, C.; Manuel, M.L.; Yuan, S.; Kevil, C.G.; McCarter, K.D.; Lu, W.; Sun, H. Role of Hydrogen Sulfide in Early Blood-Brain Barrier Disruption following Transient Focal Cerebral Ischemia. PLoS ONE 2015, 10, e0117982. [Google Scholar] [CrossRef]
- Zhang, M.; Shan, H.; Wang, T.; Liu, W.; Wang, Y.; Wang, L.; Zhang, L.; Chang, P.; Dong, W.; Chen, X.; et al. Dynamic Change of Hydrogen Sulfide After Traumatic Brain Injury and its Effect in Mice. Neurochem. Res. 2013, 38, 714–725. [Google Scholar] [CrossRef]
- Huerta de la Cruz, S.; Rodríguez-Palma, E.J.; Santiago-Castañeda, C.L.; Beltrán-Ornelas, J.H.; Sánchez-López, A.; Rocha, L.; Centurión, D. Exogenous hydrogen sulfide restores CSE and CBS but no 3-MST protein expression in the hypothalamus and brainstem after severe traumatic brain injury. Metab. Brain Dis. 2022, 37, 1863–1874. [Google Scholar] [CrossRef]
- Pushchina, E.V.; Stukaneva, M.E.; Varaksin, A.A. Hydrogen Sulfide Modulates Adult and Reparative Neurogenesis in the Cerebellum of Juvenile Masu Salmon, Oncorhynchus masou. Int. J. Mol. Sci. 2020, 21, 9638. [Google Scholar] [CrossRef] [PubMed]
- Pushchina, E.V.; Zharikova, E.I.; Varaksin, A.A. Mechanical Brain Injury Increases Cells’ Production of Cystathionine β-Synthase and Glutamine Synthetase, but Reduces Pax2 Expression in the Telencephalon of Juvenile Chum Salmon, Oncorhynchus keta. Int. J. Mol. Sci. 2021, 22, 1279. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Cao, L.; Feng, X.; Zhou, B.; Li, L. Octreotide-mediated neurofunctional recovery in rats following traumatic brain injury. Role of H2S, Nrf2 and TNF-α. Acta Cirúrgica Bras. 2021, 36, e361204. [Google Scholar] [CrossRef]
- Denoix, N.; Merz, T.; Unmuth, S.; Hoffmann, A.; Nespoli, E.; Scheuerle, A.; Huber-Lang, M.; Gündel, H.; Waller, C.; Radermacher, P.; et al. Cerebral Immunohistochemical Characterization of the H2S and the Oxytocin Systems in a Porcine Model of Acute Subdural Hematoma. Front. Neurol. 2020, 11, 649. [Google Scholar] [CrossRef]
- Greco, V.; Neri, C.; Pieragostino, D.; Spalloni, A.; Persichilli, S.; Gastaldi, M.; Mercuri, N.B.; Longone, P.; Urbani, A. Investigating Different Forms of Hydrogen Sulfide in Cerebrospinal Fluid of Various Neurological Disorders. Metabolites 2021, 11, 152. [Google Scholar] [CrossRef]
- Zhang, X.; Bian, J.-S. Hydrogen Sulfide: A Neuromodulator and Neuroprotectant in the Central Nervous System. ACS Chem. Neurosci. 2014, 5, 876–883. [Google Scholar] [CrossRef]
- Jiang, X.; Huang, Y.; Lin, W.; Gao, D.; Fei, Z. Protective effects of hydrogen sulfide in a rat model of traumatic brain injury via activation of mitochondrial adenosine triphosphate–sensitive potassium channels and reduction of oxidative stress. J. Surg. Res. 2013, 184, e27–e35. [Google Scholar] [CrossRef]
- Xu, K.; Wu, F.; Xu, K.; Li, Z.; Wei, X.; Lu, Q.; Jiang, T.; Wu, F.; Xu, X.; Xiao, J.; et al. NaHS restores mitochondrial function and inhibits autophagy by activating the PI3K/Akt/mTOR signalling pathway to improve functional recovery after traumatic brain injury. Chem. Biol. Interact. 2018, 286, 96–105. [Google Scholar] [CrossRef]
- Wang, R.; Wu, X.-X.; Tian, Z.; Hu, T.; Cai, C.; Wu, G.-P.; Jiang, G.-B.; Liu, B. Sustained release of hydrogen sulfide from anisotropic ferrofluid hydrogel for the repair of spinal cord injury. Bioact. Mater. 2023, 23, 118–128. [Google Scholar] [CrossRef]
- Chen, X.; Huang, X.; Liu, C.; Li, S.; Yang, Z.; Zhang, F.; Chen, X.; Shan, H.; Tao, L.; Zhang, M. Surface-fill H2S-releasing silk fibroin hydrogel for brain repair through the repression of neuronal pyroptosis. Acta Biomater. 2022, 154, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pan, L.; Jiang, A.; Yin, M. Hydrogen sulfide upregulated lncRNA CasC7 to reduce neuronal cell apoptosis in spinal cord ischemia-reperfusion injury rat. Biomed. Pharmacother. 2018, 98, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhou, H.; Zhang, X. SAM, a cystathionine beta-synthase activator, promotes hydrogen sulfide to promote neural repair resulting from massive cerebral infarction induced by middle cerebral artery occlusion. Metab. Brain Dis. 2022, 37, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhang, M.; Zhang, G.; Yan, S.; Yan, W. Hydrogen Sulfide Improves Functional Recovery in Rat Traumatic Spinal Cord Injury Model by Inducing Nuclear Translocation of NF-E2-Related Factor 2. Biol. Pharm. Bull. 2021, 44, b21-00259. [Google Scholar] [CrossRef]
- Liu, H.; Tong, K.; Zhong, Z.; Wang, G. Mechanism of Hydrogen Sulfide Drug-Loaded Nanoparticles Promoting the Repair of Spinal Cord Injury in Rats Through Mammalian Target of Rapamycin/Signal Transducer and Activator of Transcription 3 Signaling Pathway. Sci. Adv. Mater. 2021, 13, 1691–1698. [Google Scholar] [CrossRef]
- Kanemaru, E.; Miyazaki, Y.; Marutani, E.; Ezaka, M.; Goto, S.; Ohshima, E.; Bloch, D.B.; Ichinose, F. Intranasal administration of polysulfide prevents neurodegeneration in spinal cord and rescues mice from delayed paraplegia after spinal cord ischemia. Redox Biol. 2023, 60, 102620. [Google Scholar] [CrossRef]
- Nii, T.; Eguchi, R.; Yamaguchi, S.; Otsuguro, K. Hydrogen sulfide induces Ca2+ release from the endoplasmic reticulum and suppresses ATP-induced Ca2+ signaling in rat spinal cord astrocytes. Eur. J. Pharmacol. 2021, 891, 173684. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fu, Z.; Wu, Y.; Hu, X.; Zhu, T.; Jin, C. Neuroprotective effect of hydrogen sulfide on acute cauda equina injury in rats. Spine J. 2016, 16, 402–407. [Google Scholar] [CrossRef]
- Kida, K.; Marutani, E.; Nguyen, R.K.; Ichinose, F. Inhaled hydrogen sulfide prevents neuropathic pain after peripheral nerve injury in mice. Nitric Oxide 2015, 46, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Jeong, N. Hydrogen sulfide controls peripheral nerve degeneration and regeneration: A novel therapeutic strategy for peripheral demyelinating disorders or nerve degenerative diseases. Neural Regen. Res. 2014, 9, 2119–2121. [Google Scholar] [CrossRef] [PubMed]
- Predmore, B.L.; Lefer, D.J.; Gojon, G. Hydrogen Sulfide in Biochemistry and Medicine. Antioxid. Redox Signal. 2012, 17, 119–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, K.R. Is hydrogen sulfide a circulating “gasotransmitter” in vertebrate blood? Biochim. Biophys. Acta–Bioenerg. 2009, 1787, 856–863. [Google Scholar] [CrossRef] [Green Version]
- Bruce King, S. Potential biological chemistry of hydrogen sulfide (H2S) with the nitrogen oxides. Free Radic. Biol. Med. 2013, 55, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Kesherwani, V.; Nelson, K.S.; Agrawal, S.K. Effect of sodium hydrosulphide after acute compression injury of spinal cord. Brain Res. 2013, 1527, 222–229. [Google Scholar] [CrossRef]
- Searcy, D.G.; Whitehead, J.P.; Maroney, M.J. Interaction of Cu, Zn Superoxide Dismutase with Hydrogen Sulfide. Arch. Biochem. Biophys. 1995, 318, 251–263. [Google Scholar] [CrossRef]
- Vega-Vela, N.E.; Osorio, D.; Avila-Rodriguez, M.; Gonzalez, J.; García-Segura, L.M.; Echeverria, V.; Barreto, G.E. L-Type Calcium Channels Modulation by Estradiol. Mol. Neurobiol. 2017, 54, 4996–5007. [Google Scholar] [CrossRef]
- Jiang, M.C.; Birch, D.V.; Heckman, C.J.; Tysseling, V.M. The Involvement of CaV1.3 Channels in Prolonged Root Reflexes and Its Potential as a Therapeutic Target in Spinal Cord Injury. Front. Neural Circuits 2021, 15, 642111. [Google Scholar] [CrossRef] [PubMed]
- Alles, S.R.; Garcia, E.; Balasubramanyan, S.; Jones, K.; Tyson, J.R.; Joy, T.; Snutch, T.P.; Smith, P.A. Peripheral nerve injury increases contribution of L-type calcium channels to synaptic transmission in spinal lamina II: Role of α2δ–1 subunits. Mol. Pain 2018, 14, 174480691876580. [Google Scholar] [CrossRef] [PubMed]
- Ihbe, N.; Le Prieult, F.; Wang, Q.; Distler, U.; Sielaff, M.; Tenzer, S.; Thal, S.C.; Mittmann, T. Adaptive Mechanisms of Somatostatin-Positive Interneurons after Traumatic Brain Injury through a Switch of α Subunits in L-Type Voltage-Gated Calcium Channels. Cereb. Cortex 2022, 32, 1093–1109. [Google Scholar] [CrossRef]
- Tang, G.; Wu, L.; Wang, R. Interaction of hydrogen sulfide with ion channels. Clin. Exp. Pharmacol. Physiol. 2010, 37, 753–763. [Google Scholar] [CrossRef]
- Nagai, Y.; Tsugane, M.; Oka, J.; Kimura, H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004, 18, 557–559. [Google Scholar] [CrossRef]
- Yong, Q.C.; Choo, C.H.; Tan, B.H.; Low, C.-M.; Bian, J.-S. Effect of hydrogen sulfide on intracellular calcium homeostasis in neuronal cells. Neurochem. Int. 2010, 56, 508–515. [Google Scholar] [CrossRef]
- García-Bereguiaín, M.A.; Samhan-Arias, A.K.; Martín-Romero, F.J.; Gutiérrez-Merino, C. Hydrogen Sulfide Raises Cytosolic Calcium in Neurons Through Activation of L-Type Ca2+ Channels. Antioxid. Redox Signal. 2008, 10, 31–42. [Google Scholar] [CrossRef]
- Okubo, K.; Takahashi, T.; Sekiguchi, F.; Kanaoka, D.; Matsunami, M.; Ohkubo, T.; Yamazaki, J.; Fukushima, N.; Yoshida, S.; Kawabata, A. Inhibition of T-type calcium channels and hydrogen sulfide-forming enzyme reverses paclitaxel-evoked neuropathic hyperalgesia in rats. Neuroscience 2011, 188, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Tarui, T.; Yoshida, S.; Sekiguchi, F.; Matsunami, M.; Ohi, A.; Fukami, K.; Ichida, S.; Nishikawa, H.; Kawabata, A. Hydrogen sulfide evokes neurite outgrowth and expression of high-voltage-activated Ca2+ currents in NG108-15 cells: Involvement of T-type Ca2+ channels. J. Neurochem. 2009, 108, 676–684. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- Yang, K.-L.; Li, W.-H.; Liu, Y.-J.; Wei, Y.-J.; Ren, Y.-K.; Mai, C.-D.; Zhang, S.-Y.; Zuo, Y.; Sun, Z.-Z.; Li, D.-L.; et al. Hydrogen Sulfide Attenuates Neuroinflammation by Inhibiting the NLRP3/Caspase-1/GSDMD Pathway in Retina or Brain Neuron following Rat Ischemia/Reperfusion. Brain Sci. 2022, 12, 1245. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Perumal, N.; Manicam, C.; Mercieca, K.; Prokosch, V. Proteomics Reveals the Potential Protective Mechanism of Hydrogen Sulfide on Retinal Ganglion Cells in an Ischemia/Reperfusion Injury Animal Model. Pharmaceuticals 2020, 13, 213. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Éva Sikura, K.; Combi, Z.; Potor, L.; Szerafin, T.; Hendrik, Z.; Méhes, G.; Gergely, P.; Whiteman, M.; Beke, L.; Fürtös, I.; et al. Hydrogen sulfide inhibits aortic valve calcification in heart via regulating RUNX2 by NF-κB, a link between inflammation and mineralization. J. Adv. Res. 2021, 27, 165–176. [Google Scholar] [CrossRef]
- Rose, P.; Zhu, Y.-Z.; Moore, P.K. Hydrogen Sulfide and the Immune System. Adv. Exp. Med. Biol. 2021, 1315, 99–128. [Google Scholar]
- Singh, A.K.; Awasthi, D.; Dubey, M.; Nagarkoti, S.; Kumar, A.; Chandra, T.; Barthwal, M.K.; Tripathi, A.K.; Dikshit, M. High oxidative stress adversely affects NFκB mediated induction of inducible nitric oxide synthase in human neutrophils: Implications in chronic myeloid leukemia. Nitric Oxide 2016, 58, 28–41. [Google Scholar] [CrossRef]
- Rodkin, S.V.; Kovaleva, V.D.; Berezhnaya, E.V.; Neginskaya, M.A.; Uzdensky, A.B. Ca2+- and NF-κB-dependent generation of NO in the photosensitized neurons and satellite glial cells. J. Photochem. Photobiol. B Biol. 2019, 199, 111603. [Google Scholar] [CrossRef]
- Qu, W.; Cheng, Y.; Peng, W.; Wu, Y.; Rui, T.; Luo, C.; Zhang, J. Targeting iNOS Alleviates Early Brain Injury After Experimental Subarachnoid Hemorrhage via Promoting Ferroptosis of M1 Microglia and Reducing Neuroinflammation. Mol. Neurobiol. 2022, 59, 3124–3139. [Google Scholar] [CrossRef]
- Kubo, S.; Kurokawa, Y.; Doe, I.; Masuko, T.; Sekiguchi, F.; Kawabata, A. Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase. Toxicology 2007, 241, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon, G.; Gojon, G.; et al. H2S Protects Against Pressure Overload–Induced Heart Failure via Upregulation of Endothelial Nitric Oxide Synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Batallé, G.; Balboni, G.; Pol, O. Hydrogen Sulfide Increases the Analgesic Effects of µ- and δ-Opioid Receptors during Neuropathic Pain: Pathways Implicated. Antioxidants 2022, 11, 1321. [Google Scholar] [CrossRef] [PubMed]
- Côté, M.-P.; Azzam, G.A.; Lemay, M.A.; Zhukareva, V.; Houlé, J.D. Activity-Dependent Increase in Neurotrophic Factors Is Associated with an Enhanced Modulation of Spinal Reflexes after Spinal Cord Injury. J. Neurotrauma 2011, 28, 299–309. [Google Scholar] [CrossRef] [Green Version]
- DeKosky, S.T.; Goss, J.R.; Miller, P.D.; Styren, S.D.; Kochanek, P.M.; Marion, D. Upregulation of Nerve Growth Factor Following Cortical Trauma. Exp. Neurol. 1994, 130, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Goss, J.R.; O’Malley, M.E.; Zou, L.; Styren, S.D.; Kochanek, P.M.; DeKosky, S.T. Astrocytes Are the Major Source of Nerve Growth Factor Upregulation Following Traumatic Brain Injury in the Rat. Exp. Neurol. 1998, 149, 301–309. [Google Scholar] [CrossRef]
- Hermann, D.M.; Kilic, E.; Kügler, S.; Isenmann, S.; Bähr, M. Adenovirus-Mediated Glial Cell Line-Derived Neurotrophic Factor (GDNF) Expression Protects against Subsequent Cortical Cold Injury in Rats. Neurobiol. Dis. 2001, 8, 964–973. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhuang, Y.-Y.; Wu, L.; Xie, M.; Gu, H.-F.; Wang, B.; Tang, X.-Q. Hydrogen Sulfide Ameliorates Cognitive Dysfunction in Formaldehyde-Exposed Rats: Involvement in the Upregulation of Brain-Derived Neurotrophic Factor. Neuropsychobiology 2020, 79, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, F.; Bagheri, F.; Rafaiee, R.; Norozi, P.; Khaksari, M. Hydrogen sulfide improves spatial memory impairment via increases of BDNF expression and hippocampal neurogenesis following early postnatal alcohol exposure. Physiol. Behav. 2020, 215, 112784. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, H.; Xue, H.; Zhang, J.; Han, X.; Yan, S.; Bo, S.; Liu, S.; Yuan, L.; Deng, L.; et al. Neuroprotective Effects of Hydrogen Sulfide Against Early Brain Injury and Secondary Cognitive Deficits Following Subarachnoid Hemorrhage. Brain Pathol. 2017, 27, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, J.; Ao, G.; Hu, L.; Liu, H.; Xiao, Y.; Du, H.; Alkayed, N.J.; Liu, C.-F.; Cheng, J. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J. Neurochem. 2014, 129, 827–838. [Google Scholar] [CrossRef]
- Kumar, M.; Sandhir, R. Hydrogen sulfide attenuates hyperhomocysteinemia-induced blood-brain barrier permeability by inhibiting MMP-9. Int. J. Neurosci. 2022, 132, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Li, Q.; Fan, J.; Zhong, H.; Cao, L.; Duan, M. Therapeutic Hypothermia Combined with Hydrogen Sulfide Treatment Attenuated Early Blood–Brain Barrier Disruption and Brain Edema Induced by Cardiac Arrest and Resuscitation in Rat Model. Neurochem. Res. 2022, 48, 967–979. [Google Scholar] [CrossRef]
- Li, H.; Zhu, L.; Feng, J.; Hu, X.; Li, C.; Zhang, B. Hydrogen Sulfide Decreases Blood-Brain Barrier Damage via Regulating Protein Kinase C and Tight Junction After Cardiac Arrest in Rats. Cell. Physiol. Biochem. 2018, 47, 994–1006. [Google Scholar] [CrossRef]
- López-Preza, F.I.; Huerta de la Cruz, S.; Santiago-Castañeda, C.; Silva-Velasco, D.L.; Beltran-Ornelas, J.H.; Tapia-Martínez, J.; Sánchez-López, A.; Rocha, L.; Centurión, D. Hydrogen sulfide prevents the vascular dysfunction induced by severe traumatic brain injury in rats by reducing reactive oxygen species and modulating eNOS and H2S-synthesizing enzyme expression. Life Sci. 2023, 312, 121218. [Google Scholar] [CrossRef]
- Haber, M.; James, J.; Kim, J.; Sangobowale, M.; Irizarry, R.; Ho, J.; Nikulina, E.; Grin’kina, N.M.; Ramadani, A.; Hartman, I.; et al. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes and modifies neuroinflammation in a rat model of mild traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1312–1326. [Google Scholar] [CrossRef]
- Mekhail, M.; Almazan, G.; Tabrizian, M. Oligodendrocyte-protection and remyelination post-spinal cord injuries: A review. Prog. Neurobiol. 2012, 96, 322–339. [Google Scholar] [CrossRef]
- Svennigsen, Å.; Dahlin, L. Repair of the Peripheral Nerve—Remyelination that Works. Brain Sci. 2013, 3, 1182–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, I.K.; Bajeli, S.; Sahu, S.; Bhat, S.A.; Kumar, A. Hydrogen sulfide-induced GAPDH sulfhydration disrupts the CCAR2-SIRT1 interaction to initiate autophagy. Autophagy 2021, 17, 3511–3529. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M.; Vega, V.S.; Works, C.; Lin, J. The chemical biology of hydrogen sulfide and related hydropersulfides: Interactions with biologically relevant metals and metalloproteins. Curr. Opin. Chem. Biol. 2020, 55, 52–58. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. p53 and mitochondrial function in neurons. Biochim. Biophys. Acta–Mol. Basis Dis. 2014, 1842, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.-Q.; Luo, T.-T.; Luo, S.-C.; Wang, J.-Q.; Wang, S.-M.; Bai, Y.-H.; Yang, Y.-L.; Wang, Y.-Y. p53 and mitochondrial dysfunction: Novel insight of neurodegenerative diseases. J. Bioenerg. Biomembr. 2016, 48, 337–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodkin, S.; Dzreyan, V.; Bibov, M.; Ermakov, A.; Derezina, T.; Kirichenko, E. NO-Dependent Mechanisms of p53 Expression and Cell Death in Rat’s Dorsal Root Ganglia after Sciatic-Nerve Transection. Biomedicines 2022, 10, 1664. [Google Scholar] [CrossRef]
- Rodkin, S.; Khaitin, A.; Pitinova, M.; Dzreyan, V.; Guzenko, V.; Rudkovskii, M.; Sharifulina, S.; Uzdensky, A. The Localization of p53 in the Crayfish Mechanoreceptor Neurons and Its Role in Axotomy-Induced Death of Satellite Glial Cells Remote from the Axon Transection Site. J. Mol. Neurosci. 2020, 70, 532–541. [Google Scholar] [CrossRef]
- Rachmany, L.; Tweedie, D.; Rubovitch, V.; Yu, Q.-S.; Li, Y.; Wang, J.-Y.; Pick, C.G.; Greig, N.H. Cognitive Impairments Accompanying Rodent Mild Traumatic Brain Injury Involve p53-Dependent Neuronal Cell Death and Are Ameliorated by the Tetrahydrobenzothiazole PFT-α. PLoS ONE 2013, 8, e79837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cui, Z.; Feng, G.; Bao, G.; Xu, G.; Sun, Y.; Wang, L.; Chen, J.; Jin, H.; Liu, J.; et al. RBM5 and p53 expression after rat spinal cord injury: Implications for neuronal apoptosis. Int. J. Biochem. Cell Biol. 2015, 60, 43–52. [Google Scholar] [CrossRef]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef] [PubMed]
- Dzreyan, V.; Rodkin, S.; Nikul, V.; Pitinova, M.; Uzdensky, A. The Expression of E2F1, p53, and Caspase 3 in the Rat Dorsal Root Ganglia After Sciatic Nerve Transection. J. Mol. Neurosci. 2021, 71, 826–835. [Google Scholar] [CrossRef]
- Yang, G.; Sun, X.; Wang, R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 2004, 18, 1782–1784. [Google Scholar] [CrossRef]
- Ye, X.; Li, Y.; Lv, B.; Qiu, B.; Zhang, S.; Peng, H.; Kong, W.; Tang, C.; Huang, Y.; Du, J.; et al. Endogenous Hydrogen Sulfide Persulfidates Caspase-3 at Cysteine 163 to Inhibit Doxorubicin-Induced Cardiomyocyte Apoptosis. Oxid. Med. Cell. Longev. 2022, 2022, 6153772. [Google Scholar] [CrossRef] [PubMed]
- Khodapasand, E.; Jafarzadeh, N.; Farrokhi, F.; Kamalidehghan, B.; Houshmand, M. Is Bax/Bcl-2 ratio considered as a prognostic marker with age and tumor location in colorectal cancer? Iran. Biomed. J. 2015, 19, 69–75. [Google Scholar] [CrossRef]
- Duan, H.-Z.; Wu, C.-W.; Shen, S.-L.; Zhang, J.-Y.; Li, L. Neuroprotective Effects of Early Brain Injury after Subarachnoid Hemorrhage in Rats by Calcium Channel Mediating Hydrogen Sulfide. Cell. Mol. Neurobiol. 2021, 41, 1707–1714. [Google Scholar] [CrossRef]
- Scheid, S.; Goeller, M.; Baar, W.; Wollborn, J.; Buerkle, H.; Schlunck, G.; Lagrèze, W.; Goebel, U.; Ulbrich, F. Hydrogen Sulfide Reduces Ischemia and Reperfusion Injury in Neuronal Cells in a Dose- and Time-Dependent Manner. Int. J. Mol. Sci. 2021, 22, 10099. [Google Scholar] [CrossRef]
- Lu, Q.-B.; Ding, Y.; Fu, X.; Sun, H.; Zhang, J.-R. Hydrogen sulfide in health and diseases: Crosstalk with noncoding RNAs. Am. J. Physiol. Physiol. 2023, 324, C856–C877. [Google Scholar] [CrossRef]
- Zhang, Q.; Yuan, L.; Liu, D.; Wang, J.; Wang, S.; Zhang, Q.; Gong, Y.; Liu, H.; Hao, A.; Wang, Z. Hydrogen sulfide attenuates hypoxia-induced neurotoxicity through inhibiting microglial activation. Pharmacol. Res. 2014, 84, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, Y.; Wang, Z.; Kong, Y.; Gao, R.; Chen, G. Hydrogen sulfide therapy in brain diseases: From bench to bedside. Med. Gas Res. 2017, 7, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen Sulfide-Linked Sulfhydration of NF-κB Mediates Its Antiapoptotic Actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Lipinski, M.M. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells 2019, 8, 693. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, C.; Zhao, Z.; Aungst, S.; Sabirzhanov, B.; Faden, A.I.; Lipinski, M.M. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 2014, 10, 2208–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shi, C.; Wang, H.; Gao, C.; Chang, P.; Chen, X.; Shan, H.; Zhang, M.; Tao, L. Hydrogen sulfide protects against cell damage through modulation of PI3K/Akt/Nrf2 signaling. Int. J. Biochem. Cell Biol. 2019, 117, 105636. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Qi, L.; Wang, Y.; Li, X.; Li, Q.; Tang, X.; Wang, X.; Wu, C. Antagonizing effects of curcumin against mercury-induced autophagic death and trace elements disorder by regulating PI3K/AKT and Nrf2 pathway in the spleen. Ecotoxicol. Environ. Saf. 2021, 222, 112529. [Google Scholar] [CrossRef]
- Luo, L.-F.; Qin, L.-Y.; Wang, J.-X.; Guan, P.; Wang, N.; Ji, E.-S. Astragaloside IV Attenuates the Myocardial Injury Caused by Adriamycin by Inhibiting Autophagy. Front. Pharmacol. 2021, 12, 669782. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, H.; Li, Y.; Guo, Y. Hydrogen sulfide protects spinal cord and induces autophagy via miR-30c in a rat model of spinal cord ischemia-reperfusion injury. J. Biomed. Sci. 2015, 22, 50. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, Y.; Lei, Y.; Liu, T.; Wan, Z.; Meng, H.; Wang, H. Ferroptosis Is Regulated by Mitochondria in Neurodegenerative Diseases. Neurodegener. Dis. 2020, 20, 20–34. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, P.; Wang, N.; Dong, W.; Chen, Z.; Wu, M.; Wang, Z.; Yu, Z.; Guan, D.; Wang, L.; et al. Neuroprotection of NRF2 against Ferroptosis after Traumatic Brain Injury in Mice. Antioxidants 2023, 12, 731. [Google Scholar] [CrossRef]
- Jin, R.; Yang, R.; Cui, C.; Zhang, H.; Cai, J.; Geng, B.; Chen, Z. Ferroptosis due to Cystathionine γ Lyase/Hydrogen Sulfide Downregulation under High Hydrostatic Pressure Exacerbates VSMC Dysfunction. Front. Cell Dev. Biol. 2022, 10, 829316. [Google Scholar] [CrossRef]
- Yu, M.; Wang, W.; Dang, J.; Liu, B.; Xu, J.; Li, J.; Liu, Y.; He, L.; Ying, Y.; Cai, J.; et al. Hydrogen sulfide protects retinal pigment epithelium cells against ferroptosis through the AMPK- and p62-dependent non-canonical NRF2-KEAP1 pathway. Exp. Cell Res. 2023, 422, 113436. [Google Scholar] [CrossRef]
- Yu, Y.; Li, X.; Wu, X.; Li, X.; Wei, J.; Chen, X.; Sun, Z.; Zhang, Q. Sodium hydrosulfide inhibits hemin-induced ferroptosis and lipid peroxidation in BV2 cells via the CBS/H2S system. Cell. Signal. 2023, 104, 110594. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, H.; Xu, H.; Wu, Y.; Wu, C.; Jia, C.; Li, Y.; Sheng, S.; Xu, C.; Xu, H.; et al. Role of Pyroptosis in Traumatic Brain and Spinal Cord Injuries. Int. J. Biol. Sci. 2020, 16, 2042–2050. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Yang, Z.; Liu, C.; Chen, X.; Zhang, Y.; Zhang, F.; Shi, H.; Chen, X.; Tao, L.; et al. Implantation of injectable SF hydrogel with sustained hydrogen sulfide delivery reduces neuronal pyroptosis and enhances functional recovery after severe intracerebral hemorrhage. Biomater. Adv. 2022, 135, 212743. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Li, L.; Shen, S.; Ma, Y.; Yin, X.; Liu, Z.; Yuan, C.; Wang, Y.; Zhang, J. Hydrogen Sulfide Reduces Cognitive Impairment in Rats After Subarachnoid Hemorrhage by Ameliorating Neuroinflammation Mediated by the TLR4/NF-κB Pathway in Microglia. Front. Cell. Neurosci. 2020, 14, 210. [Google Scholar] [CrossRef]
- Chu, Q.-J.; He, L.; Zhang, W.; Liu, C.-L.; Ai, Y.-Q.; Zhang, Q. Hydrogen sulfide attenuates surgical trauma-induced inflammatory response and cognitive deficits in mice. J. Surg. Res. 2013, 183, 330–336. [Google Scholar] [CrossRef]
- Yin, L.; Gao, S.; Li, C. Exogenous hydrogen sulfide alleviates surgery-induced neuroinflammatory cognitive impairment in adult mice by inhibiting NO signaling. BMC Anesthesiol. 2020, 20, 12. [Google Scholar] [CrossRef]
- Bas-Orth, C.; Tan, Y.-W.; Lau, D.; Bading, H. Synaptic Activity Drives a Genomic Program That Promotes a Neuronal Warburg Effect. J. Biol. Chem. 2017, 292, 5183–5194. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-M.; Li, M.; Xie, J.; Li, S.; Xiang, S.-S.; Liu, H.-Y.; Chen, Z.; Zhang, P.; Kuang, X.; Tang, X.-Q. Hydrogen sulfide attenuates postoperative cognitive dysfunction through promoting the pathway of Warburg effect-synaptic plasticity in hippocampus. Toxicol. Appl. Pharmacol. 2020, 409, 115286. [Google Scholar] [CrossRef]
- Mostafa, D.K.; El Azhary, N.M.; Nasra, R.A. The hydrogen sulfide releasing compounds ATB-346 and diallyl trisulfide attenuate streptozotocin-induced cognitive impairment, neuroinflammation, and oxidative stress in rats: Involvement of asymmetric dimethylarginine. Can. J. Physiol. Pharmacol. 2016, 94, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, P.O.; Mehta, M.A.; Sharp, D.J. Catecholamines and cognition after traumatic brain injury. Brain 2016, 139, 2345–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Modi, M.; Sandhir, R. Hydrogen sulfide attenuates homocysteine-induced cognitive deficits and neurochemical alterations by improving endogenous hydrogen sulfide levels. BioFactors 2017, 43, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Chen, D.; Wu, F.; Zhang, L.; Huang, Y.; Lin, Z.; Wang, X.; Wang, R.; Xu, L.; Chen, Y. Hydrogen Sulfide Protects Against Ammonia-Induced Neurotoxicity Through Activation of Nrf2/ARE Signaling in Astrocytic Model of Hepatic Encephalopathy. Front. Cell. Neurosci. 2020, 14, 573422. [Google Scholar] [CrossRef]
- Cardoso, G.M.F.; Pletsch, J.T.; Parmeggiani, B.; Grings, M.; Glanzel, N.M.; Bobermin, L.D.; Amaral, A.U.; Wajner, M.; Leipnitz, G. Bioenergetics dysfunction, mitochondrial permeability transition pore opening and lipid peroxidation induced by hydrogen sulfide as relevant pathomechanisms underlying the neurological dysfunction characteristic of ethylmalonic encephalopathy. Biochim. Biophys. Acta–Mol. Basis Dis. 2017, 1863, 2192–2201. [Google Scholar] [CrossRef]
- Li, X.; Yu, P.; Yu, Y.; Xu, T.; Liu, J.; Cheng, Y.; Yang, X.; Cui, X.; Yin, C.; Liu, Y. Hydrogen sulfide ameliorates high glucose-induced pro-inflammation factors in HT-22 cells: Involvement of SIRT1-mTOR/NF-κB signaling pathway. Int. Immunopharmacol. 2021, 95, 107545. [Google Scholar] [CrossRef]
- Kwon, K.W.; Nam, Y.; Choi, W.S.; Kim, T.W.; Kim, G.M.; Sohn, U.D. Hepatoprotective effect of sodium hydrosulfide on hepatic encephalopathy in rats. Korean J. Physiol. Pharmacol. 2019, 23, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-L.; Xie, B.; Zhang, C.; Xu, K.-L.; Niu, Y.-Y.; Tang, X.-Q.; Zhang, P.; Zou, W.; Hu, B.; Tian, Y. Antidepressant-like and anxiolytic-like effects of hydrogen sulfide in behavioral models of depression and anxiety. Behav. Pharmacol. 2013, 24, 590–597. [Google Scholar] [CrossRef]
- Kang, X.; Jiang, L.; Lan, F.; Tang, Y.-Y.; Zhang, P.; Zou, W.; Chen, Y.-J.; Tang, X.-Q. Hydrogen sulfide antagonizes sleep deprivation-induced depression- and anxiety-like behaviors by inhibiting neuroinflammation in a hippocampal Sirt1-dependent manner. Brain Res. Bull. 2021, 177, 194–202. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Li, D.; Zeng, H.-Y.; Kan, L.-Y.; Zou, W.; Zhang, P.; Gu, H.-F.; Tang, X.-Q. Hydrogen Sulfide Inhibits Chronic Unpredictable Mild Stress-Induced Depressive-Like Behavior by Upregulation of Sirt-1: Involvement in Suppression of Hippocampal Endoplasmic Reticulum Stress. Int. J. Neuropsychopharmacol. 2017, 20, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, S.; Xin, Y.; Zhang, J.; Wang, S.; Yang, Z.; Liu, C. Hydrogen sulfide alleviates the anxiety-like and depressive-like behaviors of type 1 diabetic mice via inhibiting inflammation and ferroptosis. Life Sci. 2021, 278, 119551. [Google Scholar] [CrossRef] [PubMed]
- Batallé, G.; Cabarga, L.; Pol, O. The Inhibitory Effects of Slow-Releasing Hydrogen Sulfide Donors in the Mechanical Allodynia, Grip Strength Deficits, and Depressive-Like Behaviors Associated with Chronic Osteoarthritis Pain. Antioxidants 2019, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Q.; Tang, H.-L.; Tang, Y.-Y.; Zhang, P.; Kang, X.; Zou, W.; Tang, X.-Q. Hydrogen Sulfide Attenuates the Cognitive Dysfunction in Parkinson’s Disease Rats via Promoting Hippocampal Microglia M2 Polarization by Enhancement of Hippocampal Warburg Effect. Oxid. Med. Cell. Longev. 2022, 2022, 2792348. [Google Scholar] [CrossRef]
- Hou, X.-Y.; Hu, Z.-L.; Zhang, D.-Z.; Lu, W.; Zhou, J.; Wu, P.-F.; Guan, X.-L.; Han, Q.-Q.; Deng, S.-L.; Zhang, H.; et al. Rapid Antidepressant Effect of Hydrogen Sulfide: Evidence for Activation of mTORC1-TrkB-AMPA Receptor Pathways. Antioxid. Redox Signal. 2017, 27, 472–488. [Google Scholar] [CrossRef]
- Jiang, W.; Tang, Y.-Y.; Zhu, W.-W.; Li, C.; Zhang, P.; Li, R.-Q.; Chen, Y.-J.; Zou, W.; Tang, X.-Q. PI3K/AKT pathway mediates the antidepressant- and anxiolytic-like roles of hydrogen sulfide in streptozotocin-induced diabetic rats via promoting hippocampal neurogenesis. Neurotoxicology 2021, 85, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Luoa, X.; Fanga, T.Y.; Yanga, X.; Panga, Y.P.; Rena, J.; Newellc, K.A.; Yubc, Y.; Huangbc, X.-F.; Liuac, Y. S-adenosyl methionine improves depression-like behaviours and synaptic markers by elevating the expression of endogenous hydrogen sulfide in the hippocampus. Neuropsychiatry 2018, 8, 495–504. [Google Scholar] [CrossRef]
- Tan, H.; Zou, W.; Jiang, J.; Tian, Y.; Xiao, Z.; Bi, L.; Zeng, H.; Tang, X. Disturbance of hippocampal H2S generation contributes to CUMS-induced depression-like behavior: Involvement in endoplasmic reticulum stress of hippocampus. Acta Biochim. Biophys. Sin. 2015, 47, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.; Zeigler, M.; Mizuno, S.; Morrison, R.S.; Totah, R.A.; Barker-Haliski, M. Reductions in Hydrogen Sulfide and Changes in Mitochondrial Quality Control Proteins Are Evident in the Early Phases of the Corneally Kindled Mouse Model of Epilepsy. Int. J. Mol. Sci. 2022, 23, 1434. [Google Scholar] [CrossRef]
- Zhu, Z.; He, Y.; Liu, Z.; Zhang, W.; Kang, Q.; Lin, Y.; Qiu, J.; Zhang, Y.; Xu, P.; Zhu, X. A hydrogen sulfide donor suppresses pentylenetetrazol-induced seizures in rats via PKC signaling. Eur. J. Pharmacol. 2021, 898, 173959. [Google Scholar] [CrossRef]
- Han, Y.; Qin, J.; Chang, X.; Yang, Z.; Bu, D.; Du, J. Modulating effect of hydrogen sulfide on gamma-aminobutyric acid B receptor in recurrent febrile seizures in rats. Neurosci. Res. 2005, 53, 216–219. [Google Scholar] [CrossRef]
- Zhuang, F.; Zhou, X.; Li, H.; Yang, X.; Dong, Z.; Zhou, W.; Chen, J. Hydrogen Sulfide Promotes Learning and Memory and Suppresses Proinflammatory Cytokines in Repetitive Febrile Seizures. Neuroimmunomodulation 2016, 23, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Sutulovic, N.; Rasic-Markovic, A.; Grubac, Ž.; Duric, E.; Hrncic, D. The effects of hydrogen sulfide synthesis inhibition in lindane-induced seizures in rats: A behavioral and EEG study. Arch. Biol. Sci. 2020, 72, 457–463. [Google Scholar] [CrossRef]
- Luo, Y.; Wu, P.-F.; Zhou, J.; Xiao, W.; He, J.-G.; Guan, X.-L.; Zhang, J.-T.; Hu, Z.-L.; Wang, F.; Chen, J.-G. Aggravation of Seizure-like Events by Hydrogen Sulfide: Involvement of Multiple Targets that Control Neuronal Excitability. CNS Neurosci. Ther. 2014, 20, 411–419. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Song, L.; Yang, S.; Wang, Q.; Zhou, Y.; Zhang, X.-B.; Qing, Z.; Yang, R. Brain-targeted Near-Infrared Nanobeacon for In Situ Monitoring H2S Fluctuation during Epileptic Seizures. Anal. Chem. 2022, 94, 15085–15092. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pourcyrous, M.; Fedinec, A.L.; Parfenova, H. Cerebroprotective actions of hydrogen sulfide in the epileptic brain in newborn pigs. Pediatr. Res. 2023. [Google Scholar] [CrossRef]
- Batallé, G.; Bai, X.; Pouso-Vázquez, E.; Roch, G.; Rodríguez, L.; Pol, O. The Recovery of Cognitive and Affective Deficiencies Linked with Chronic Osteoarthritis Pain and Implicated Pathways by Slow-Releasing Hydrogen Sulfide Treatment. Antioxidants 2021, 10, 1632. [Google Scholar] [CrossRef]
- Batallé, G.; Bai, X.; Pol, O. The Interaction between Carbon Monoxide and Hydrogen Sulfide during Chronic Joint Pain in Young Female Mice. Antioxidants 2022, 11, 1271. [Google Scholar] [CrossRef]
- Cabarga, L.; Batallé, G.; Pol, O. Treatment with slow-releasing hydrogen sulfide donors inhibits the nociceptive and depressive-like behaviours accompanying chronic neuropathic pain: Endogenous antioxidant system activation. J. Psychopharmacol. 2020, 34, 737–749. [Google Scholar] [CrossRef]
- Chen, H.; Xie, K.; Chen, Y.; Wang, Y.; Wang, Y.; Lian, N.; Zhang, K.; Yu, Y. Nrf2/HO-1 signaling pathway participated in the protection of hydrogen sulfide on neuropathic pain in rats. Int. Immunopharmacol. 2019, 75, 105746. [Google Scholar] [CrossRef]
- Dichiara, M.; Artacho-Cordón, A.; Turnaturi, R.; Santos-Caballero, M.; González-Cano, R.; Pasquinucci, L.; Barbaraci, C.; Rodríguez-Gómez, I.; Gómez-Guzmán, M.; Marrazzo, A.; et al. Dual Sigma-1 receptor antagonists and hydrogen sulfide-releasing compounds for pain treatment: Design, synthesis, and pharmacological evaluation. Eur. J. Med. Chem. 2022, 230, 114091. [Google Scholar] [CrossRef]
- Vandini, E.; Ottani, A.; Zaffe, D.; Calevro, A.; Canalini, F.; Cavallini, G.M.; Rossi, R.; Guarini, S.; Giuliani, D. Mechanisms of Hydrogen Sulfide against the Progression of Severe Alzheimer’s Disease in Transgenic Mice at Different Ages. Pharmacology 2019, 103, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Gu, J.; Pang, Y.; Liu, J.; Xu, T.; Li, X.; Hua, Y.; Newell, K.A.; Huang, X.-F.; Yu, Y.; et al. Tacrine–Hydrogen Sulfide Donor Hybrid Ameliorates Cognitive Impairment in the Aluminum Chloride Mouse Model of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 3500–3509. [Google Scholar] [CrossRef]
- Aboulhoda, B.E.; Rashed, L.A.; Ahmed, H.; Obaya, E.M.M.; Ibrahim, W.; Alkafass, M.A.L.; Abd El-Aal, S.A.; ShamsEldeen, A.M. Hydrogen sulfide and mesenchymal stem cells-extracted microvesicles attenuate LPS-induced Alzheimer’s disease. J. Cell. Physiol. 2021, 236, 5994–6010. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kyles, P.; Kalani, A.; Tyagi, N. Hydrogen Sulfide Ameliorates Homocysteine-Induced Alzheimer’s Disease-Like Pathology, Blood–Brain Barrier Disruption, and Synaptic Disorder. Mol. Neurobiol. 2016, 53, 2451–2467. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.; Long, D.; Li, J.; Ji, W.; Zhang, M.; Hong, L.; Liu, J. Hydrogen sulfide attenuates spatial memory impairment and hippocampal neuroinflammation in beta-amyloid rat model of Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 687. [Google Scholar] [CrossRef] [Green Version]
- Aschner, M.; Skalny, A.V.; Ke, T.; da Rocha, J.B.; Paoliello, M.M.; Santamaria, A.; Bornhorst, J.; Rongzhu, L.; Svistunov, A.A.; Djordevic, A.B.; et al. Hydrogen Sulfide (H2S) Signaling as a Protective Mechanism against Endogenous and Exogenous Neurotoxicants. Curr. Neuropharmacol. 2022, 20, 1908–1924. [Google Scholar] [CrossRef]
- Cao, X.; Cao, L.; Ding, L.; Bian, J. A New Hope for a Devastating Disease: Hydrogen Sulfide in Parkinson’s Disease. Mol. Neurobiol. 2017, 55, 3789–3799. [Google Scholar] [CrossRef]
- Xue, X.; Bian, J.-S. Neuroprotective Effects of Hydrogen Sulfide in Parkinson’s Disease Animal Models. Methods Enzymol. 2015, 554, 169–186. [Google Scholar]
- Xie, L.; Hu, L.-F.; Teo, X.Q.; Tiong, C.X.; Tazzari, V.; Sparatore, A.; del Soldato, P.; Dawe, G.S.; Bian, J.-S. Therapeutic Effect of Hydrogen Sulfide-Releasing L-Dopa Derivative ACS84 on 6-OHDA-Induced Parkinson’s Disease Rat Model. PLoS ONE 2013, 8, e60200. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Tazzari, V.; Giustarini, D.; Rossi, R.; Sparatore, A.; Del Soldato, P.; McGeer, E.; McGeer, P.L. Effects of Hydrogen Sulfide-releasing l-DOPA Derivatives on Glial Activation. J. Biol. Chem. 2010, 285, 17318–17328. [Google Scholar] [CrossRef] [Green Version]
- Sarukhani, M.; Haghdoost-Yazdi, H.; Sarbazi Golezari, A.; Babayan-Tazehkand, A.; Dargahi, T.; Rastgoo, N. Evaluation of the antiparkinsonism and neuroprotective effects of hydrogen sulfide in acute 6-hydroxydopamine-induced animal model of Parkinson’s disease: Behavioral, histological and biochemical studies. Neurol. Res. 2018, 40, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, D.; Su, Y.; Zhao, H.; Pang, W.; Zhao, W.; Wu, S. Protective effect of hydrogen sulfide is mediated by negative regulation of epigenetic histone acetylation in Parkinson’s disease. Arch. Med. Sci. 2020, 19. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, J.; Pan, Y.; Dong, J.; Zhang, L.; Zhang, X.; Zhang, L. Hydrogen sulfide functions as a neuromodulator to regulate striatal neurotransmission in a mouse model of Parkinson’s disease. J. Neurosci. Res. 2015, 93, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Hacioglu, G.; Cirrik, S.; Tezcan Yavuz, B.; Tomruk, C.; Keskin, A.; Uzunoglu, E.; Takir, S. The BDNF-TrkB signaling pathway is partially involved in the neuroprotective effects of hydrogen sulfide in Parkinson’s disease. Eur. J. Pharmacol. 2023, 944, 175595. [Google Scholar] [CrossRef]
- Liu, Y.; Liao, S.; Quan, H.; Lin, Y.; Li, J.; Yang, Q. Involvement of microRNA-135a-5p in the Protective Effects of Hydrogen Sulfide Against Parkinson’s Disease. Cell. Physiol. Biochem. 2016, 40, 18–26. [Google Scholar] [CrossRef]
- Huang, Y.; Omorou, M.; Gao, M.; Mu, C.; Xu, W.; Xu, H. Hydrogen sulfide and its donors for the treatment of cerebral ischaemia-reperfusion injury: A comprehensive review. Biomed. Pharmacother. 2023, 161, 114506. [Google Scholar] [CrossRef]
- Liu, J.; Mesfin, F.M.; Hunter, C.E.; Olson, K.R.; Shelley, W.C.; Brokaw, J.P.; Manohar, K.; Markel, T.A. Recent Development of the Molecular and Cellular Mechanisms of Hydrogen Sulfide Gasotransmitter. Antioxidants 2022, 11, 1788. [Google Scholar] [CrossRef] [PubMed]
- Shayea, A.M.F.; Mousa, A.M.A.; Renno, W.M.; Nadar, M.S.; Qabazard, B.; Yousif, M.H.M. Chronic Treatment With Hydrogen Sulfide Donor GYY4137 Mitigates Microglial and Astrocyte Activation in the Spinal Cord of Streptozotocin-Induced Diabetic Rats. J. Neuropathol. Exp. Neurol. 2020, 79, 1320–1343. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Anders, F.; Thanos, S.; Mann, C.; Liu, A.; Grus, F.H.; Pfeiffer, N.; Prokosch-Willing, V. Hydrogen Sulfide Protects Retinal Ganglion Cells Against Glaucomatous Injury In Vitro and In Vivo. Investig. Opthalmology Vis. Sci. 2017, 58, 5129–5141. [Google Scholar] [CrossRef] [Green Version]
- Lazarević, M.; Battaglia, G.; Jevtić, B.; Djedovic, N.; Bruno, V.; Cavalli, E.; Miljković, Đ.; Nicoletti, F.; Momčilović, M.; Fagone, P. Upregulation of Tolerogenic Pathways by the Hydrogen Sulfide Donor GYY4137 and Impaired Expression of H2S-Producing Enzymes in Multiple Sclerosis. Antioxidants 2020, 9, 608. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Perry, A.; Zhou, Z.; Bucci, M.; Papapetropoulos, A.; Cirino, G.; Wood, M.E. Phosphinodithioate and Phosphoramidodithioate Hydrogen Sulfide Donors. In Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide; Springer: Berlin/Heidelberg, Germany, 2015; pp. 337–363. [Google Scholar]
- Huang, C.W.; Feng, W.; Peh, M.T.; Peh, K.; Dymock, B.W.; Moore, P.K. A novel slow-releasing hydrogen sulfide donor, FW1256, exerts anti-inflammatory effects in mouse macrophages and in vivo. Pharmacol. Res. 2016, 113, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; McGeer, E.; Kodela, R.; Kashfi, K.; McGeer, P.L. NOSH-aspirin (NBS-1120), a novel nitric oxide and hydrogen sulfide releasing hybrid, attenuates neuroinflammation induced by microglial and astrocytic activation: A new candidate for treatment of neurodegenerative disorders. Glia 2013, 61, 1724–1734. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Casili, G.; Randi, E.; Panagaki, T.; Zuhra, K.; Petrosino, M.; Szabo, C. Inhibition of the 3-mercaptopyruvate sulfurtransferase—Hydrogen sulfide system promotes cellular lipid accumulation. GeroScience 2022, 44, 2271–2289. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, M.; Zuhra, K.; Kopec, J.; Hutchin, A.; Szabo, C.; Majtan, T. H2S biogenesis by cystathionine beta-synthase: Mechanism of inhibition by aminooxyacetic acid and unexpected role of serine. Cell. Mol. Life Sci. 2022, 79, 438. [Google Scholar] [CrossRef]












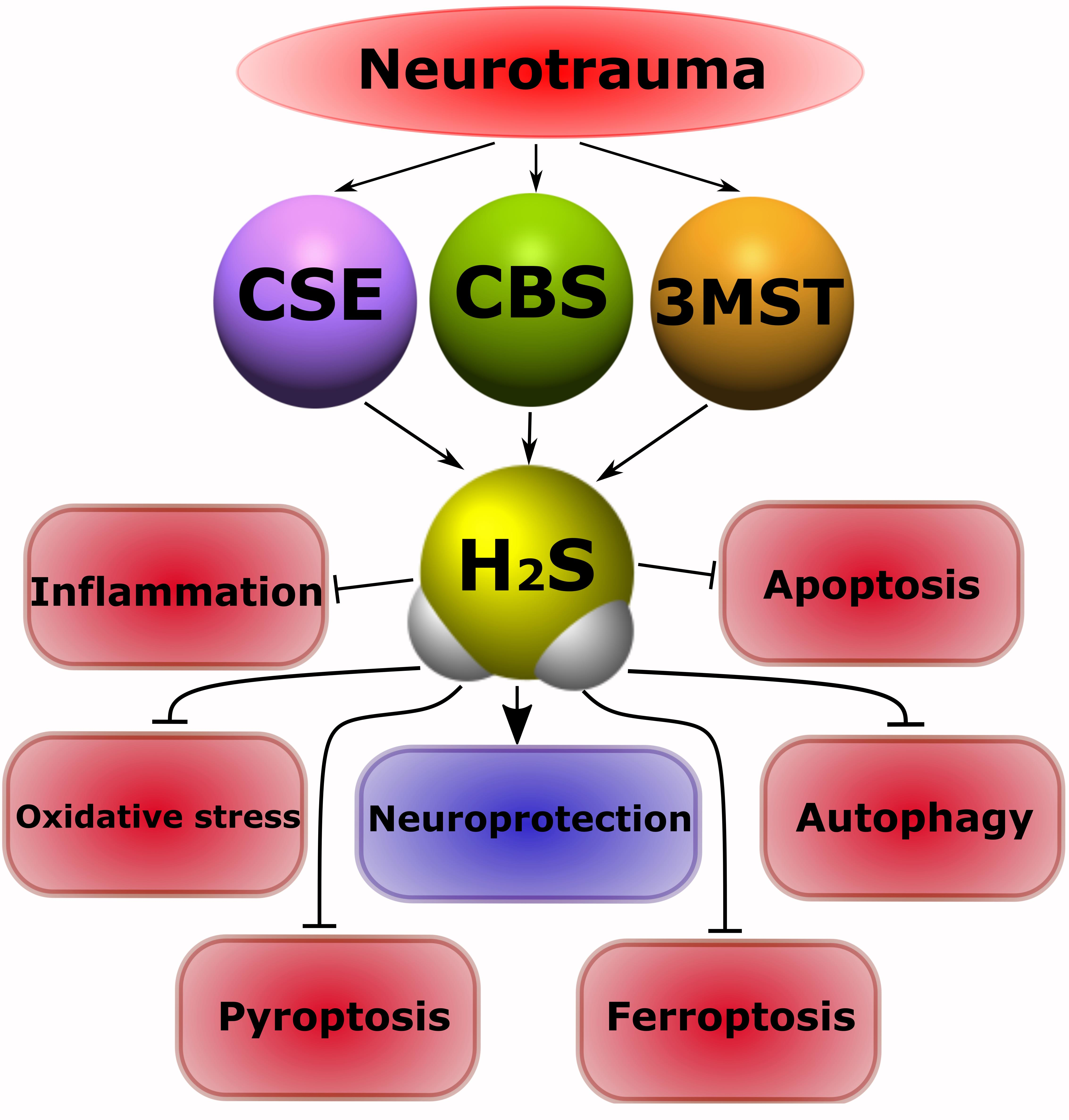{kind=link}
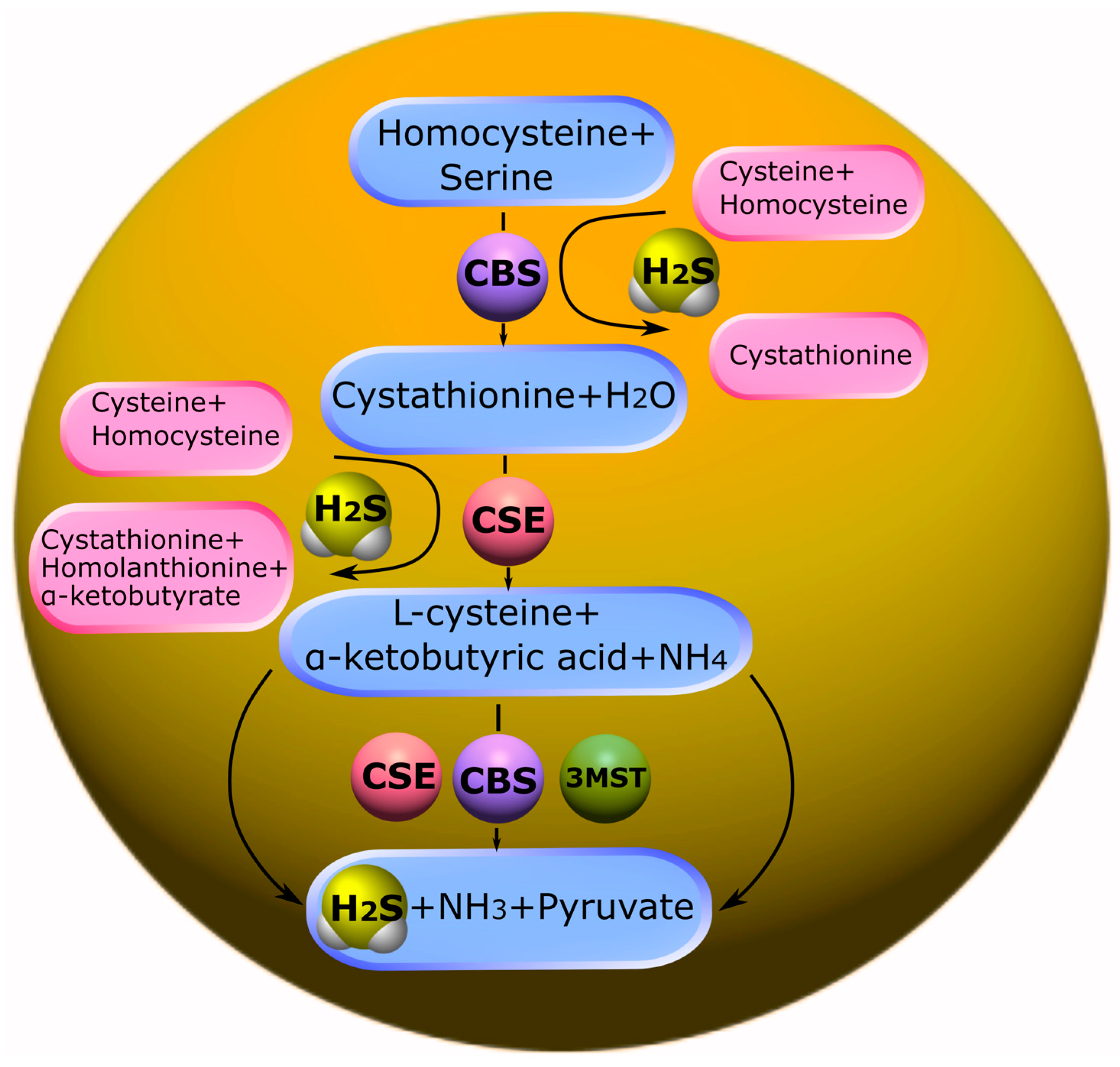{kind=link}
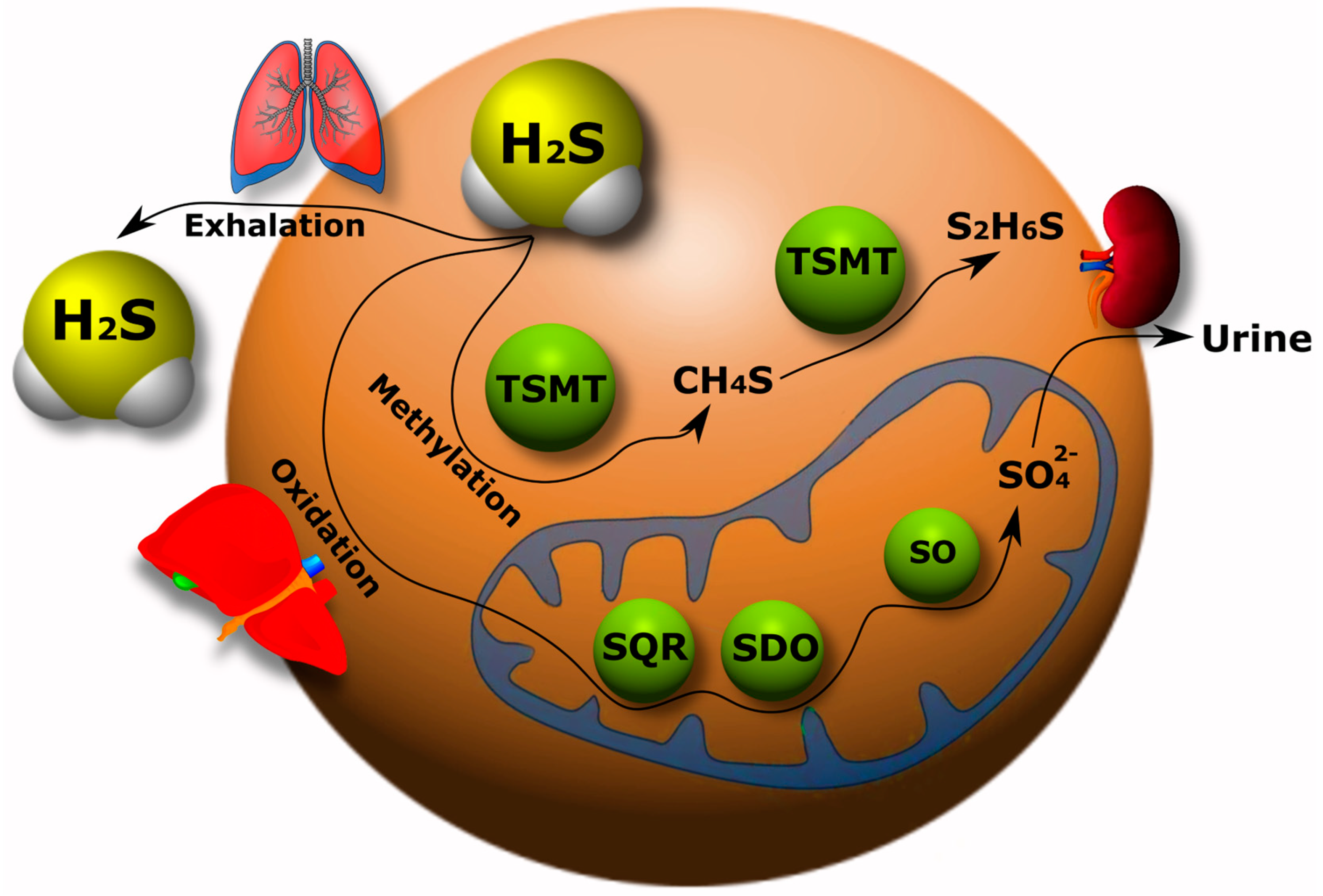{kind=link}
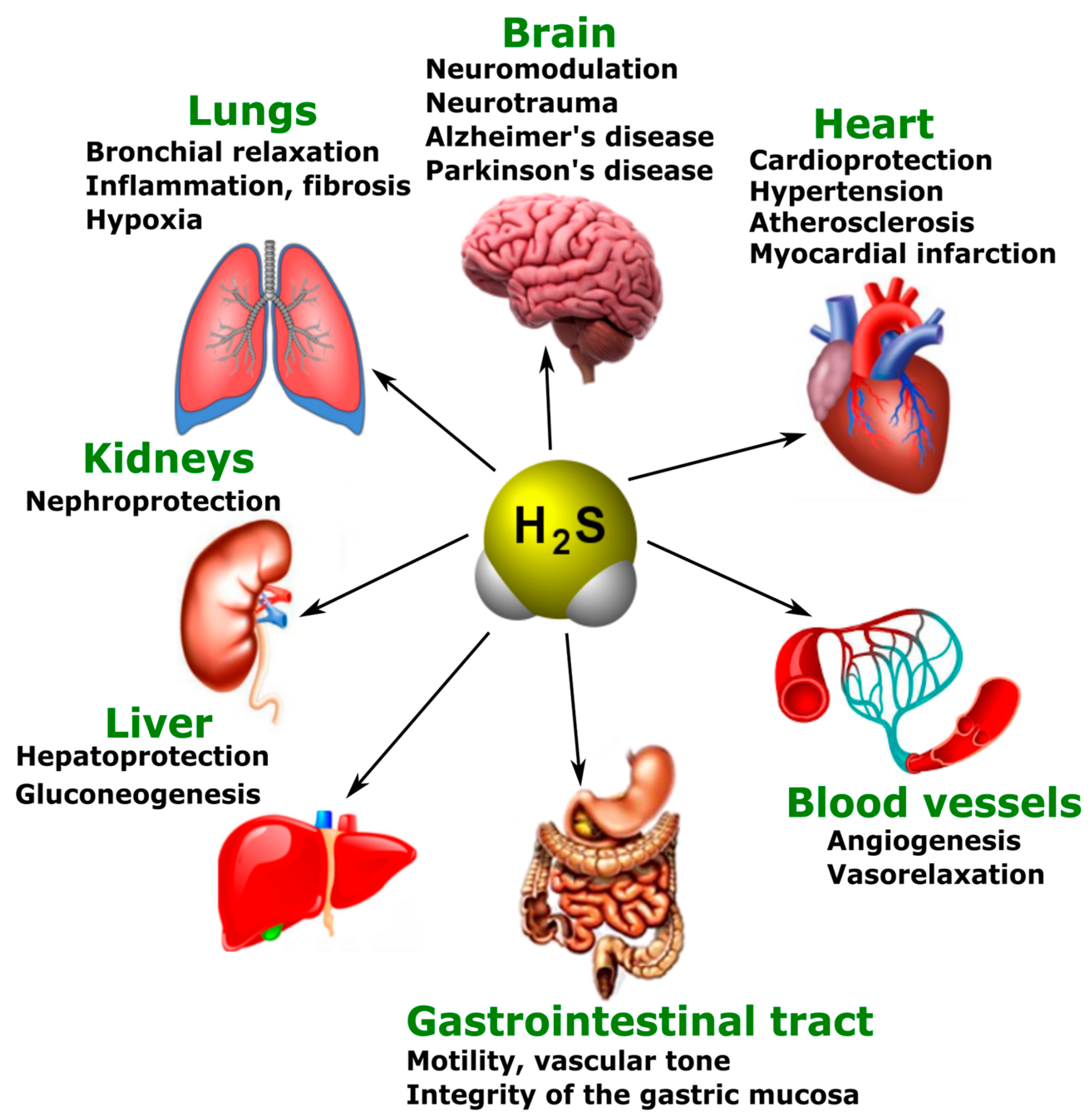{kind=link}
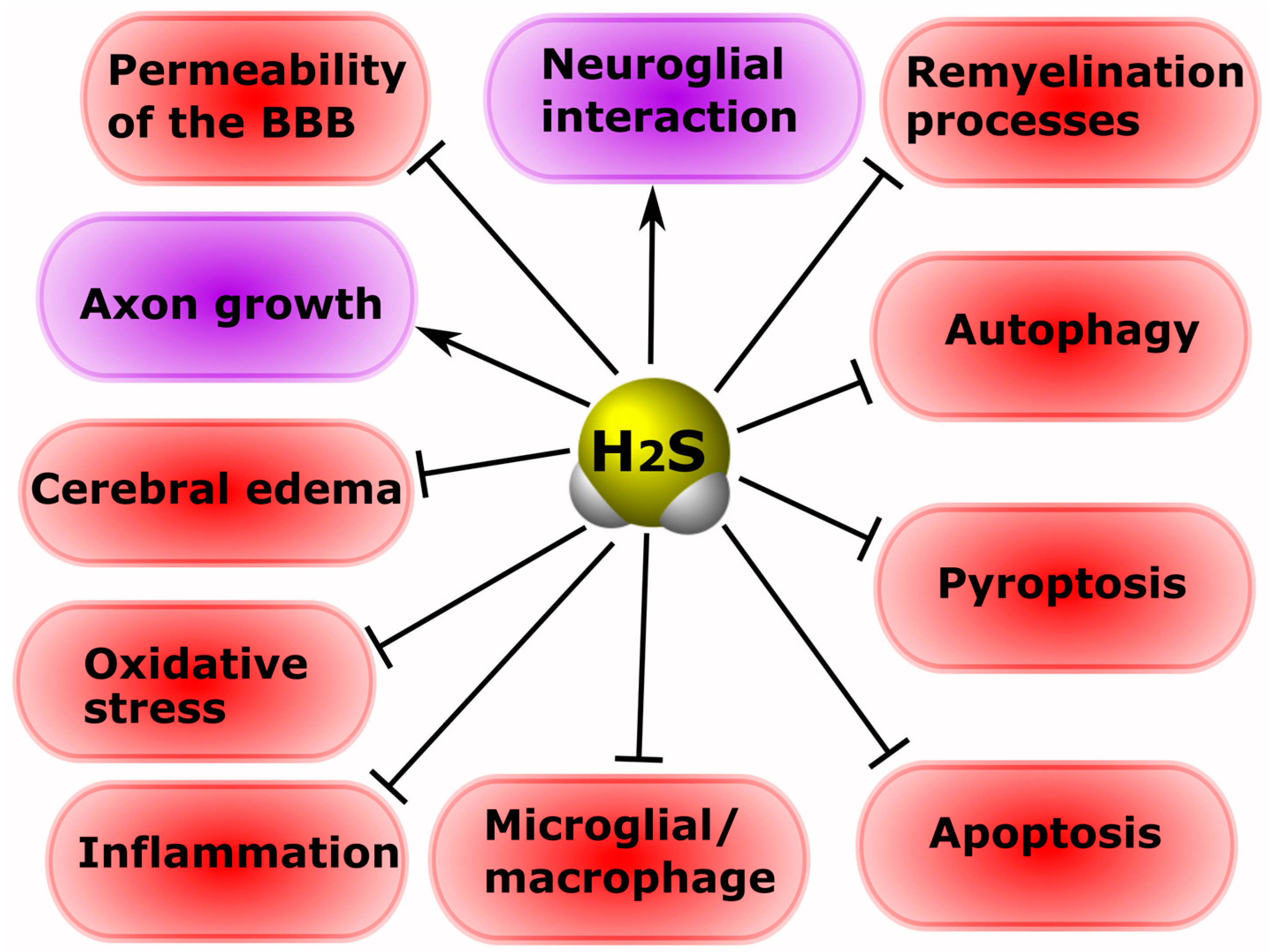{kind=link}
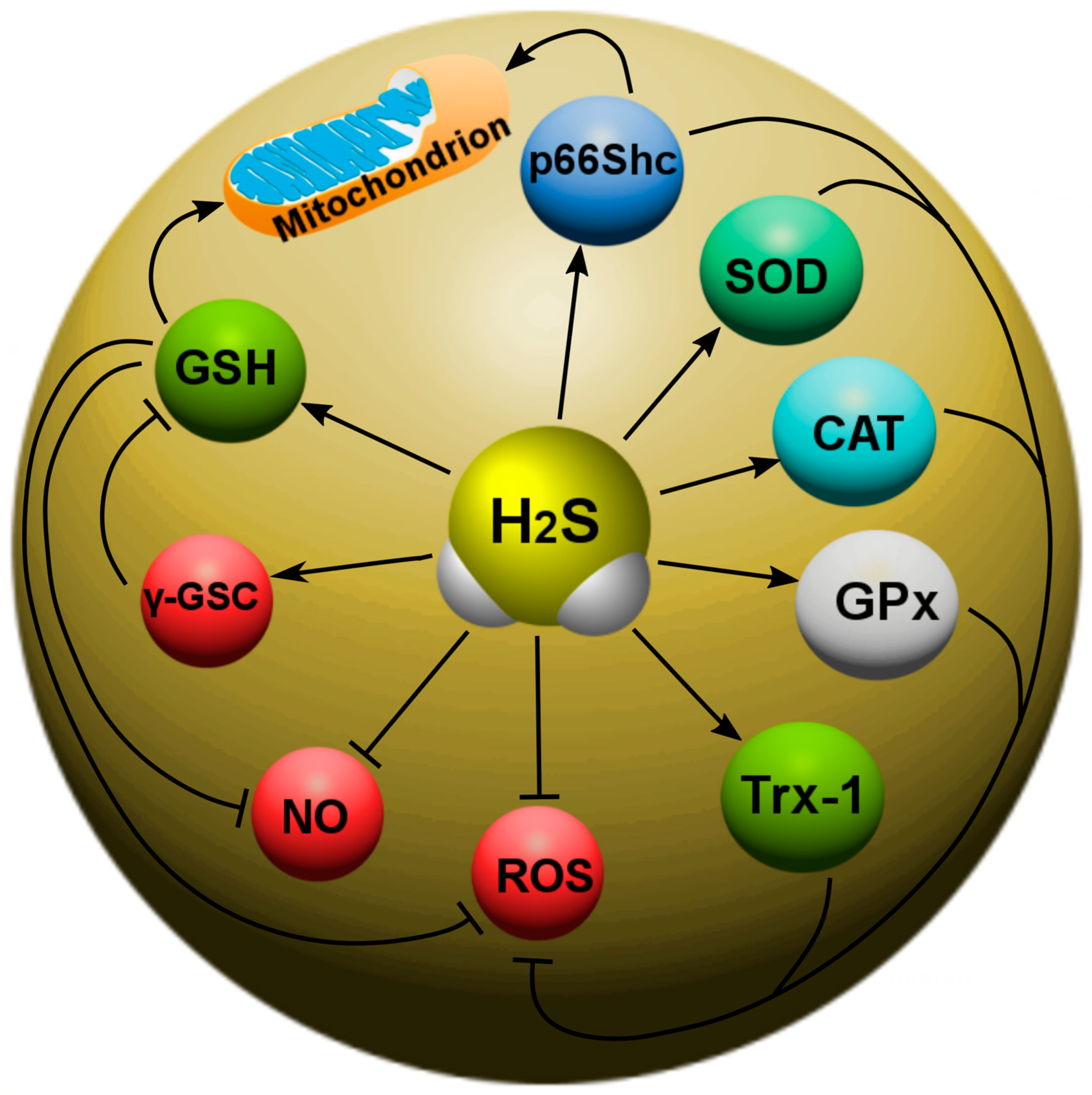{kind=link}
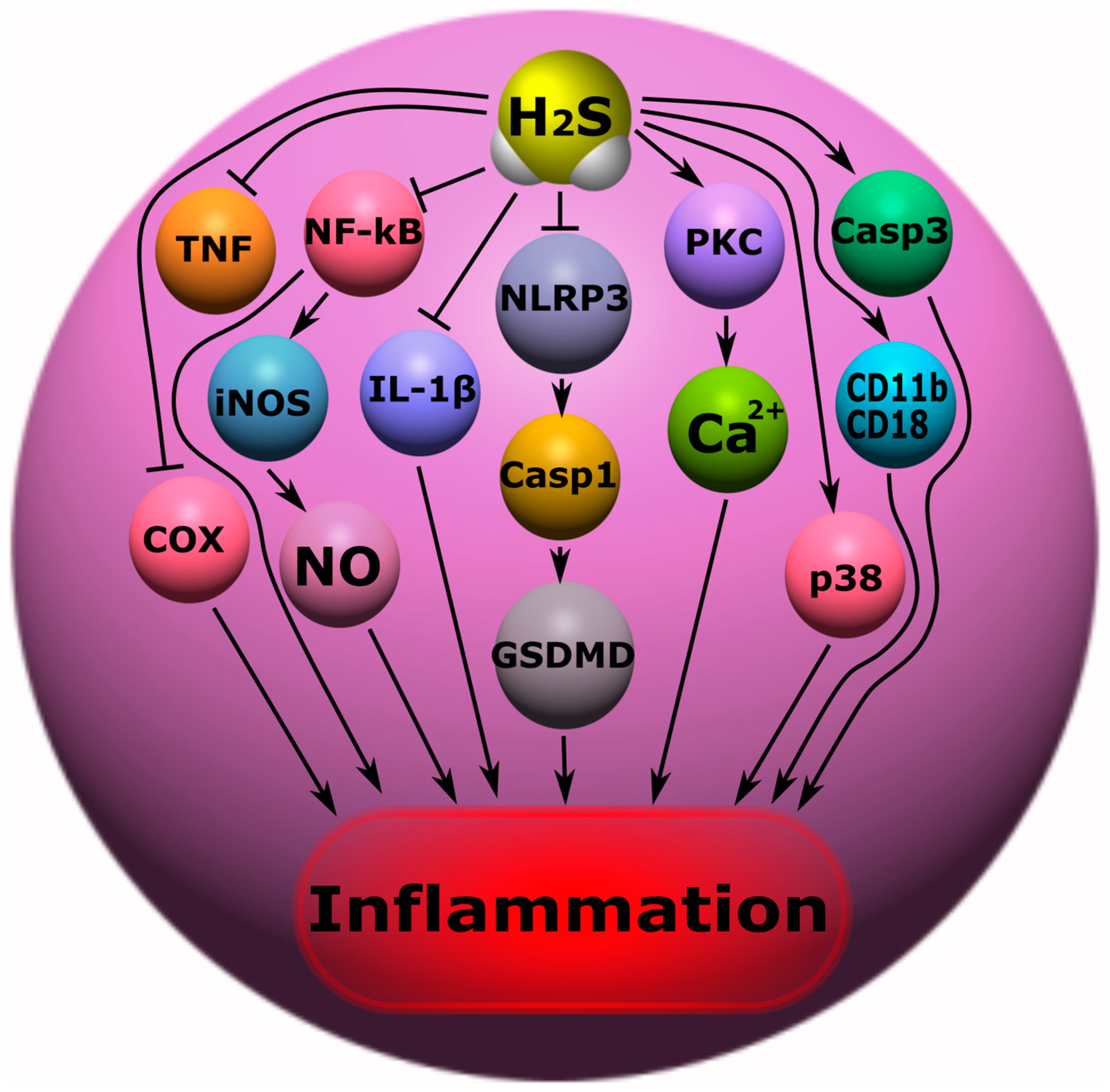{kind=link}
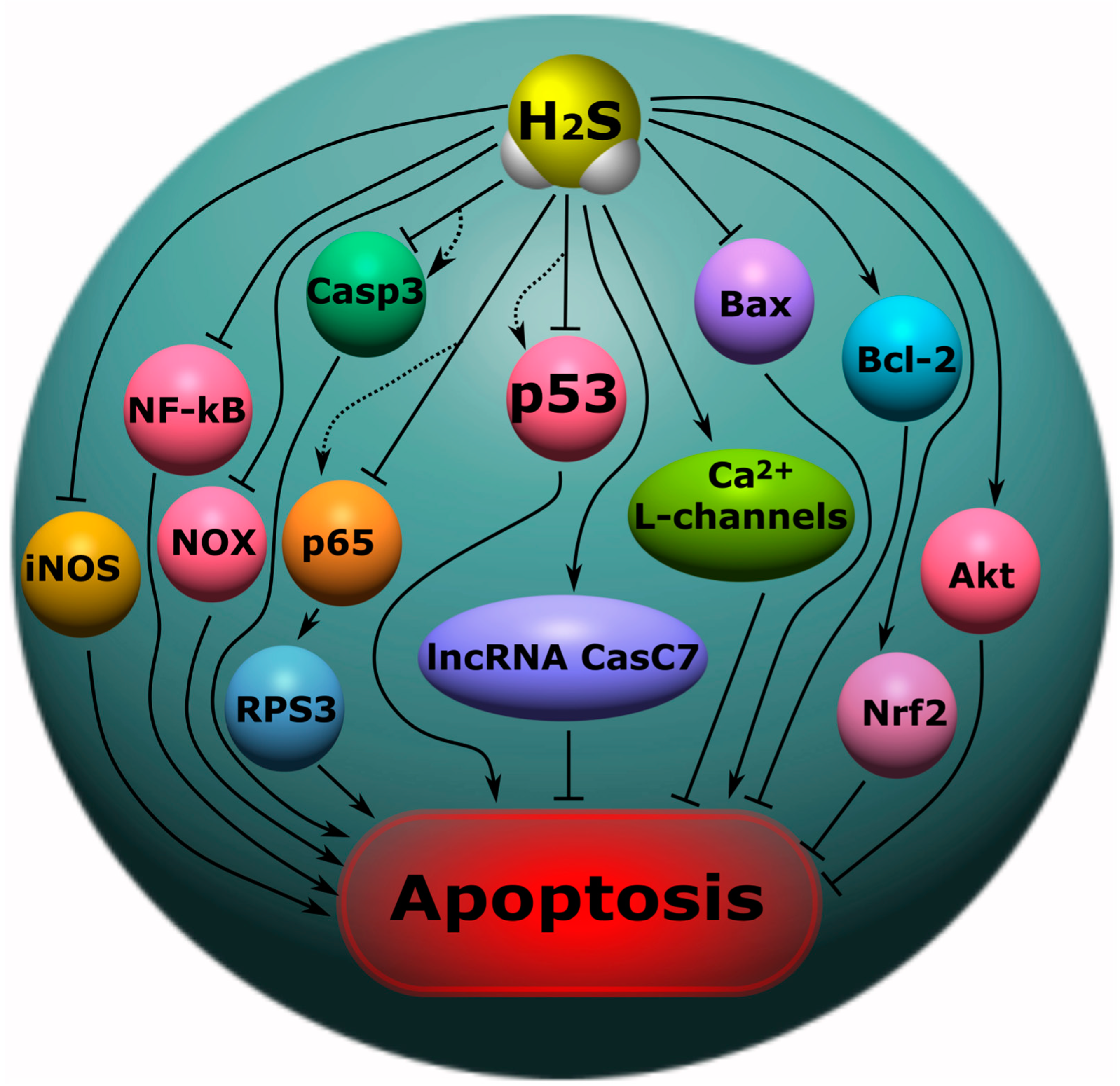{kind=link}
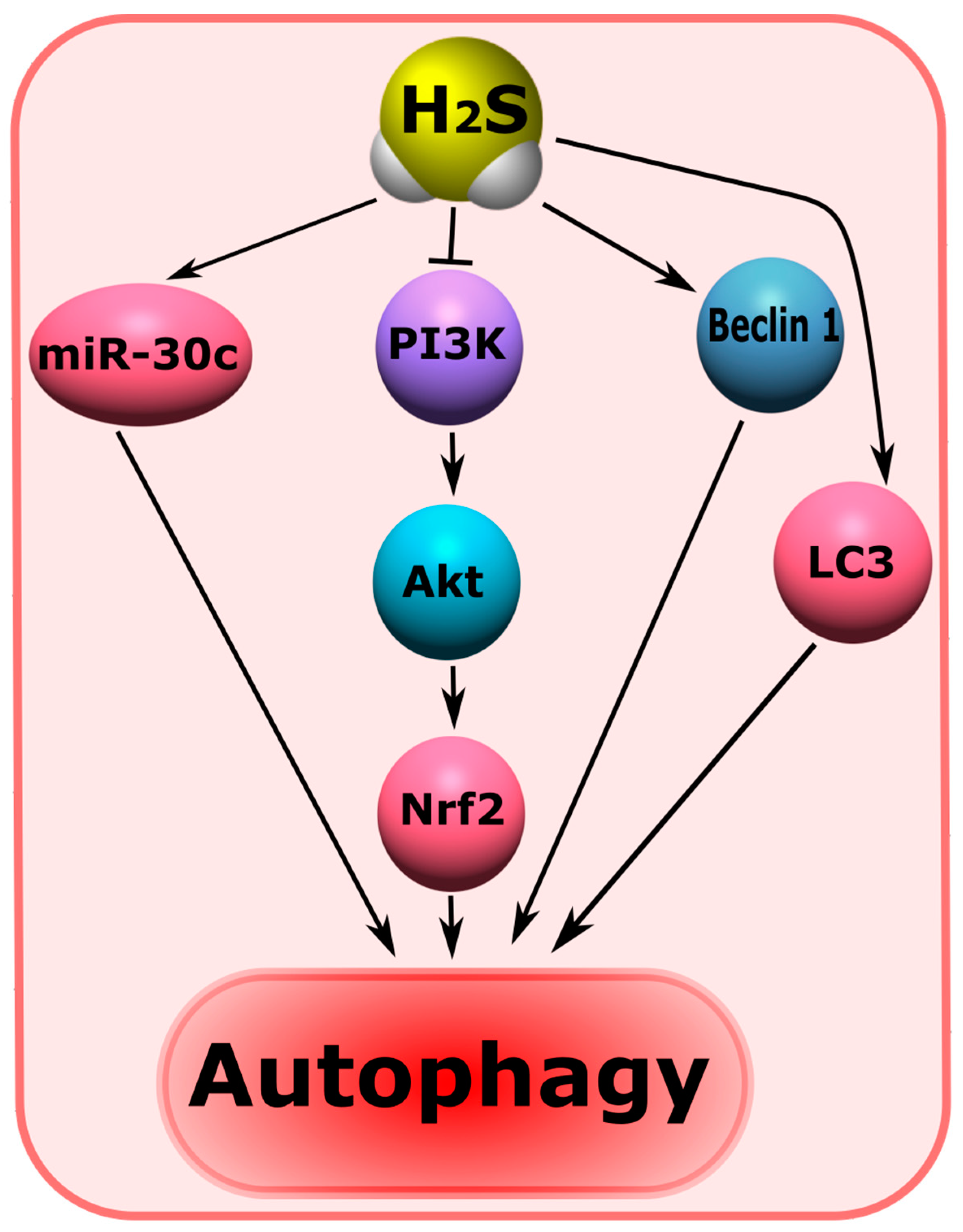{kind=link}
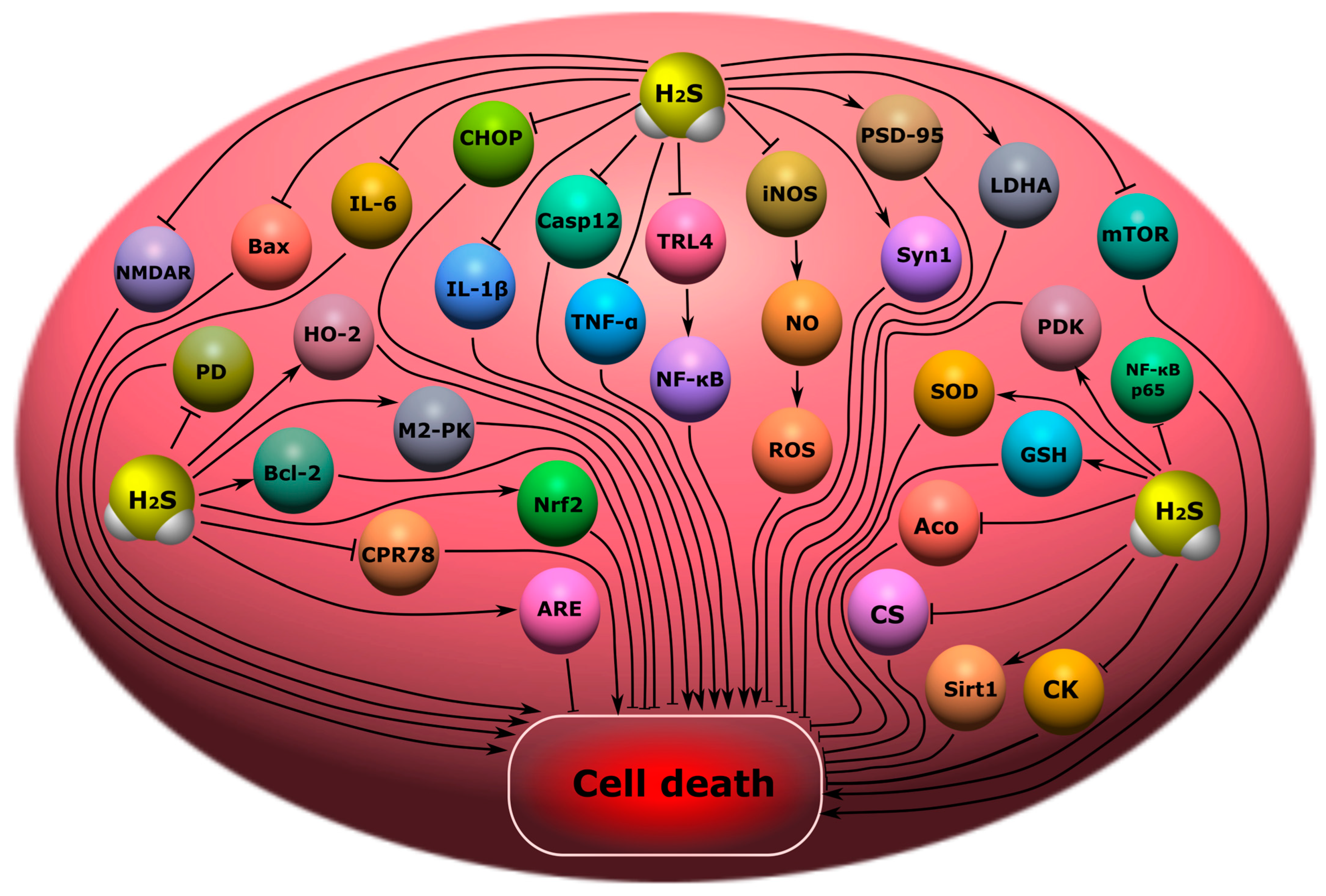{kind=link}
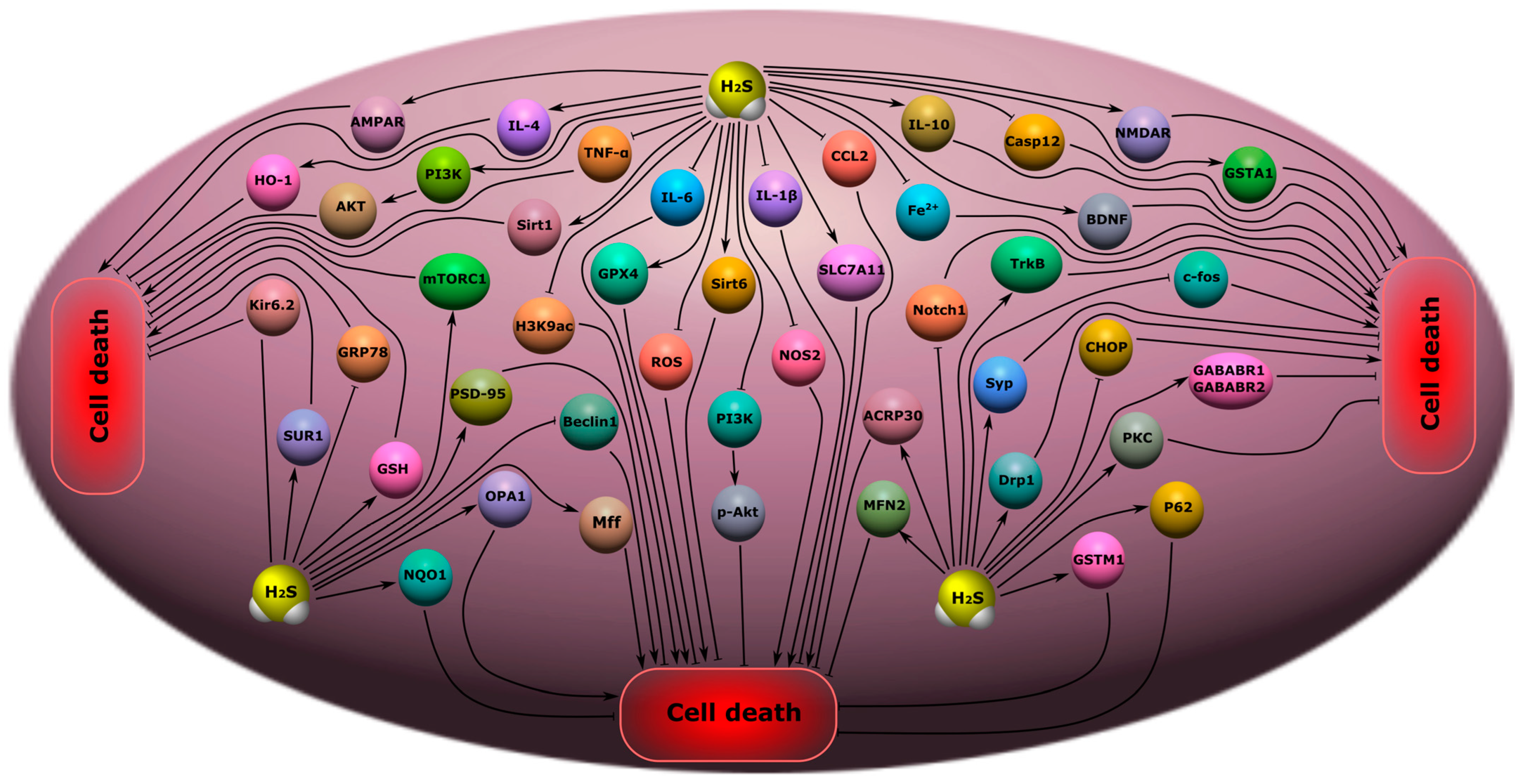{kind=link}
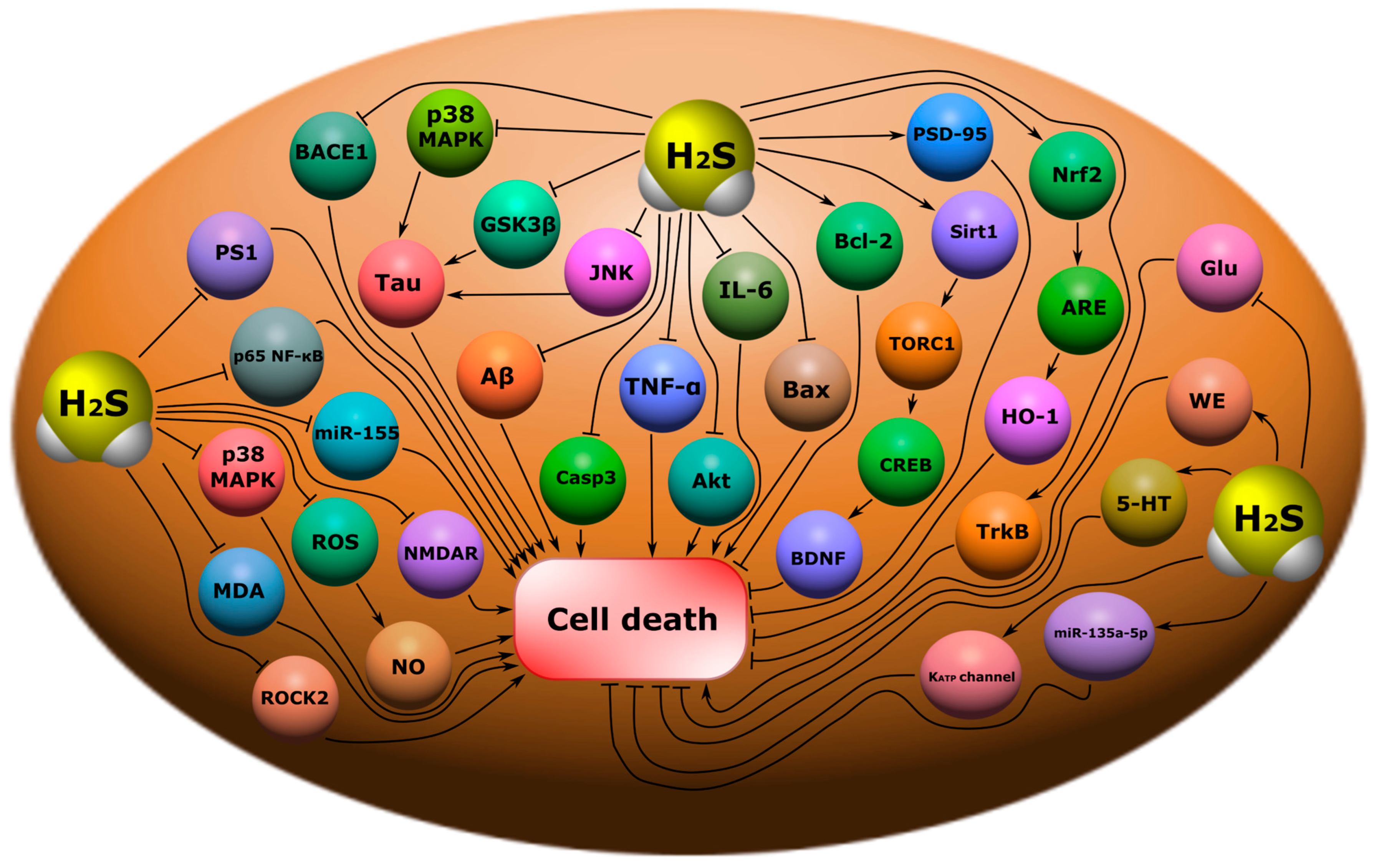{kind=link}
| Database | Search Strategy |
|---|---|
| PubMed | (hydrogen sulfide OR gasotransmitters OR cystathionine-β-synthase OR cystathionine-γ-lyase OR 3-mercaptopyruvate sulfurtransferase OR hydrogen sulfide donors) AND (neurotrauma OR traumatic brain injury OR spinal cord injury OR peripheral nerve injury OR axotomy OR apoptosis OR autophagy OR ferroptosis OR pyroptosis OR cell death OR axon OR dendrites OR dendritic spines OR cognitive impairment OR depression OR anxiety disorders OR epilepsy OR encephalopathy OR chronic pain OR oxidative stress OR inflammation OR neuroinflammation OR brain OR spinal cord OR neurotrophic factors OR cytokines OR neurodegeneration OR Alzheimer’s disease OR Parkinson’s disease OR protein folding) |
| Scopus | TITLE-ABS-KEY (“hydrogen sulfide” OR “gasotransmitters”) AND TITLE-ABS-KEY (“brain” OR “spinal cord” OR “nerves” OR “neurotrophic factors” OR “mental disorders” OR “neurodegenerative diseases”) |
| Web of Science | TOPIC (*hydrogen sulfide* OR *gasotransmitters*) AND TOPIC (*neurotrauma* OR *apoptosis* OR *autophagy* OR *pyroptosis* OR *ferroptosis* OR *axon* OR *dendrites* OR *donors* OR *mental disorders* OR *neurodegenerative diseases*) |
| The Role of H2S in Cell Death | ||
|---|---|---|
| Process | Effects of H2S on signaling pathways/molecular targets | Effect |
| Oxidative stress | Increasing GSH, Trx-1, COD, CAT, GPx, p66Shcis, and ROS uptake | Reducing oxidative stress levels |
| Ca2+-homeostasis | Modulation of NMDAR activity via PKA-dependent and independent pathways; regulation of L-type Ca2+ channels and fast T-type Ca-type CaV 3.2 channels | Increase/decrease in intracellular Ca2+ levels |
| Inflammation | Inhibition of ROS, NF-κB, leukocyte endothelial adhesion, TNF, IL-1 β, iNOS, NLRP3/caspase-1/GSDMD, cytochrome c oxidase | Reducing the level of inflammation |
| Oxidation of H2S to form sulfite leads to leukocyte adhesion and neutrophil activation via CD11b/CD18 and PKC/CaM; inhibition of H2S cleavage of caspase-3 and p38 in granulocytes | Increased levels of inflammation | |
| Remyelination | PI3K/AKT/mTOR activation | Remyelination and repair of axons |
| Regulation of LAMP1, p75NTR, c-Jun, and p-ERK1/2 | Increased dedifferentiation and proliferation of Schwann cells in Wallerian degeneration | |
| Apoptosis | Decreased expression of p53, caspase-3, Bax, NF-κB p65, NOX4, iNOS, COX-2; increased levels of Bcl-2, ncRNA CasC7 and Akt phosphorylation | Reducing the level of apoptosis |
| Autophagy | Inhibition of the PI3K/Akt/Nrf2 and ROS signaling pathways | Decreased autophagy |
| Increasing miR-30c, Beclin 1, and LC3 levels | Increase in autophagy | |
| Ferroptosis | Inhibition of ROS, LDH, and Fe2+ accumulation; increased GSH levels, NRF2/KEAP1 and AMPK; activation of p62 phosphorylation | Reducing ferroptosis |
| Pyroptosis | Inhibition of NOD-, LRR-, and NLRP3, GSDMD, caspase-1, and ASC | Reducing pyroptosis |
| The role of H2S in psychiatric disorders | ||
| Cognitive impairment | Inhibition of endoplasmic reticulum stress, caspase-12, CHOP and TLR4/NF-κB; decrease in the level of TNF-α, IL-1β and IL-6, Sirt1, ROS, LP, CPR78, CHOP, caspase-12, Bax; increase in synapsin-1 and PSD-95, Bcl-2, HO-2, M2-RK, LDHA, PDK in the hippocampus; catecholamine level modulation | Reducing the symptoms of cognitive impairment; improving spatial memory, learning, memorization; reducing the level of apoptosis in the cerebral cortex and hippocampus |
| Encephalopathy | Nrf2/ARE activation; decrease in IL-1β, IL-6, TNF-α levels; restoration of SIRT1 levels and phosphorylation of mTOR and NF-κB p65 | Reducing the symptoms of encephalopathy; reducing the level of neuroinflammation and apoptosis |
| Decreased activity of CS, Aco, and CK; increased LP in the brain with a high level of H2S | Increased neuroinflammation and apoptosis | |
| Depression and anxiety disorders | Increased expression of Sirt1, Sirt6, IL-4, IL-10, NOS2, PI3K/p-Akt, mTORC1, TrkB PSD-95, synaptophysin, and AMPA receptor GluR1/2 subunit; decreased levels of IL-1β, IL-6, TNF-α; deposition of Fe2+, ROS, H3K9ac, Notch1, Beclin 1, GRP78 | Antidepressant and anxiolytic effect; improvement of memory, learning ability; reduction of apoptosis; loss of dendritic spines |
| Epilepsy | Decreased levels of aquaporin 4, 1β, IL-6, TNF-α, c-fos; increased expression of PKC, Kir6.2 and SUR1, GABABR1 and GABABR2 | Reducing epileptic seizures |
| NMDAR and AMPAR activation | Increase in epileptic seizures | |
| Chronic pain | Decrease in PI3K, TNF-α, IL-1β, IL-6; increase in Nrf2, HO-1, NQO1 and GSTM1 | Reduced symptoms of chronic pain, neuroinflammation and apoptosis |
| The role of H2S in neurodegenerative disorders | ||
| Alzheimer’s disease | Inhibition of Tau hyperphosphorylation; expression of JNK, p38, TNF-α, IL-6, IL-1β, miR-155, pAkt, Bax, caspase-3, Aβ1-40, Aβ42; phosphorylation of p38 MAPK, p65 NF-κB, BACE1; increased levels of synaptophysin, PSD-95, Bcl-2, Sirt1, Nrf2 | Improving memory, learning; reducing the number of amyloid β-plaques in the hippocampal cortex, the level of neuroinflammation and oxidative stress |
| Parkinson’s disease | Increased expression of Nrf-2, dopamine, GSH; activation of BDNF/TrkB, miR-135a-5p; inhibition of ROS/NO, LP, ROCK2 | Reducing the progression of Parkinson’s disease; decreased death of dopaminergic neurons in the SN; neuroinflammation, oxidative stress |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodkin, S.; Nwosu, C.; Sannikov, A.; Raevskaya, M.; Tushev, A.; Vasilieva, I.; Gasanov, M. The Role of Hydrogen Sulfide in Regulation of Cell Death following Neurotrauma and Related Neurodegenerative and Psychiatric Diseases. Int. J. Mol. Sci. 2023, 24, 10742. https://doi.org/10.3390/ijms241310742
Rodkin S, Nwosu C, Sannikov A, Raevskaya M, Tushev A, Vasilieva I, Gasanov M. The Role of Hydrogen Sulfide in Regulation of Cell Death following Neurotrauma and Related Neurodegenerative and Psychiatric Diseases. International Journal of Molecular Sciences. 2023; 24(13):10742. https://doi.org/10.3390/ijms241310742
Chicago/Turabian StyleRodkin, Stanislav, Chizaram Nwosu, Alexander Sannikov, Margarita Raevskaya, Alexander Tushev, Inna Vasilieva, and Mitkhat Gasanov. 2023. "The Role of Hydrogen Sulfide in Regulation of Cell Death following Neurotrauma and Related Neurodegenerative and Psychiatric Diseases" International Journal of Molecular Sciences 24, no. 13: 10742. https://doi.org/10.3390/ijms241310742