
The Role of Gasotransmitter-Dependent Signaling Mechanisms in Apoptotic Cell Death in Cardiovascular, Rheumatic, Kidney, and Neurodegenerative Diseases and Mental Disorders
 , , , , ,
, , , , ,
Abstract
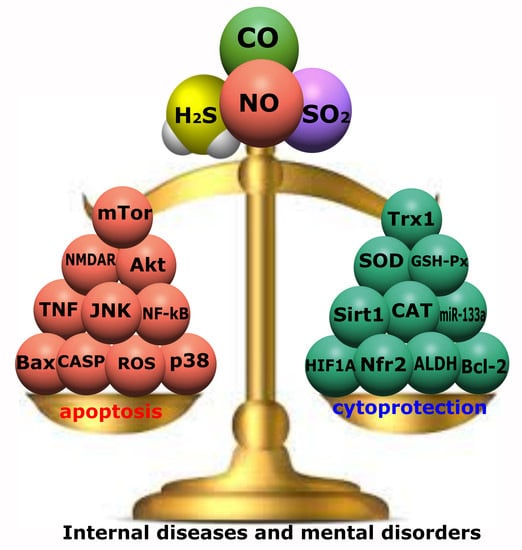
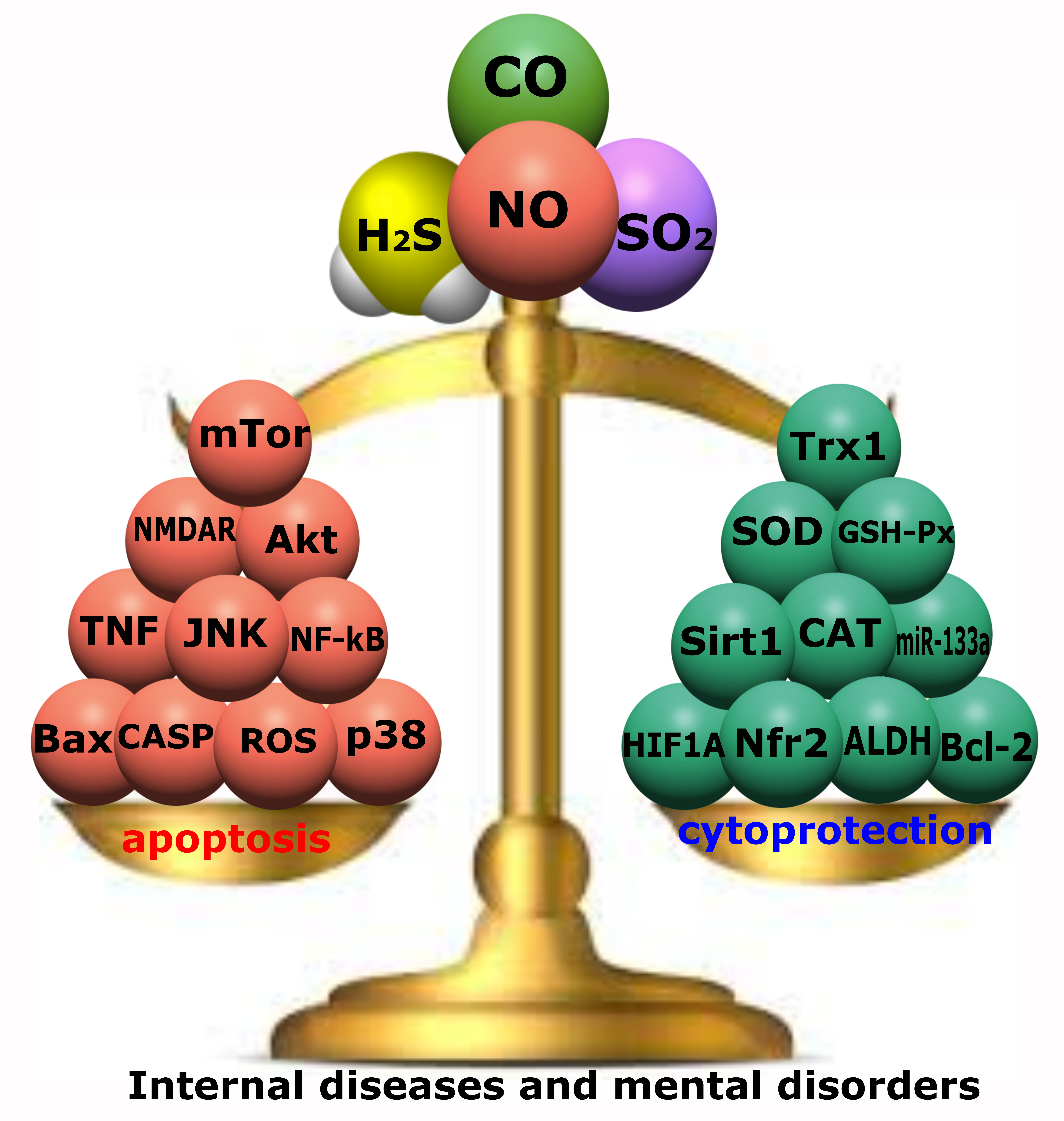
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
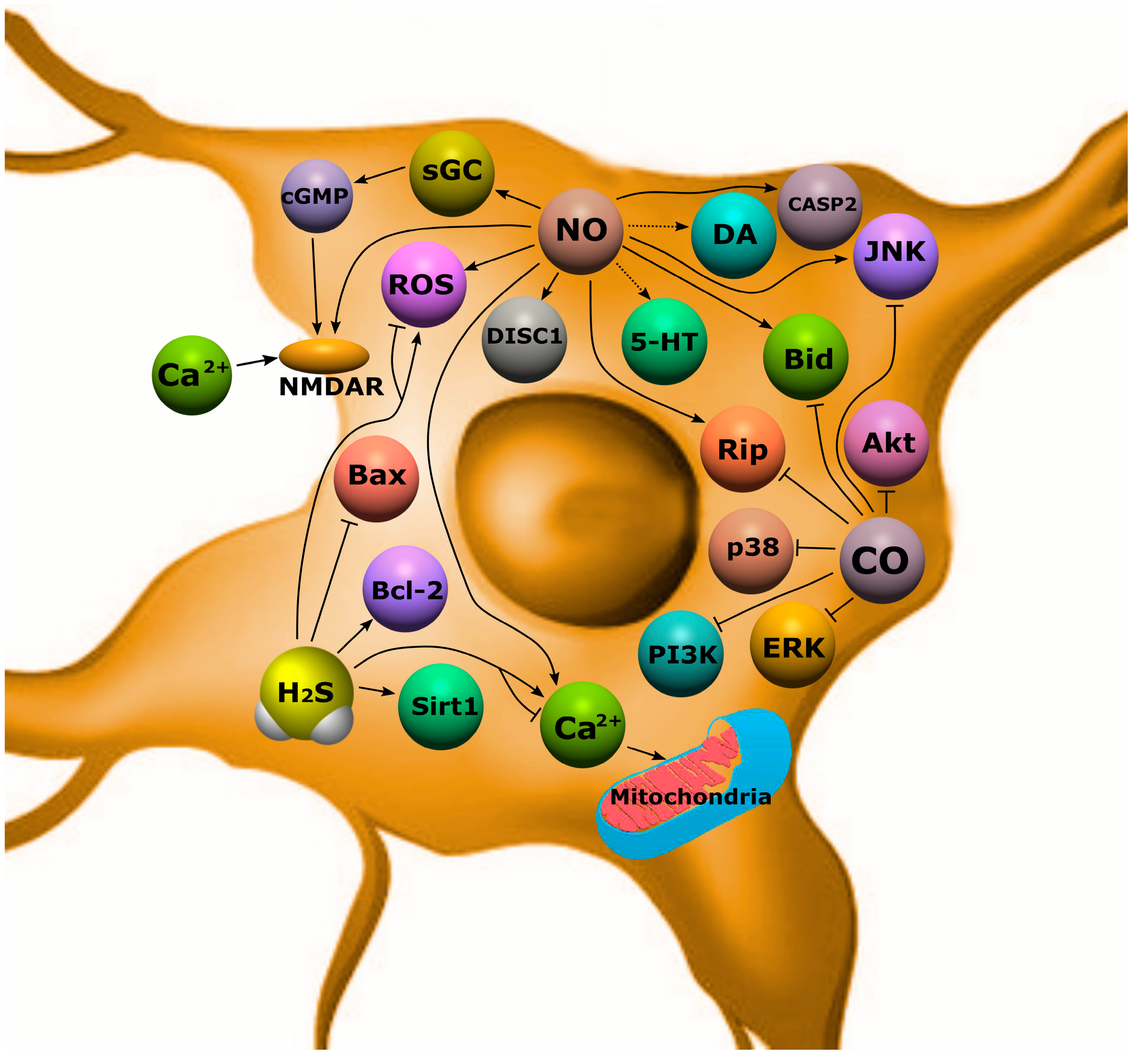{kind=link}
1. Introduction
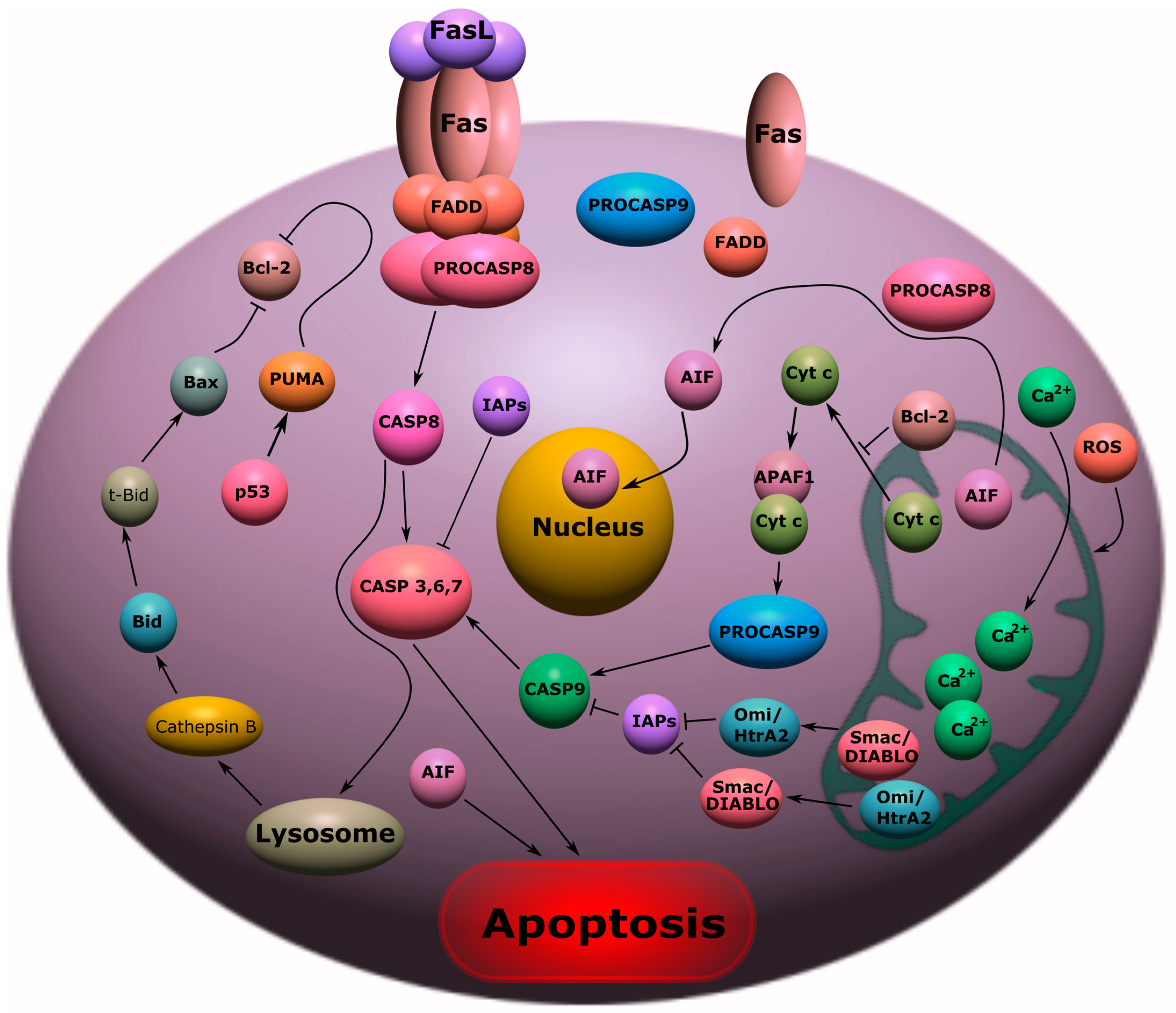
2. Apoptosis
2.1. Receptor-Dependent Pathway of Apoptosis
2.2. Mitochondrial Pathway of Apoptosis
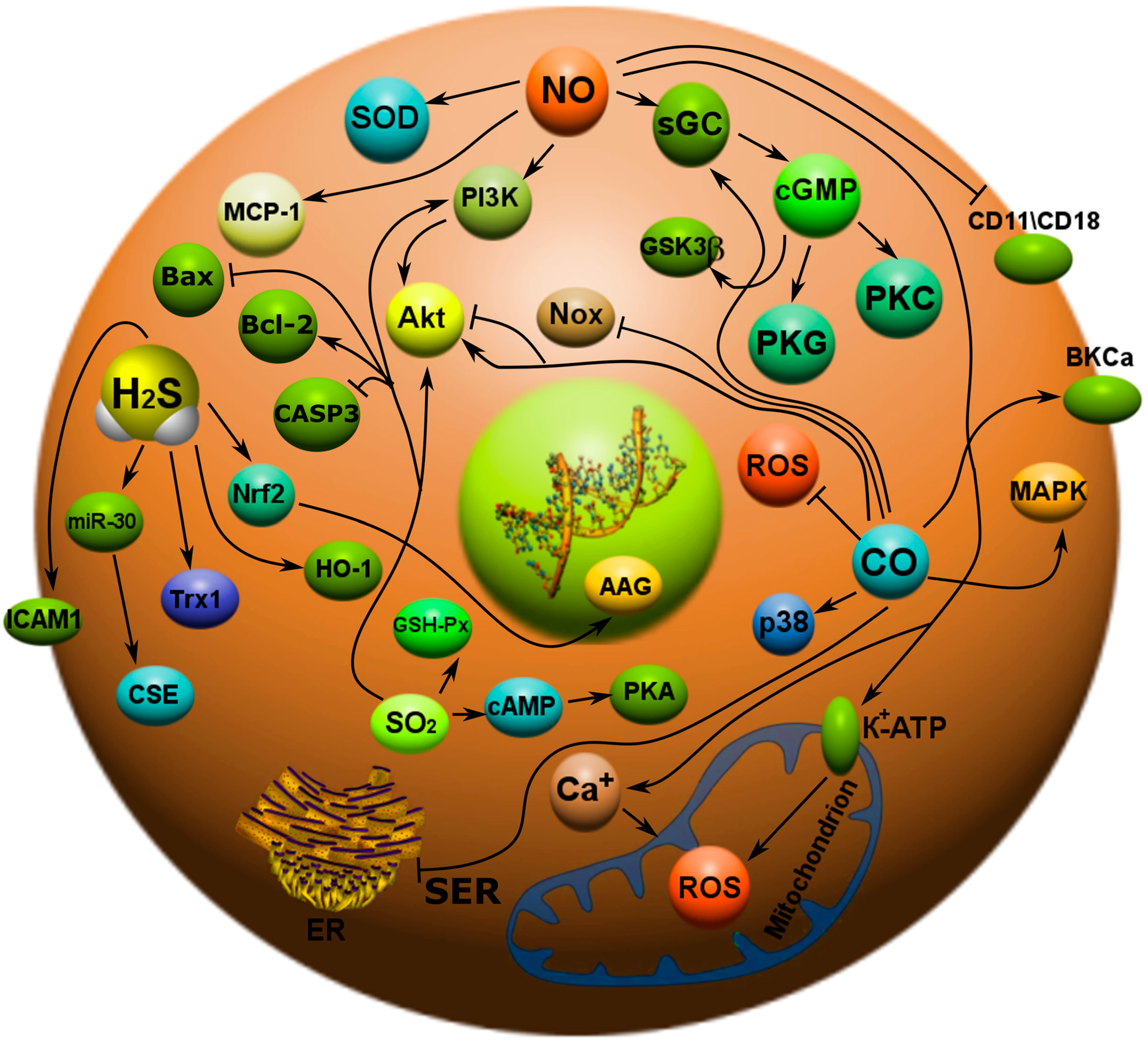
3. Gasotransmitters and Their Role in Apoptosis
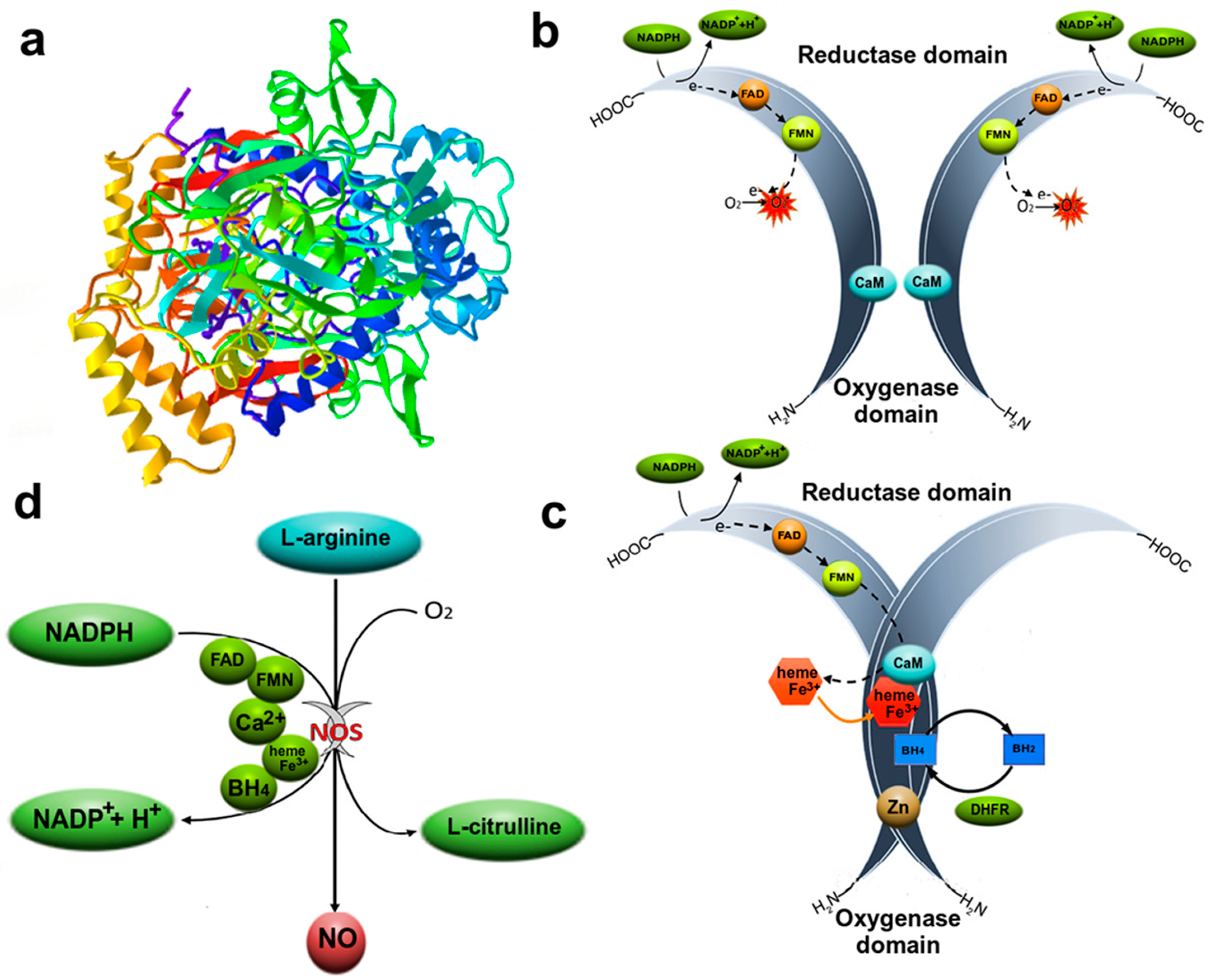
3.1. Nitric Oxide
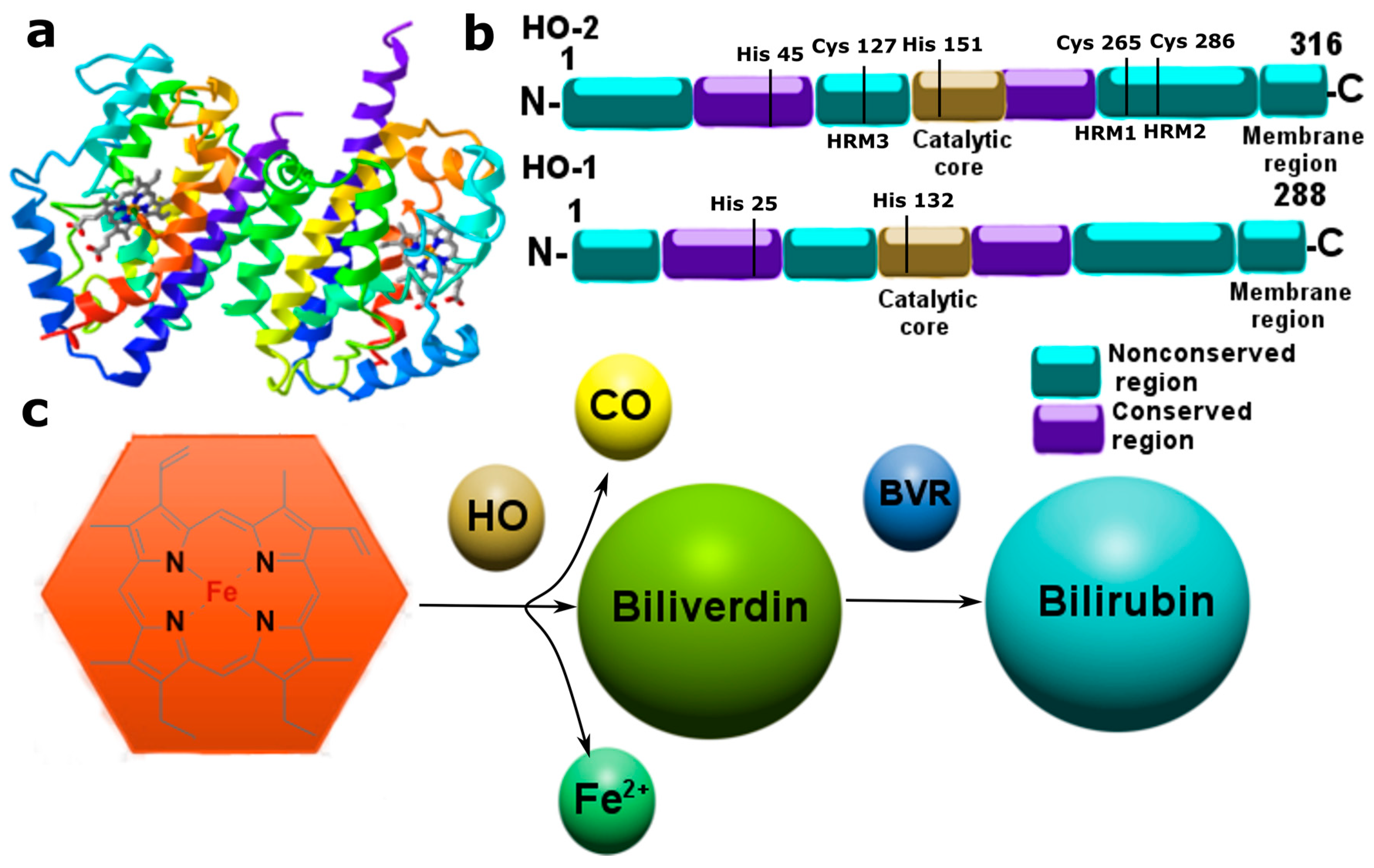
3.2. Carbon Monoxide
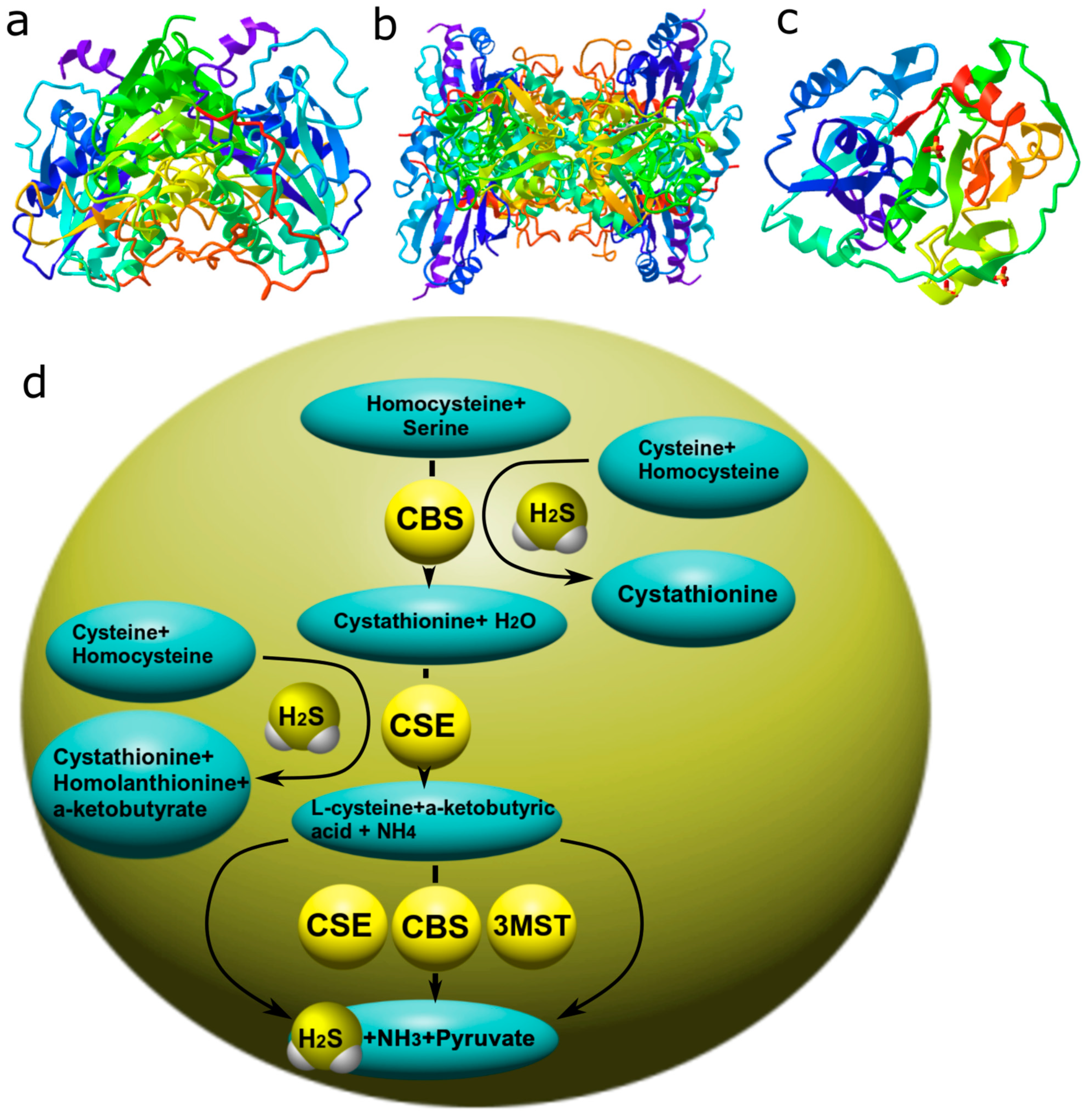
3.3. Hydrogen Sulfide
3.4. Sulfur Dioxide
4. Molecular Mechanisms of Gasotransmitter-Dependent Apoptosis in Internal Diseases
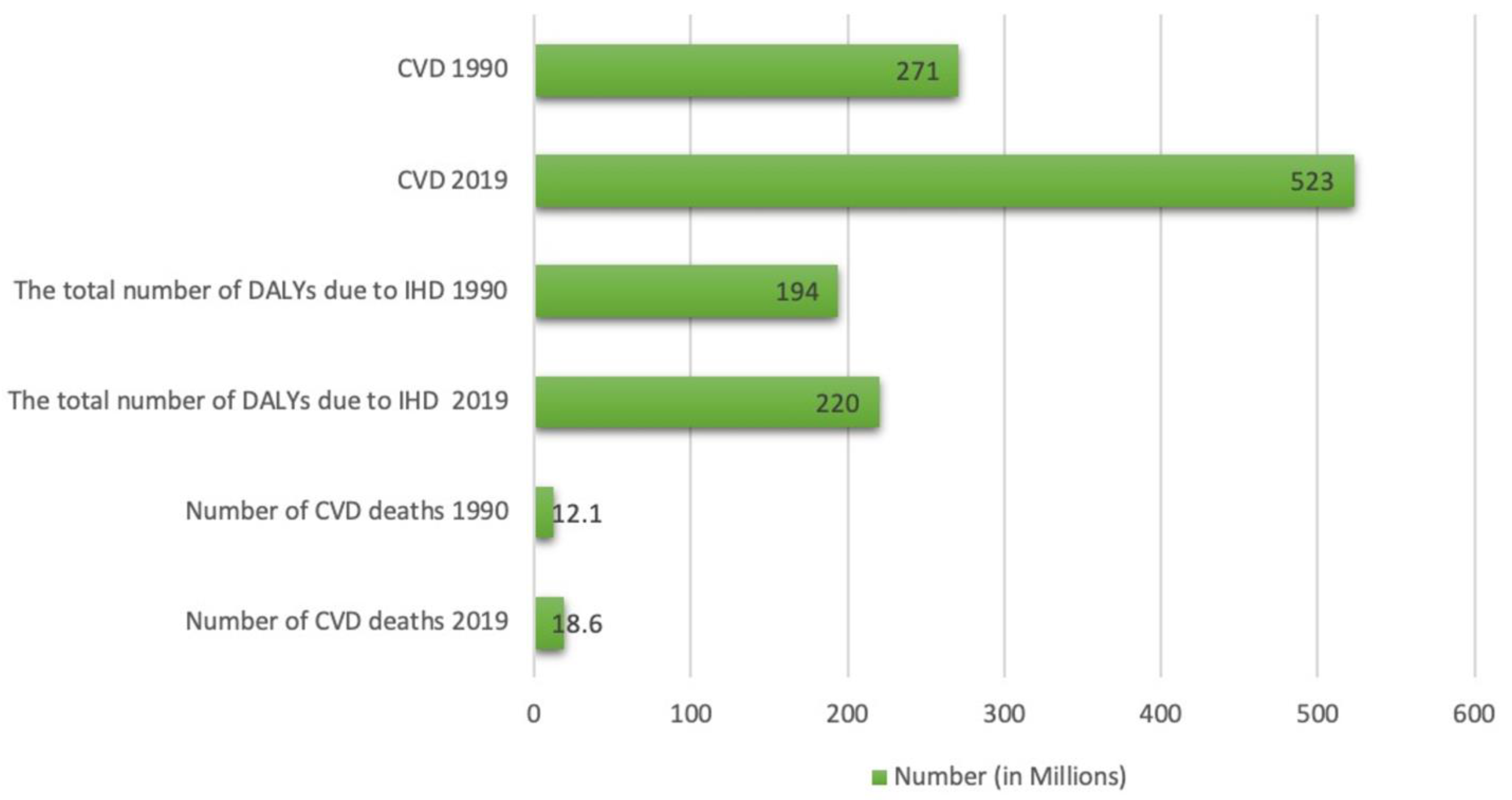
4.1. Cardiovascular Diseases
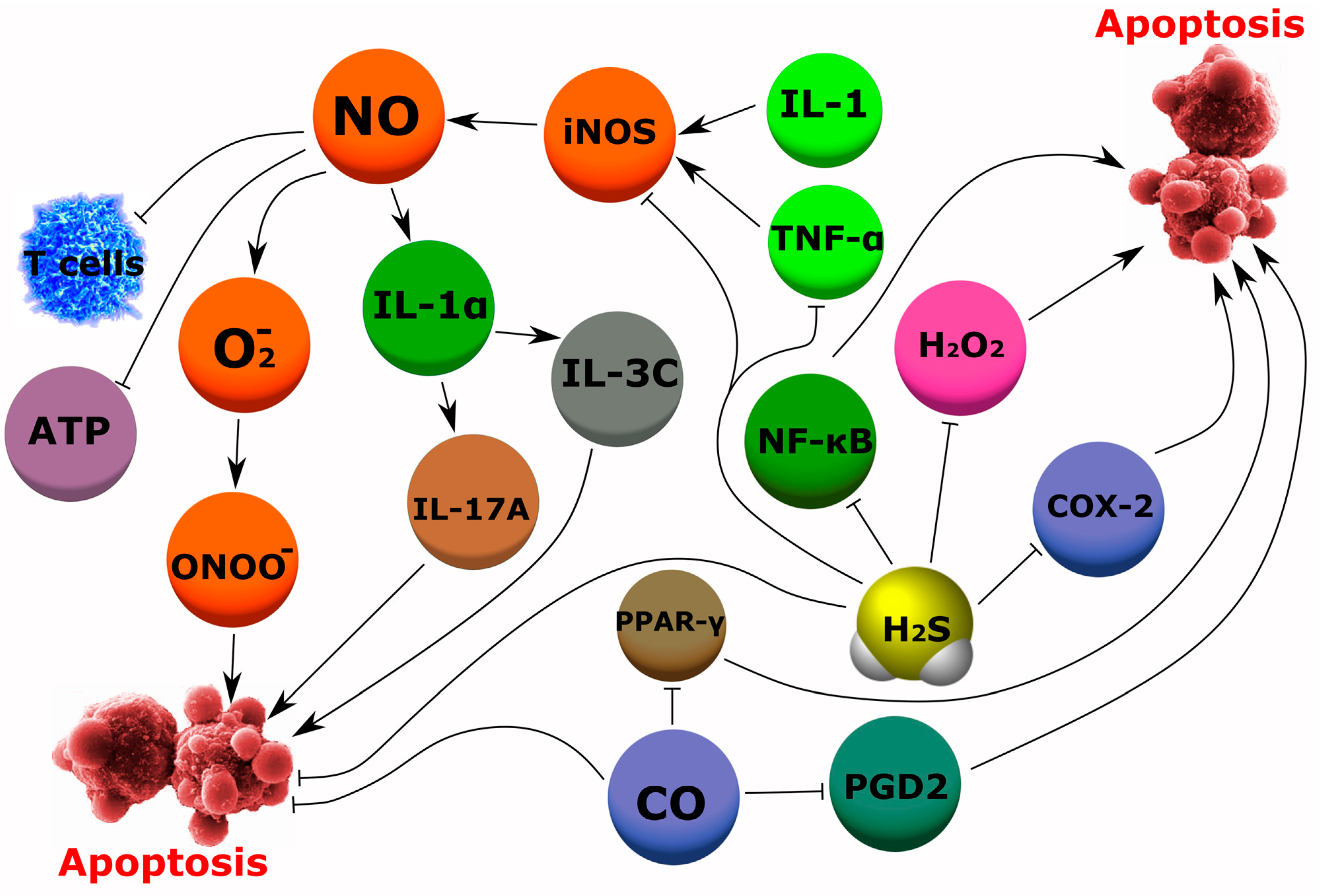
4.2. Rheumatic Diseases
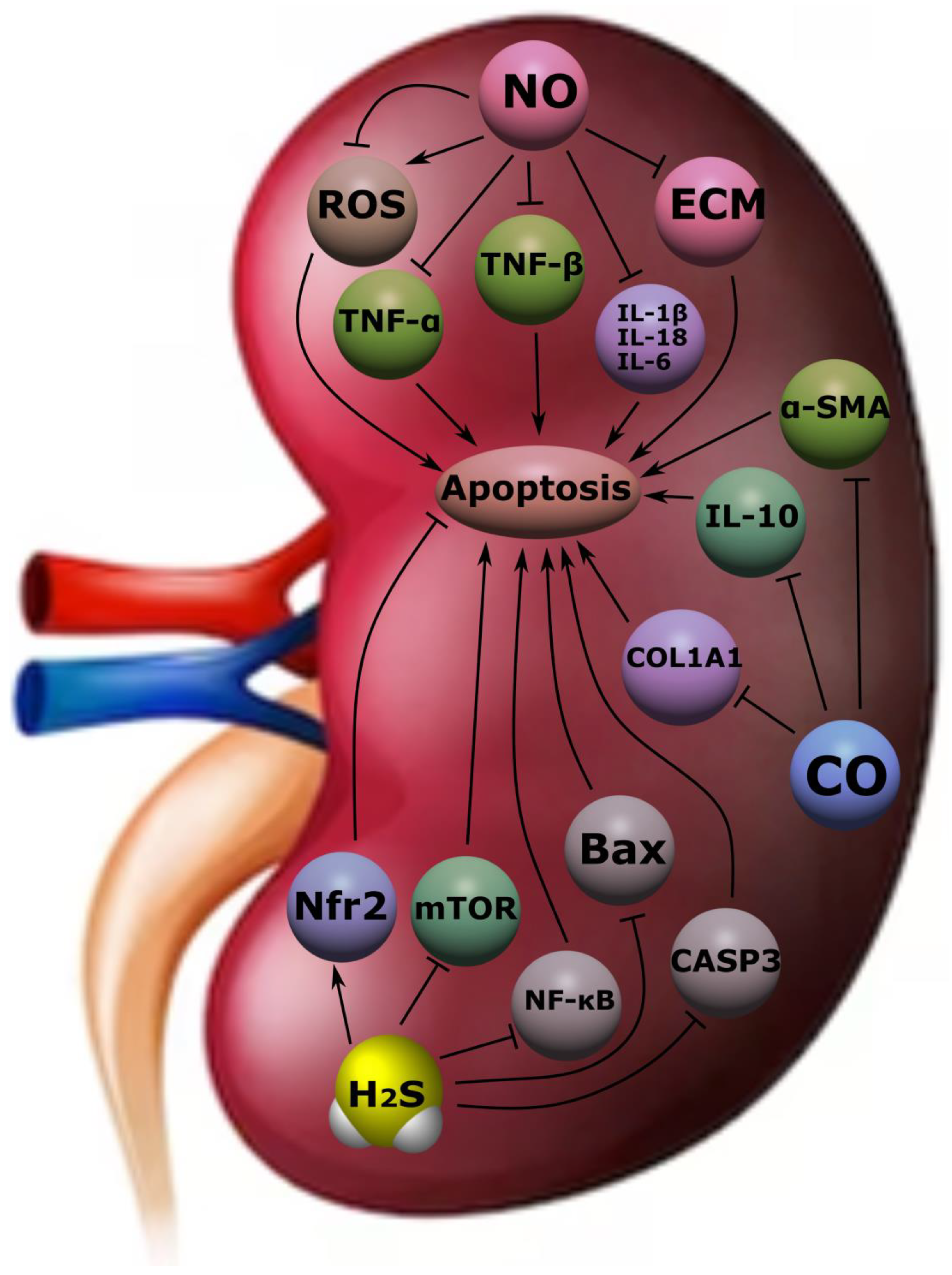
4.3. Kidney Diseases
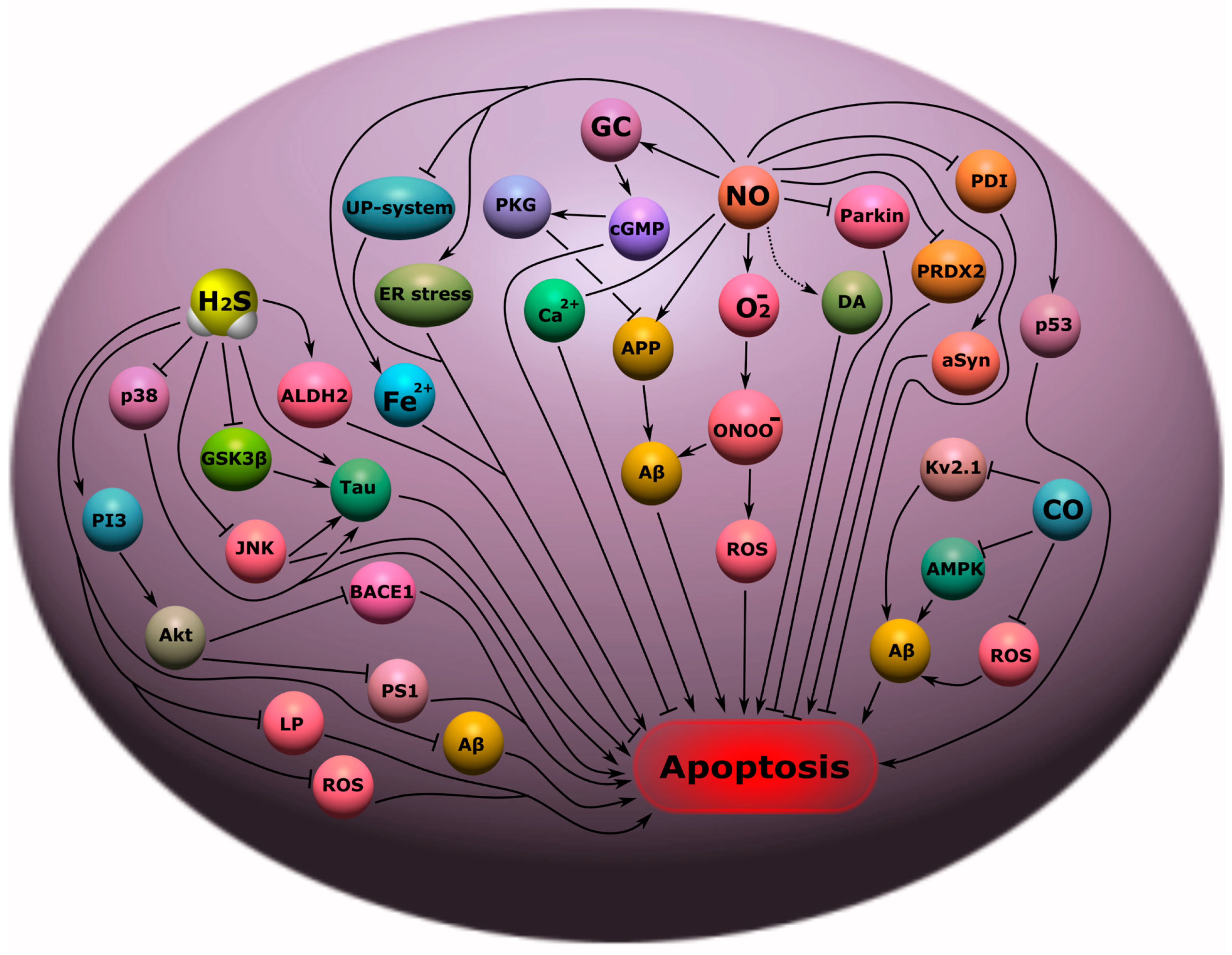
4.4. Neurodegenerative Diseases
5. Molecular Mechanisms of Gasotransmitter-Dependent Apoptosis in Neuropsychiatric Diseases
5.1. Schizophrenia
5.2. Depression
5.3. Bipolar Disorder
5.4. Anxiety Disorders
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balez, R.; Ooi, L. Getting to NO Alzheimer’s Disease: Neuroprotection versus Neurotoxicity Mediated by Nitric Oxide. Oxid. Med. Cell. Longev. 2016, 2016, 3806157. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of Nitric Oxide Synthases in Parkinson’s Disease: A Review on the Antioxidant and Anti-inflammatory Activity of Polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.-G.; Stanarius, A.; Baumann, B.; Henning, H.; Krell, D.; Danos, P.; Falkai, P.; Bogerts, B. Nitric oxide synthase-containing neurons in the human hypothalamus: Reduced number of immunoreactive cells in the paraventricular nucleus of depressive patients and schizophrenics. Neuroscience 1998, 83, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, J.; Wang, H. Hydrogen sulfide alleviates oxidative stress injury and reduces apoptosis induced by MPP+ in Parkinson’s disease cell model. Mol. Cell. Biochem. 2020, 472, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Nasyrova, R.F.; Ivashchenko, D.V.; Ivanov, M.V.; Neznanov, N.G. Role of nitric oxide and related molecules in schizophrenia pathogenesis: Biochemical, genetic and clinical aspects. Front. Physiol. 2015, 6, 139. [Google Scholar] [CrossRef]
- Wang, X.-B.; Du, J.-B.; Cui, H. Sulfur dioxide, a double-faced molecule in mammals. Life Sci. 2014, 98, 63–67. [Google Scholar] [CrossRef]
- Hendriks, K.D.; Maassen, H.; van Dijk, P.R.; Henning, R.H.; van Goor, H.; Hillebrands, J.-L. Gasotransmitters in health and disease: A mitochondria-centered view. Curr. Opin. Pharmacol. 2019, 45, 87–93. [Google Scholar] [CrossRef]
- Chen, Y.; Yuan, S.; Cao, Y.; Kong, G.; Jiang, F.; Li, Y.; Wang, Q.; Tang, M.; Zhang, Q.; Wang, Q.; et al. Gasotransmitters: Potential Therapeutic Molecules of Fibrotic Diseases. Oxid. Med. Cell. Longev. 2021, 2021, 3206982. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, Y.; Du, J.; Jin, H.; Tang, C.; Huang, Y. Sulfur Dioxide: An Endogenous Protector Against Myocardial Injury. J. Cardiovasc. Pharmacol. 2020, 76, 389–396. [Google Scholar] [CrossRef]
- Rodkin, S.; Dzreyan, V.; Bibov, M.; Ermakov, A.; Derezina, T.; Kirichenko, E. NO-Dependent Mechanisms of p53 Expression and Cell Death in Rat’s Dorsal Root Ganglia after Sciatic-Nerve Transection. Biomedicines 2022, 10, 1664. [Google Scholar] [CrossRef]
- Rodkin, S.V.; Kovaleva, V.D.; Berezhnaya, E.V.; Neginskaya, M.A.; Uzdensky, A.B. Ca2+- and NF-κB-dependent generation of NO in the photosensitized neurons and satellite glial cells. J. Photochem. Photobiol. B Biol. 2019, 199, 111603. [Google Scholar] [CrossRef]
- Obeng, E. Apoptosis (programmed cell death) and its signals—A review. Braz. J. Biol. 2021, 81, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L. The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, M.; Lv, Z.; Zhang, W.; Shao, Y.; Zhao, X.; Li, C. Fas-associated death domain (FADD) in sea cucumber (Apostichopus japonicus): Molecular cloning, characterization and pro-apoptotic function analysis. Dev. Comp. Immunol. 2020, 108, 103673. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gu, Y.; Xiong, X.; Zheng, Y.; Liu, X.; Wang, W.; Meng, G. Roles of the adaptor protein tumor necrosis factor receptor type 1-associated death domain protein (TRADD) in human diseases. Biomed. Pharmacother. 2022, 153, 113467. [Google Scholar] [CrossRef]
- Green, D.R. Caspase Activation and Inhibition. Cold Spring Harb. Perspect. Biol. 2022, 14, a041020. [Google Scholar] [CrossRef]
- Luo, X.; O’Neill, K.L.; Huang, K. The third model of Bax/Bak activation: A Bcl-2 family feud finally resolved? F1000Research 2020, 9, 935. [Google Scholar] [CrossRef]
- Green, D.R. The Mitochondrial Pathway of Apoptosis. Cold Spring Harb. Perspect. Biol. 2022, 14, a041038. [Google Scholar] [CrossRef] [PubMed]
- Bian, C.; Su, J.; Zheng, Z.; Wei, J.; Wang, H.; Meng, L.; Xin, Y.; Jiang, X. ARTS, an unusual septin, regulates tumorigenesis by promoting apoptosis. Biomed. Pharmacother. 2022, 152, 113281. [Google Scholar] [CrossRef]
- Wu, L.; Li, X.; Li, Z.; Cheng, Y.; Wu, F.; Lv, C.; Zhang, W.; Tang, W. HtrA serine proteases in cancers: A target of interest for cancer therapy. Biomed. Pharmacother. 2021, 139, 111603. [Google Scholar] [CrossRef]
- Cetraro, P.; Plaza-Diaz, J.; MacKenzie, A.; Abadía-Molina, F. A Review of the Current Impact of Inhibitors of Apoptosis Proteins and Their Repression in Cancer. Cancers 2022, 14, 1671. [Google Scholar] [CrossRef]
- Bajt, M.L.; Cover, C.; Lemasters, J.J.; Jaeschke, H. Nuclear Translocation of Endonuclease G and Apoptosis-Inducing Factor during Acetaminophen-Induced Liver Cell Injury. Toxicol. Sci. 2006, 94, 217–225. [Google Scholar] [CrossRef]
- Li, M. The role of P53 up-regulated modulator of apoptosis (PUMA) in ovarian development, cardiovascular and neurodegenerative diseases. Apoptosis 2021, 26, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Keszler, A.; Lindemer, B.; Hogg, N.; Weihrauch, D.; Lohr, N.L. Wavelength-dependence of vasodilation and NO release from S-nitrosothiols and dinitrosyl iron complexes by far red/near infrared light. Arch. Biochem. Biophys. 2018, 649, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Pandareesh, M.D.; Anand, T. Neuroprotective and Anti-Apoptotic Propensity of Bacopa monniera Extract Against Sodium Nitroprusside Induced Activation of iNOS, Heat Shock Proteins and Apoptotic Markers in PC12 Cells. Neurochem. Res. 2014, 39, 800–814. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and Oxidative Stress in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2014, 42, S125–S152. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, F.; Yang, S. Roles of Nitric Oxide in Brain Ischemia and Reperfusion. Int. J. Mol. Sci. 2022, 23, 4243. [Google Scholar] [CrossRef]
- Yakovlev, V.A.; Bayden, A.S.; Graves, P.R.; Kellogg, G.E.; Mikkelsen, R.B. Nitration of the Tumor Suppressor Protein p53 at Tyrosine 327 Promotes p53 Oligomerization and Activation. Biochemistry 2010, 49, 5331–5339. [Google Scholar] [CrossRef]
- Nakaya, N.; Lowe, S.W.; Taya, Y.; Chenchik, A.; Enikolopov, G. Specific pattern of p53 phosphorylation during nitric oxide-induced cell cycle arrest. Oncogene 2000, 19, 6369–6375. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, J.; Ma, P.; Myers, D.E.; Goldberg, I.G.; Sittler, K.J.; Barb, J.J.; Munson, P.J.; del Pilar Cintron, A.; McCoy, J.P.; et al. cGMP-independent nitric oxide signaling and regulation of the cell cycle. BMC Genom. 2005, 6, 151. [Google Scholar] [CrossRef]
- Tejedo, J.; Bernabé, J.; Ramírez, R.; Sobrino, F.; Bedoya, F. NO induces a cGMP-independent release of cytochrome c from mitochondria which precedes caspase 3 activation in insulin producing RINm5F cells. FEBS Lett. 1999, 459, 238–243. [Google Scholar] [CrossRef]
- Dubey, M.; Nagarkoti, S.; Awasthi, D.; Singh, A.K.; Chandra, T.; Kumaravelu, J.; Barthwal, M.K.; Dikshit, M. Nitric oxide-mediated apoptosis of neutrophils through caspase-8 and caspase-3-dependent mechanism. Cell Death Dis. 2016, 7, e2348. [Google Scholar] [CrossRef]
- Brune, B.; Mohr, S. Protein Thiol Modification of Glyceraldehyde-3-phosphate Dehydrogenase and Caspase-3 by Nitric Oxide. Curr. Protein Pept. Sci. 2001, 2, 61–72. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Inflammatory Neurodegeneration and Mechanisms of Microglial Killing of Neurons. Mol. Neurobiol. 2010, 41, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Q.; Robles, A.I.; Hanigan, C.L.; Hofseth, L.J.; Trudel, L.J.; Harris, C.C.; Wogan, G.N. Apoptotic Signaling Pathways Induced by Nitric Oxide in Human Lymphoblastoid Cells Expressing Wild-Type or Mutant p53. Cancer Res. 2004, 64, 3022–3029. [Google Scholar] [CrossRef]
- Mijatović, S.; Savić-Radojević, A.; Plješa-Ercegovac, M.; Simić, T.; Nicoletti, F.; Maksimović-Ivanić, D. The Double-Faced Role of Nitric Oxide and Reactive Oxygen Species in Solid Tumors. Antioxidants 2020, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, R. Carbon Monoxide: Endogenous Production, Physiological Functions, and Pharmacological Applications. Pharmacol. Rev. 2005, 57, 585–630. [Google Scholar] [CrossRef]
- Yang, X.; Caestecker, M.; Otterbein, L.E.; Wang, B. Carbon monoxide: An emerging therapy for acute kidney injury. Med. Res. Rev. 2020, 40, 1147–1177. [Google Scholar] [CrossRef] [PubMed]
- Corona, D.; Ekser, B.; Gioco, R.; Caruso, M.; Schipa, C.; Veroux, P.; Giaquinta, A.; Granata, A.; Veroux, M. Heme-Oxygenase and Kidney Transplantation: A Potential for Target Therapy? Biomolecules 2020, 10, 840. [Google Scholar] [CrossRef]
- Piantadosi, C.A. Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic. Biol. Med. 2008, 45, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovasc. Res. 2002, 55, 396–405. [Google Scholar] [CrossRef]
- Brouard, S.; Berberat, P.O.; Tobiasch, E.; Seldon, M.P.; Bach, F.H.; Soares, M.P. Heme Oxygenase-1-derived Carbon Monoxide Requires the Activation of Transcription Factor NF-κB to Protect Endothelial Cells from Tumor Necrosis Factor-α-mediated Apoptosis. J. Biol. Chem. 2002, 277, 17950–17961. [Google Scholar] [CrossRef] [PubMed]
- Inguaggiato, P.; Gonzalez-Michaca, L.; Croatt, A.J.; Haggard, J.J.; Alam, J.; Nath, K.A. Cellular overexpression of heme oxygenase-1 up-regulates p21 and confers resistance to apoptosis. Kidney Int. 2001, 60, 2181–2191. [Google Scholar] [CrossRef]
- Petrache, I.; Otterbein, L.E.; Alam, J.; Wiegand, G.W.; Choi, A.M.K. Heme oxygenase-1 inhibits TNF-α-induced apoptosis in cultured fibroblasts. Am. J. Physiol. Cell. Mol. Physiol. 2000, 278, L312–L319. [Google Scholar] [CrossRef]
- Zuckerbraun, B.S.; Billiar, T.R.; Otterbein, S.L.; Kim, P.K.M.; Liu, F.; Choi, A.M.K.; Bach, F.H.; Otterbein, L.E. Carbon Monoxide Protects against Liver Failure through Nitric Oxide–induced Heme Oxygenase 1. J. Exp. Med. 2003, 198, 1707–1716. [Google Scholar] [CrossRef]
- Dallas, M.L.; Boyle, J.P.; Milligan, C.J.; Sayer, R.; Kerrigan, T.L.; McKinstry, C.; Lu, P.; Mankouri, J.; Harris, M.; Scragg, J.L.; et al. Carbon monoxide protects against oxidant-induced apoptosis via inhibition of K v 2.1. FASEB J. 2011, 25, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Korbut, E.; Brzozowski, T.; Magierowski, M. Carbon Monoxide Being Hydrogen Sulfide and Nitric Oxide Molecular Sibling, as Endogenous and Exogenous Modulator of Oxidative Stress and Antioxidative Mechanisms in the Digestive System. Oxid. Med. Cell. Longev. 2020, 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.S.; Queiroga, C.S.F.; Sousa, M.F.Q.; Alves, P.M.; Vieira, H.L.A. Carbon Monoxide Modulates Apoptosis by Reinforcing Oxidative Metabolism in Astrocytes. J. Biol. Chem. 2012, 287, 10761–10770. [Google Scholar] [CrossRef]
- Thom, S.R.; Fisher, D.; Xu, Y.A.; Notarfrancesco, K.; Ischiropoulos, H. Adaptive responses and apoptosis in endothelial cells exposed to carbon monoxide. Proc. Natl. Acad. Sci. USA 2000, 97, 1305–1310. [Google Scholar] [CrossRef]
- Lundquist, I.; Alm, P.; Salehi, A.; Henningsson, R.; Grapengiesser, E.; Hellman, B. Carbon monoxide stimulates insulin release and propagates Ca 2+ signals between pancreatic β-cells. Am. J. Physiol. Metab. 2003, 285, E1055–E1063. [Google Scholar] [CrossRef]
- Xiao, Q.; Ying, J.; Xiang, L.; Zhang, C. The biologic effect of hydrogen sulfide and its function in various diseases. Medicine 2018, 97, e13065. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Wang, L.; Liu, G.; Wang, S.; Wang, Y.; Wu, Y.; Wang, J.; Sun, X. Role of hydrogen sulfide in subarachnoid hemorrhage. CNS Neurosci. Ther. 2022, 28, 805–817. [Google Scholar] [CrossRef]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Jurkowska, H.; Kaczor-Kamińska, M.; Bronowicka-Adamska, P.; Wróbel, M. Cystathionine γ-lyase. Postepy Hig. Med. Dosw. 2014, 68, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, S.; Shan, H.; Zhang, M. Biologic Effect of Hydrogen Sulfide and Its Role in Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, G.; Zhou, Z.; Tang, Z.; Zhang, N.; Zhu, X.; Ni, X. Endogenous Hydrogen Sulfide Is an Important Factor in Maintaining Arterial Oxygen Saturation. Front. Pharmacol. 2021, 12, 677110. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Munaron, L.; Avanzato, D.; Moccia, F.; Mancardi, D. Hydrogen sulfide as a regulator of calcium channels. Cell Calcium 2013, 53, 77–84. [Google Scholar] [CrossRef]
- Sun, J.; Li, X.; Gu, X.; Du, H.; Zhang, G.; Wu, J.; Wang, F. Neuroprotective effect of hydrogen sulfide against glutamate-induced oxidative stress is mediated via the p53/glutaminase 2 pathway after traumatic brain injury. Aging 2021, 13, 7180–7189. [Google Scholar] [CrossRef] [PubMed]
- Calenic, B.; Yaegaki, K.; Kozhuharova, A.; Imai, T. Oral Malodorous Compound Causes Oxidative Stress and p53-Mediated Programmed Cell Death in Keratinocyte Stem Cells. J. Periodontol. 2010, 81, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Chen, S.; Jin, H.; Tang, C.; Du, J. Endogenous generation of sulfur dioxide in rat tissues. Biochem. Biophys. Res. Commun. 2011, 415, 61–67. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, H.; Lv, B.; Tang, C.; Du, J.; Jin, H. Sulfur Dioxide: Endogenous Generation, Biological Effects, Detection, and Therapeutic Potential. Antioxid. Redox Signal. 2022, 36, 256–274. [Google Scholar] [CrossRef]
- Huang, Y.; Tang, C.; Du, J.; Jin, H. Endogenous Sulfur Dioxide: A New Member of Gasotransmitter Family in the Cardiovascular System. Oxid. Med. Cell. Longev. 2016, 2016, 8961951. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Liu, D.; Ochs, T.; Tang, C.; Chen, S.; Zhang, S.; Geng, B.; Jin, H.; Du, J. Endogenous sulfur dioxide protects against isoproterenol-induced myocardial injury and increases myocardial antioxidant capacity in rats. Lab. Investig. 2011, 91, 12–23. [Google Scholar] [CrossRef]
- Jin, H.; Liu, A.; Holmberg, L.; Zhao, M.; Chen, S.; Yang, J.; Sun, Y.; Chen, S.; Tang, C.; Du, J. The Role of Sulfur Dioxide in the Regulation of Mitochondrion-Related Cardiomyocyte Apoptosis in Rats with Isopropylarterenol-Induced Myocardial Injury. Int. J. Mol. Sci. 2013, 14, 10465–10482. [Google Scholar] [CrossRef]
- Andry, J.F.; Tannady, H.; Rembulan, G.D.; Rianto, A. The importance of big data for healthcare and its usage in clinical statistics of cardiovascular disease. J. Popul. Ther. Clin. Pharmacol. 2022, 29, 107–115. [Google Scholar] [CrossRef]
- Kleinert, H.; Pautz, A.; Linker, K.; Schwarz, P.M. Regulation of the expression of inducible nitric oxide synthase. Eur. J. Pharmacol. 2004, 500, 255–266. [Google Scholar] [CrossRef]
- Jones, S.P.; Girod, W.G.; Palazzo, A.J.; Granger, D.N.; Grisham, M.B.; Jourd’Heuil, D.; Huang, P.L.; Lefer, D.J. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am. J. Physiol. Circ. Physiol. 1999, 276, H1567–H1573. [Google Scholar] [CrossRef]
- Zhang, D.-M.; Chai, Y.; Erickson, J.R.; Brown, J.H.; Bers, D.M.; Lin, Y.-F. Intracellular signalling mechanism responsible for modulation of sarcolemmal ATP-sensitive potassium channels by nitric oxide in ventricular cardiomyocytes. J. Physiol. 2014, 592, 971–990. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.E.; Lu, X.; Lei, M.; Xiang, F.-L.; Hammoud, L.; Jiang, M.; Wang, H.; Jones, D.L.; Sims, S.M.; Feng, Q. Neuronal Nitric Oxide Synthase Protects Against Myocardial Infarction-Induced Ventricular Arrhythmia and Mortality in Mice. Circulation 2009, 120, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zhang, S.; Zhang, Z.; Wang, X.; Tian, X.; Zhang, L.; Du, J.; Huang, Y.; Jin, H. Nitric oxide inhibits endothelial cell apoptosis by inhibiting cysteine-dependent SOD1 monomerization. FEBS Open Bio 2022, 12, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Choi, A.M.K. A New Road to Induce Heme Oxygenase-1 Expression by Carbon Monoxide. Circ. Res. 2007, 101, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.E.; Naughton, P.; Shurey, S.; Green, C.J.; Johnson, T.R.; Mann, B.E.; Foresti, R.; Motterlini, R. Cardioprotective Actions by a Water-Soluble Carbon Monoxide–Releasing Molecule. Circ. Res. 2003, 93, e2–e8. [Google Scholar] [CrossRef]
- Kim, K.M.; Pae, H.-O.; Zheng, M.; Park, R.; Kim, Y.-M.; Chung, H.-T. Carbon Monoxide Induces Heme Oxygenase-1 via Activation of Protein Kinase R–Like Endoplasmic Reticulum Kinase and Inhibits Endothelial Cell Apoptosis Triggered by Endoplasmic Reticulum Stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [CrossRef]
- Kim, H.S.; Loughran, P.A.; Rao, J.; Billiar, T.R.; Zuckerbraun, B.S. Carbon monoxide activates NF-κB via ROS generation and Akt pathways to protect against cell death of hepatocytes. Am. J. Physiol. Liver Physiol. 2008, 295, G146–G152. [Google Scholar] [CrossRef]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.K.; Soares, M.P. Carbon Monoxide Generated by Heme Oxygenase 1 Suppresses Endothelial Cell Apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef]
- Wen, Y.-D.; Wang, H.; Zhu, Y.-Z. The Drug Developments of Hydrogen Sulfide on Cardiovascular Disease. Oxid. Med. Cell. Longev. 2018, 2018, 4010395. [Google Scholar] [CrossRef]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The Role of Nrf2 in Cardiovascular Function and Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef]
- Shefa, U.; Kim, M.-S.; Jeong, N.Y.; Jung, J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxid. Med. Cell. Longev. 2018, 2018, 1873962. [Google Scholar] [CrossRef]
- Liu, J.; Wu, J.; Sun, A.; Sun, Y.; Yu, X.; Liu, N.; Dong, S.; Yang, F.; Zhang, L.; Zhong, X.; et al. Hydrogen sulfide decreases high glucose/palmitate-induced autophagy in endothelial cells by the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 2016, 6, 33. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen Sulfide Mediates Cardioprotection Through Nrf2 Signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Hai, Z.; Zuo, W. Aberrant DNA methylation in the pathogenesis of atherosclerosis. Clin. Chim. Acta 2016, 456, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Shen, Z.; Miao, L.; Xin, X.; Lin, S.; Zhu, Y.; Guo, W.; Zhu, Y.Z. miRNA-30 Family Inhibition Protects Against Cardiac Ischemic Injury by Regulating Cystathionine-γ-Lyase Expression. Antioxid. Redox Signal. 2015, 22, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hao, D.-D.; Zhang, J.-S.; Zhu, Y.-C. Hydrogen sulphide inhibits cardiomyocyte hypertrophy by up-regulating miR-133a. Biochem. Biophys. Res. Commun. 2011, 413, 342–347. [Google Scholar] [CrossRef]
- Kesherwani, V.; Nandi, S.S.; Sharawat, S.K.; Shahshahan, H.R.; Mishra, P.K. Hydrogen sulfide mitigates homocysteine-mediated pathological remodeling by inducing miR-133a in cardiomyocytes. Mol. Cell. Biochem. 2015, 404, 241–250. [Google Scholar] [CrossRef]
- Ren, L.; Wang, Q.; Chen, Y.; Ma, Y.; Wang, D. Involvement of MicroRNA-133a in the Protective Effect of Hydrogen Sulfide against Ischemia/Reperfusion-Induced Endoplasmic Reticulum Stress and Cardiomyocyte Apoptosis. Pharmacology 2019, 103, 1–9. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, X.; Chen, H.; Li, L.; Xie, W.; Lan, G.; Zhao, Z.; Zheng, X.-L.; Wang, Z.; Tang, C. MicroRNA-186 promotes macrophage lipid accumulation and secretion of pro-inflammatory cytokines by targeting cystathionine γ-lyase in THP-1 macrophages. Atherosclerosis 2016, 250, 122–132. [Google Scholar] [CrossRef]
- Gong, D.; Cheng, H.; Xie, W.; Zhang, M.; Liu, D.; Lan, G.; Huang, C.; Zhao, Z.; Chen, L.; Yao, F.; et al. Cystathionine γ-lyase(CSE)/hydrogen sulfide system is regulated by miR-216a and influences cholesterol efflux in macrophages via the PI3K/AKT/ABCA1 pathway. Biochem. Biophys. Res. Commun. 2016, 470, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Pei, Y.; Cao, Q.; Wang, R. MicroRNA-21 represses human cystathionine gamma-lyase expression by targeting at specificity protein-1 in smooth muscle cells. J. Cell. Physiol. 2012, 227, 3192–3200. [Google Scholar] [CrossRef]
- Toldo, S.; Das, A.; Mezzaroma, E.; Chau, V.Q.; Marchetti, C.; Durrant, D.; Samidurai, A.; Van Tassell, B.W.; Yin, C.; Ockaili, R.A.; et al. Induction of MicroRNA-21 With Exogenous Hydrogen Sulfide Attenuates Myocardial Ischemic and Inflammatory Injury in Mice. Circ. Cardiovasc. Genet. 2014, 7, 311–320. [Google Scholar] [CrossRef]
- Kang, B.; Hong, J.; Xiao, J.; Zhu, X.; Ni, X.; Zhang, Y.; He, B.; Wang, Z. Involvement of miR-1 in the protective effect of hydrogen sulfide against cardiomyocyte apoptosis induced by ischemia/reperfusion. Mol. Biol. Rep. 2014, 41, 6845–6853. [Google Scholar] [CrossRef]
- Zhao, M.; Yang, J.; Wang, X.; Tang, C.; Du, J.; Jin, H. The PI3K/Akt pathway mediates the protection of SO2 preconditioning against myocardial ischemia/reperfusion injury in rats. Acta Pharmacol. Sin. 2013, 34, 501–506. [Google Scholar] [CrossRef]
- Belin de Chantemele, E.J.; Stepp, D.W. Influence of obesity and metabolic dysfunction on the endothelial control in the coronary circulation. J. Mol. Cell. Cardiol. 2012, 52, 840–847. [Google Scholar] [CrossRef]
- Ruiz-Hurtado, G.; Delgado, C. Nitric oxide pathway in hypertrophied heart: New therapeutic uses of nitric oxide donors. J. Hypertens. 2010, 28, S56–S61. [Google Scholar] [CrossRef]
- Wegiel, B.; Gallo, D.J.; Raman, K.G.; Karlsson, J.M.; Ozanich, B.; Chin, B.Y.; Tzeng, E.; Ahmad, S.; Ahmed, A.; Baty, C.J.; et al. Nitric Oxide–Dependent Bone Marrow Progenitor Mobilization by Carbon Monoxide Enhances Endothelial Repair After Vascular Injury. Circulation 2010, 121, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Ishikawa, K.; Matsumoto, H.; Kimura, S.; Kamiyama, Y.; Maruyama, Y. Synergetic Antioxidant and Vasodilatory Action of Carbon Monoxide in Angiotensin II–Induced Cardiac Hypertrophy. Hypertension 2007, 50, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Ndisang, J.F.; Zhao, W.; Wang, R. Selective Regulation of Blood Pressure by Heme Oxygenase-1 in Hypertension. Hypertension 2002, 40, 315–321. [Google Scholar] [CrossRef]
- Shi, Y.-X.; Chen, Y.; Zhu, Y.-Z.; Huang, G.-Y.; Moore, P.K.; Huang, S.-H.; Yao, T.; Zhu, Y.-C. Chronic sodium hydrosulfide treatment decreases medial thickening of intramyocardial coronary arterioles, interstitial fibrosis, and ROS production in spontaneously hypertensive rats. Am. J. Physiol. Circ. Physiol. 2007, 293, H2093–H2100. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine γ-Lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- CHEN, L.; INGRID, S.; DING, Y.; LIU, Y.; QI, J.; TANG, C.; DU, J. Imbalance of endogenous homocysteine and hydrogen sulfide metabolic pathway in essential hypertensive children. Chin. Med. J. 2007, 120, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Sun, Y.; Tang, C.; Ochs, T.; Qi, J.; Du, J.; Jin, H. Sulfur dioxide derivatives improve the vasorelaxation in the spontaneously hypertensive rat by enhancing the vasorelaxant response to nitric oxide. Exp. Biol. Med. 2012, 237, 867–872. [Google Scholar] [CrossRef]
- Liu, D.; Huang, Y.; Bu, D.; Liu, A.D.; Holmberg, L.; Jia, Y.; Tang, C.; Du, J.; Jin, H. Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the Erk/MAP kinase pathway mediated by cAMP/PKA signaling. Cell Death Dis. 2014, 5, e1251. [Google Scholar] [CrossRef]
- Zeiher, A.M.; Fisslthaler, B.; Schray-Utz, B.; Busse, R. Nitric Oxide Modulates the Expression of Monocyte Chemoattractant Protein 1 in Cultured Human Endothelial Cells. Circ. Res. 1995, 76, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Arndt, H.; Smith, C.W.; Granger, D.N. Leukocyte-endothelial cell adhesion in spontaneously hypertensive and normotensive rats. Hypertension 1993, 21, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Zeiher, A.M. Nitric oxide–an endothelial cell survival factor. Cell Death Differ. 1999, 6, 964–968. [Google Scholar] [CrossRef]
- Hogan, M.; Cerami, A.; Bucala, R. Advanced glycosylation endproducts block the antiproliferative effect of nitric oxide. Role in the vascular and renal complications of diabetes mellitus. J. Clin. Investig. 1992, 90, 1110–1115. [Google Scholar] [CrossRef]
- Southgate, K.; Newby, A. Serum-induced proliferation of rabbit aortic smooth muscle cells from the contractile state is inhibited by 8-Br-CAMP but not 8-Br-cGMP. Atherosclerosis 1990, 82, 113–123. [Google Scholar] [CrossRef]
- Förstermann, U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, X.; Jin, H.; Wei, H.; Li, W.; Bu, D.; Tang, X.; Ren, Y.; Tang, C.; Du, J. Role of Hydrogen Sulfide in the Development of Atherosclerotic Lesions in Apolipoprotein E Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tang, C.; Jin, H.; Du, J. Regulatory effects of sulfur dioxide on the development of atherosclerotic lesions and vascular hydrogen sulfide in atherosclerotic rats. Atherosclerosis 2011, 215, 323–330. [Google Scholar] [CrossRef]
- Janssens, S.; Pokreisz, P.; Schoonjans, L.; Pellens, M.; Vermeersch, P.; Tjwa, M.; Jans, P.; Scherrer-Crosbie, M.; Picard, M.H.; Szelid, Z.; et al. Cardiomyocyte-Specific Overexpression of Nitric Oxide Synthase 3 Improves Left Ventricular Performance and Reduces Compensatory Hypertrophy After Myocardial Infarction. Circ. Res. 2004, 94, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ji, X.; Boysen, P.G. Exogenous nitric oxide generates ROS and induces cardioprotection: Involvement of PKG, mitochondrial K ATP channels, and ERK. Am. J. Physiol. Circ. Physiol. 2004, 286, H1433–H1440. [Google Scholar] [CrossRef]
- Bhushan, S.; Kondo, K.; Polhemus, D.J.; Otsuka, H.; Nicholson, C.K.; Tao, Y.-X.; Huang, H.; Georgiopoulou, V.V.; Murohara, T.; Calvert, J.W.; et al. Nitrite Therapy Improves Left Ventricular Function During Heart Failure via Restoration of Nitric Oxide–Mediated Cytoprotective Signaling. Circ. Res. 2014, 114, 1281–1291. [Google Scholar] [CrossRef]
- Couto, G.K.; Britto, L.R.G.; Mill, J.G.; Rossoni, L.V. Enhanced nitric oxide bioavailability in coronary arteries prevents the onset of heart failure in rats with myocardial infarction. J. Mol. Cell. Cardiol. 2015, 86, 110–120. [Google Scholar] [CrossRef]
- King, A.L.; Polhemus, D.J.; Bhushan, S.; Otsuka, H.; Kondo, K.; Nicholson, C.K.; Bradley, J.M.; Islam, K.N.; Calvert, J.W.; Tao, Y.-X.; et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 3182–3187. [Google Scholar] [CrossRef]
- Sivarajah, A.; Collino, M.; Yasin, M.; Benetti, E.; Gallicchio, M.; Mazzon, E.; Cuzzocrea, S.; Fantozzi, R.; Thiemermann, C. anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 2009, 31, 267–274. [Google Scholar] [CrossRef]
- Wang, X.-B.; Huang, X.-M.; Ochs, T.; Li, X.-Y.; Jin, H.-F.; Tang, C.-S.; Du, J.-B. Effect of sulfur dioxide preconditioning on rat myocardial ischemia/reperfusion injury by inducing endoplasmic reticulum stress. Basic Res. Cardiol. 2011, 106, 865–878. [Google Scholar] [CrossRef]
- Nie, A.; Meng, Z. Modulation of L-type calcium current in rat cardiac myocytes by sulfur dioxide derivatives. Food Chem. Toxicol. 2006, 44, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Koncz, A.; Telarico, T.; Fernandez, D.; Érsek, B.; Buzás, E.; Perl, A. Central role of nitric oxide in the pathogenesis of rheumatoid arthritis and sysemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, 210. [Google Scholar] [CrossRef]
- Spiller, F.; Oliveira Formiga, R.; Fernandes da Silva Coimbra, J.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q. Targeting nitric oxide as a key modulator of sepsis, arthritis and pain. Nitric Oxide 2019, 89, 32–40. [Google Scholar] [CrossRef] [PubMed]
- van’T Hof, R.J.; Ralston, S.H. Nitric oxide and bone. Immunology 2001, 103, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Scholz, T.; Yau, A.C.Y.; Guerard, S.; Hüffmeier, U.; Burkhardt, H.; Holmdahl, R. Mannan-induced Nos2 in macrophages enhances IL-17-driven psoriatic arthritis by innate lymphocytes. Sci. Adv. 2018, 4, eaas9864. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Yang, S.; Wang, J.; Xu, M.; Wang, S.; Yi, H. Inducible nitric oxide synthase and systemic lupus erythematosus: A systematic review and meta-analysis. BMC Immunol. 2020, 21, 6. [Google Scholar] [CrossRef]
- Assavarittirong, C.; Samborski, W.; Grygiel-Górniak, B. Oxidative Stress in Fibromyalgia: From Pathology to Treatment. Oxid. Med. Cell. Longev. 2022, 2022, 1582432. [Google Scholar] [CrossRef]
- Rahmati, M.; Mobasheri, A.; Mozafari, M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone 2016, 85, 81–90. [Google Scholar] [CrossRef]
- Karan, A.; Karan, M.A.; Vural, P.; Erten, N.; Taşçioglu, C.; Aksoy, C.; Canbaz, M.; Öncel, A. Synovial fluid nitric oxide levels in patients with knee osteoarthritis. Clin. Rheumatol. 2003, 22, 397–399. [Google Scholar] [CrossRef]
- Leonidou, A.; Lepetsos, P.; Kenanidis, E.; Macheras, G.; Tzetis, M.; Potoupnis, M.; Tsiridis, E. Association of Polymorphisms in the Promoter Region of NOS2A Gene with Primary Knee Osteoarthritis in the Greek Population. Cureus 2020, 12, e6780. [Google Scholar] [CrossRef]
- Yunus, M.H.M.; Nordin, A.; Kamal, H. Pathophysiological Perspective of Osteoarthritis. Medicina 2020, 56, 614. [Google Scholar] [CrossRef] [PubMed]
- Spreng, D.; Sigrist, N.; Schweighauser, A.; Busato, A.; Schawalder, P. Endogenous nitric oxide production in canine osteoarthritis: Detection in urine, serum, and synovial fluid specimens. Vet. Surg. 2001, 30, ajvet0300191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, M.; Shi, T.; Gao, M.; Lv, Y.; Zhao, Y.; Li, J.; Zhang, M.; Zhang, H.; Guan, F.; et al. Analysis of Serum Metabolomics in Rats with Osteoarthritis by Mass Spectrometry. Molecules 2021, 26, 7181. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.J.; Wong, P.T.-H. Hydrogen sulfide in stroke: Protective or deleterious? Neurochem. Int. 2017, 105, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Burguera, E.F.; Meijide-Failde, R.; Blanco, F.J. Hydrogen Sulfide and Inflammatory Joint Diseases. Curr. Drug Targets 2017, 18, 1641–1652. [Google Scholar] [CrossRef]
- Li, M.; Mao, J.C.; Zhu, Y.Z. Hydrogen Sulfide: A Novel Immunoinflammatory Regulator in Rheumatoid Arthritis. Adv. Exp. Med. Biol. 2021, 1315, 161–179. [Google Scholar] [CrossRef]
- Sunzini, F.; De Stefano, S.; Chimenti, M.S.; Melino, S. Hydrogen Sulfide as Potential Regulatory Gasotransmitter in Arthritic Diseases. Int. J. Mol. Sci. 2020, 21, 1180. [Google Scholar] [CrossRef]
- Nasi, S.; Ehirchiou, D.; Bertrand, J.; Castelblanco, M.; Mitchell, J.; Ishii, I.; So, A.; Busso, N. The Gasotransmitter Hydrogen Sulfide (H2S) Prevents Pathologic Calcification (PC) in Cartilage. Antioxidants 2021, 10, 1433. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef]
- Wu, W.J.; Jia, W.W.; Liu, X.H.; Pan, L.L.; Zhang, Q.Y.; Yang, D.; Shen, X.Y.; Liu, L.; Zhu, Y.Z. S-propargyl-cysteine attenuates inflammatory response in rheumatoid arthritis by modulating the Nrf2-ARE signaling pathway. Redox Biol. 2016, 10, 157–167. [Google Scholar] [CrossRef]
- Gambari, L.; Lisignoli, G.; Gabusi, E.; Manferdini, C.; Paolella, F.; Piacentini, A.; Grassi, F. Distinctive expression pattern of cystathionine-β-synthase and cystathionine-γ-lyase identifies mesenchymal stromal cells transition to mineralizing osteoblasts. J. Cell. Physiol. 2017, 232, 3574–3585. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liao, F.; Lin, X.; Zheng, F.; Fan, J.; Cui, Q.; Yang, J.; Geng, B.; Cai, J. Cystathionine γ-Lyase–Hydrogen Sulfide Induces Runt-Related Transcription Factor 2 Sulfhydration, Thereby Increasing Osteoblast Activity to Promote Bone Fracture Healing. Antioxid. Redox Signal. 2017, 27, 742–753. [Google Scholar] [CrossRef]
- Zhai, Y.; Behera, J.; Tyagi, S.C.; Tyagi, N. Hydrogen sulfide attenuates homocysteine-induced osteoblast dysfunction by inhibiting mitochondrial toxicity. J. Cell. Physiol. 2019, 234, 18602–18614. [Google Scholar] [CrossRef]
- Lv, M.; Liu, Y.; Xiao, T.H.; Jiang, W.; Lin, B.W.; Zhang, X.M.; Lin, Y.M.; Xu, Z.S. GYY4137 stimulates osteoblastic cell proliferation and differentiation via an ERK1/2-dependent anti-oxidant mechanism. Am. J. Transl. Res. 2017, 9, 1183–1192. [Google Scholar]
- Bitar, M.S.; Nader, J.; Al-Ali, W.; Al Madhoun, A.; Arefanian, H.; Al-Mulla, F. Hydrogen Sulfide Donor NaHS Improves Metabolism and Reduces Muscle Atrophy in Type 2 Diabetes: Implication for Understanding Sarcopenic Pathophysiology. Oxid. Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ellwood, R.A.; Slade, L.; Lewis, J.; Torregrossa, R.; Sudevan, S.; Piasecki, M.; Whiteman, M.; Etheridge, T.; Szewczyk, N.J. Sulfur amino acid supplementation displays therapeutic potential in a C. elegans model of Duchenne muscular dystrophy. Commun. Biol. 2022, 5, 1255. [Google Scholar] [CrossRef]
- Alcaraz, M.; Guillen, M.; Ferrandiz, M.; Megias, J.; Motterlini, R. Carbon Monoxide-Releasing Molecules: A Pharmacological Expedient to Counteract Inflammation. Curr. Pharm. Des. 2008, 14, 465–472. [Google Scholar] [CrossRef]
- Tseng, F.J.; Chia, W.T.; Wang, C.H.; Shyu, J.F.; Gou, G.H.; Shui, H.A.; Sytwu, H.K.; Pan, R.Y.; Weng, C.F. Carbon Monoxide Inhibits Receptor Activator of NF-κB (RANKL)-Induced Osteoclastogenesis. Cell. Physiol. Biochem. 2015, 36, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Savitskaya, A.; Steiner, C.W.; Rath, E.; Bilban, M.; Wagner, O.; Bach, F.H.; Smolen, J.S.; Scheinecker, C. Heme oxygenase-1 end-products carbon monoxide and biliverdin ameliorate murine collagen-induced arthritis. Clin. Exp. Rheumatol. 2012, 30, 73–78. [Google Scholar]
- Maicas, N.; Ferrándiz, M.L.; Devesa, I.; Motterlini, R.; Koenders, M.I.; van den Berg, W.B.; Alcaraz, M.J. The CO-releasing molecule CORM-3 protects against articular degradation in the K/BxN serum transfer arthritis model. Eur. J. Pharmacol. 2010, 634, 184–191. [Google Scholar] [CrossRef]
- Pan, Y.; Song, J.; Ma, L.; Zong, X.; Chen, H.; Zhao, B.; Yu, Q.; Song, H. Carbon Monoxide Releasing Molecule 3 Inhibits Osteoclastogenic Differentiation of RAW264.7 Cells by Heme Oxygenase-1. Cell. Physiol. Biochem. 2018, 50, 1988–2003. [Google Scholar] [CrossRef] [PubMed]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication—Worldwide more than 850 million individuals have kidney diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef]
- Ngowi, E.E.; Sarfraz, M.; Afzal, A.; Khan, N.H.; Khattak, S.; Zhang, X.; Li, T.; Duan, S.-F.; Ji, X.-Y.; Wu, D.-D. Roles of Hydrogen Sulfide Donors in Common Kidney Diseases. Front. Pharmacol. 2020, 11, 564281. [Google Scholar] [CrossRef]
- Abe, T.; Yazawa, K.; Fujino, M.; Imamura, R.; Hatayama, N.; Kakuta, Y.; Tsutahara, K.; Okumi, M.; Ichimaru, N.; Kaimori, J.; et al. High-pressure carbon monoxide preserves rat kidney grafts from apoptosis and inflammation. Lab. Investig. 2017, 97, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Bian, J.-S. The Role of Hydrogen Sulfide in Renal System. Front. Pharmacol. 2016, 7, 385. [Google Scholar] [CrossRef]
- Wallace, J.L.; Ferraz, J.G.P.; Muscara, M.N. Hydrogen Sulfide: An Endogenous Mediator of Resolution of Inflammation and Injury. Antioxid. Redox Signal. 2012, 17, 58–67. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.-F.; Pfeilschifter, J. The Pathophysiology of H2S in Renal Glomerular Diseases. Biomolecules 2022, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Li, L.; Rose, P.; Tan, C.-H.; Parkinson, D.B.; Moore, P.K. The Effect of Hydrogen Sulfide Donors on Lipopolysaccharide-Induced Formation of Inflammatory Mediators in Macrophages. Antioxid. Redox Signal. 2010, 12, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, J.C.; Junho, C.V.C.; Carneiro-Ramos, M.S.; Seabra, A.B. H2S- and NO-releasing gasotransmitter platform: A crosstalk signaling pathway in the treatment of acute kidney injury. Pharmacol. Res. 2020, 161, 105121. [Google Scholar] [CrossRef]
- Szabó, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management. JAMA 2019, 322, 1294. [Google Scholar] [CrossRef]
- Shirazi, M.K.; Azarnezhad, A.; Abazari, M.F.; Poorebrahim, M.; Ghoraeian, P.; Sanadgol, N.; Bokharaie, H.; Heydari, S.; Abbasi, A.; Kabiri, S.; et al. The role of nitric oxide signaling in renoprotective effects of hydrogen sulfide against chronic kidney disease in rats: Involvement of oxidative stress, autophagy and apoptosis. J. Cell. Physiol. 2019, 234, 11411–11423. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Luo, N.; Wang, L.; Zhao, Z.; Bu, H.; Xu, G.; Yan, Y.; Che, X.; Jiao, Z.; Zhao, T.; et al. Hydrogen sulfide ameliorates chronic renal failure in rats by inhibiting apoptosis and inflammation through ROS/MAPK and NF-κB signaling pathways. Sci. Rep. 2017, 7, 455. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, H.; Guo, N. Protective effect of hydrogen sulfide on the kidney (Review). Mol. Med. Rep. 2021, 24, 696. [Google Scholar] [CrossRef]
- Zeng, O.; Li, F.; Li, Y.; Li, L.; Xiao, T.; Chu, C.; Yang, J. Effect of Novel Gasotransmitter hydrogen sulfide on renal fibrosis and connexins expression in diabetic rats. Bioengineered 2016, 7, 314–320. [Google Scholar] [CrossRef]
- Jia, Q.; Wang, L.; Wang, Q.Y.; Liu, X.F.; Ma, S.F.; Yang, R. Effects of hydrogen sulfide on renal fibrosis in diabetic rats and its mechanism. Chin. J. Appl. Physiol. 2018, 34, 572–576. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, X.; Wang, X.; Peng, Y.; Du, J.; Yin, H.; Yang, H.; Ni, X.; Zhang, W. H2S alleviates renal injury and fibrosis in response to unilateral ureteral obstruction by regulating macrophage infiltration via inhibition of NLRP3 signaling. Exp. Cell Res. 2020, 387, 111779. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Noh, M.R.; Jung, J.-M.; Ishii, I.; Yoo, J.; Kim, J.I.; Park, K.M. Hydrogen sulfide-producing cystathionine γ-lyase is critical in the progression of kidney fibrosis. Free Radic. Biol. Med. 2017, 112, 423–432. [Google Scholar] [CrossRef]
- Sen, U.; Munjal, C.; Qipshidze, N.; Abe, O.; Gargoum, R.; Tyagi, S.C. Hydrogen Sulfide Regulates Homocysteine-Mediated Glomerulosclerosis. Am. J. Nephrol. 2010, 31, 442–455. [Google Scholar] [CrossRef]
- Elkhoely, A.; Kamel, R. Diallyl sulfide alleviates cisplatin-induced nephrotoxicity in rats via suppressing NF-κB downstream inflammatory proteins and p53/Puma signalling pathway. Clin. Exp. Pharmacol. Physiol. 2018, 45, 591–601. [Google Scholar] [CrossRef]
- Ko, J.-W.; Shin, J.-Y.; Kim, J.-W.; Park, S.-H.; Shin, N.-R.; Lee, I.-C.; Shin, I.-S.; Moon, C.; Kim, S.-H.; Kim, S.-H.; et al. Protective effects of diallyl disulfide against acetaminophen-induced nephrotoxicity: A possible role of CYP2E1 and NF-κB. Food Chem. Toxicol. 2017, 102, 156–165. [Google Scholar] [CrossRef]
- Otunctemur, A.; Ozbek, E.; Dursun, M.; Sahin, S.; Besiroglu, H.; Ozsoy, O.D.; Cekmen, M.; Somay, A.; Ozbay, N. Protective effect of hydrogen sulfide on gentamicin-induced renal injury. Ren. Fail. 2014, 36, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Wani, J.; Carl, M.; Henger, A.; Nelson, P.J.; Rupprecht, H. Nitric oxide modulates expression of extracellular matrix genes linked to fibrosis in kidney mesangial cells. bchm 2007, 388, 497–506. [Google Scholar] [CrossRef]
- Akool, E.-S.; Doller, A.; Müller, R.; Gutwein, P.; Xin, C.; Huwiler, A.; Pfeilschifter, J.; Eberhardt, W. Nitric Oxide Induces TIMP-1 Expression by Activating the Transforming Growth Factor β-Smad Signaling Pathway. J. Biol. Chem. 2005, 280, 39403–39416. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, W.; Beeg, T.; Beck, K.-F.; Walpen, S.; Gauer, S.; Böhles, H.; Pfeilschifter, J. Nitric oxide modulates expression of matrix metalloproteinase-9 in rat mesangial cells. Kidney Int. 2000, 57, 59–69. [Google Scholar] [CrossRef]
- Aksu, U.; Demirci, C.; Ince, C. The Pathogenesis of Acute Kidney Injury and the Toxic Triangle of Oxygen, Reactive Oxygen Species and Nitric Oxide. Controv. Acute Kidney Inj. 2011, 174, 119–128. [Google Scholar]
- Chiazza, F.; Chegaev, K.; Rogazzo, M.; Cutrin, J.C.; Benetti, E.; Lazzarato, L.; Fruttero, R.; Collino, M. A Nitric Oxide-Donor Furoxan Moiety Improves the Efficacy of Edaravone against Early Renal Dysfunction and Injury Evoked by Ischemia/Reperfusion. Oxid. Med. Cell. Longev. 2015, 2015, 804659. [Google Scholar] [CrossRef]
- Wang, L.; Lee, J.-Y.S.; Kwak, J.H.; He, Y.; Kim, S.I.; Choi, M.E. Protective effects of low-dose carbon monoxide against renal fibrosis induced by unilateral ureteral obstruction. Am. J. Physiol. Physiol. 2008, 294, F508–F517. [Google Scholar] [CrossRef] [PubMed]
- Mistry, R.K.; Brewer, A.C. Redox regulation of gasotransmission in the vascular system: A focus on angiogenesis. Free Radic. Biol. Med. 2017, 108, 500–516. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M.K. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Haschemi, A.; Chin, B.Y.; Jeitler, M.; Esterbauer, H.; Wagner, O.; Bilban, M.; Otterbein, L.E. Carbon Monoxide Induced PPARγ SUMOylation and UCP2 Block Inflammatory Gene Expression in Macrophages. PLoS ONE 2011, 6, e26376. [Google Scholar] [CrossRef]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef]
- Malinski, T. Nitric Oxide and Nitroxidative Stress in Alzheimer’s Disease. J. Alzheimer’s Dis. 2007, 11, 207–218. [Google Scholar] [CrossRef]
- Cai, Z.-X.; Guo, H.-S.; Wang, C.; Wei, M.; Cheng, C.; Yang, Z.-F.; Chen, Y.-W.; Le, W.-D.; Li, S. Double-Edged Roles of Nitric Oxide Signaling on APP Processing and Amyloid-β Production In Vitro: Preliminary Evidence from Sodium Nitroprusside. Neurotox. Res. 2016, 29, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Calingasan, N.; Nezezon, J.; Ding, A.; Lucia, M.S.; La Perle, K.; Fuortes, M.; Lin, M.; Ehrt, S.; Kwon, N.S.; et al. Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J. Exp. Med. 2005, 202, 1163–1169. [Google Scholar] [CrossRef]
- Nunes, C.; Laranjinha, J. Nitric oxide and dopamine metabolism converge via mitochondrial dysfunction in the mechanisms of neurodegeneration in Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108877. [Google Scholar] [CrossRef]
- Tiwari, S.; Singh, S. Reciprocal Upshot of Nitric Oxide, Endoplasmic Reticulum Stress, and Ubiquitin Proteasome System in Parkinson’s Disease Pathology. Neurosci. 2021, 27, 340–354. [Google Scholar] [CrossRef]
- Xiong, Z.-K.; Lang, J.; Xu, G.; Li, H.-Y.; Zhang, Y.; Wang, L.; Su, Y.; Sun, A.-J. Excessive levels of nitric oxide in rat model of Parkinson’s disease induced by rotenone. Exp. Ther. Med. 2015, 9, 553–558. [Google Scholar] [CrossRef]
- Liu, C.; Liang, M.C.; Soong, T.W. Nitric Oxide, Iron and Neurodegeneration. Front. Neurosci. 2019, 13, 114. [Google Scholar] [CrossRef]
- Latif, S.; Kang, Y.-S. Differences of Transport Activity of Arginine and Regulation on Neuronal Nitric Oxide Synthase and Oxidative Stress in Amyotrophic Lateral Sclerosis Model Cell Lines. Cells 2021, 10, 3554. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Shibata, N.; Iwata, M. Neuronal nitric oxide synthase immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol. 2001, 101, 351–357. [Google Scholar] [CrossRef]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Liu, X.Q.; Jiang, P.; Huang, H.; Yan, Y. Plasma levels of endogenous hydrogen sulfide and homocysteine in patients with Alzheimer’s disease and vascular dementia and the significance thereof. Zhonghua Yi Xue Za Zhi. 2008, 88, 2246–2249. [Google Scholar] [PubMed]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Xu, J.; Li, M.; Tang, J.; Zou, W.; Zhang, P.; Wang, L.; Wang, C.; Tang, X. Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol. Sin. 2014, 35, 707–715. [Google Scholar] [CrossRef]
- He, X.; Yan, N.; Chen, X.; Qi, Y.; Yan, Y.; Cai, Z. Hydrogen sulfide down-regulates BACE1 and PS1 via activating PI3K/Akt pathway in the brain of APP/PS1 transgenic mouse. Pharmacol. Rep. 2016, 68, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Seok Ko, H.; Il Lee, Y.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef]
- Wang, M.; Tang, J.-J.; Wang, L.-X.; Yu, J.; Zhang, L.; Qiao, C. Hydrogen sulfide enhances adult neurogenesis in a mouse model of Parkinson’s disease. Neural Regen. Res. 2021, 16, 1353. [Google Scholar] [CrossRef]
- Hu, L.-F.; Lu, M.; Tiong, C.X.; Dawe, G.S.; Hu, G.; Bian, J.-S. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 2010, 9, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Kida, K.; Yamada, M.; Tokuda, K.; Marutani, E.; Kakinohana, M.; Kaneki, M.; Ichinose, F. Inhaled Hydrogen Sulfide Prevents Neurodegeneration and Movement Disorder in a Mouse Model of Parkinson’s Disease. Antioxid. Redox Signal. 2011, 15, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Davoli, A.; Greco, V.; Spalloni, A.; Guatteo, E.; Neri, C.; Rizzo, G.R.; Cordella, A.; Romigi, A.; Cortese, C.; Bernardini, S.; et al. Evidence of hydrogen sulfide involvement in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 77, 697–709. [Google Scholar] [CrossRef]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-β1-42-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef]
- Hettiarachchi, N.T.; Boyle, J.P.; Dallas, M.L.; Al-Owais, M.M.; Scragg, J.L.; Peers, C. Heme oxygenase-1 derived carbon monoxide suppresses Aβ1–42 toxicity in astrocytes. Cell Death Dis. 2017, 8, e2884. [Google Scholar] [CrossRef]
- Khaitin, A. Calcium in Neuronal and Glial Response to Axotomy. Int. J. Mol. Sci. 2021, 22, 13344. [Google Scholar] [CrossRef]
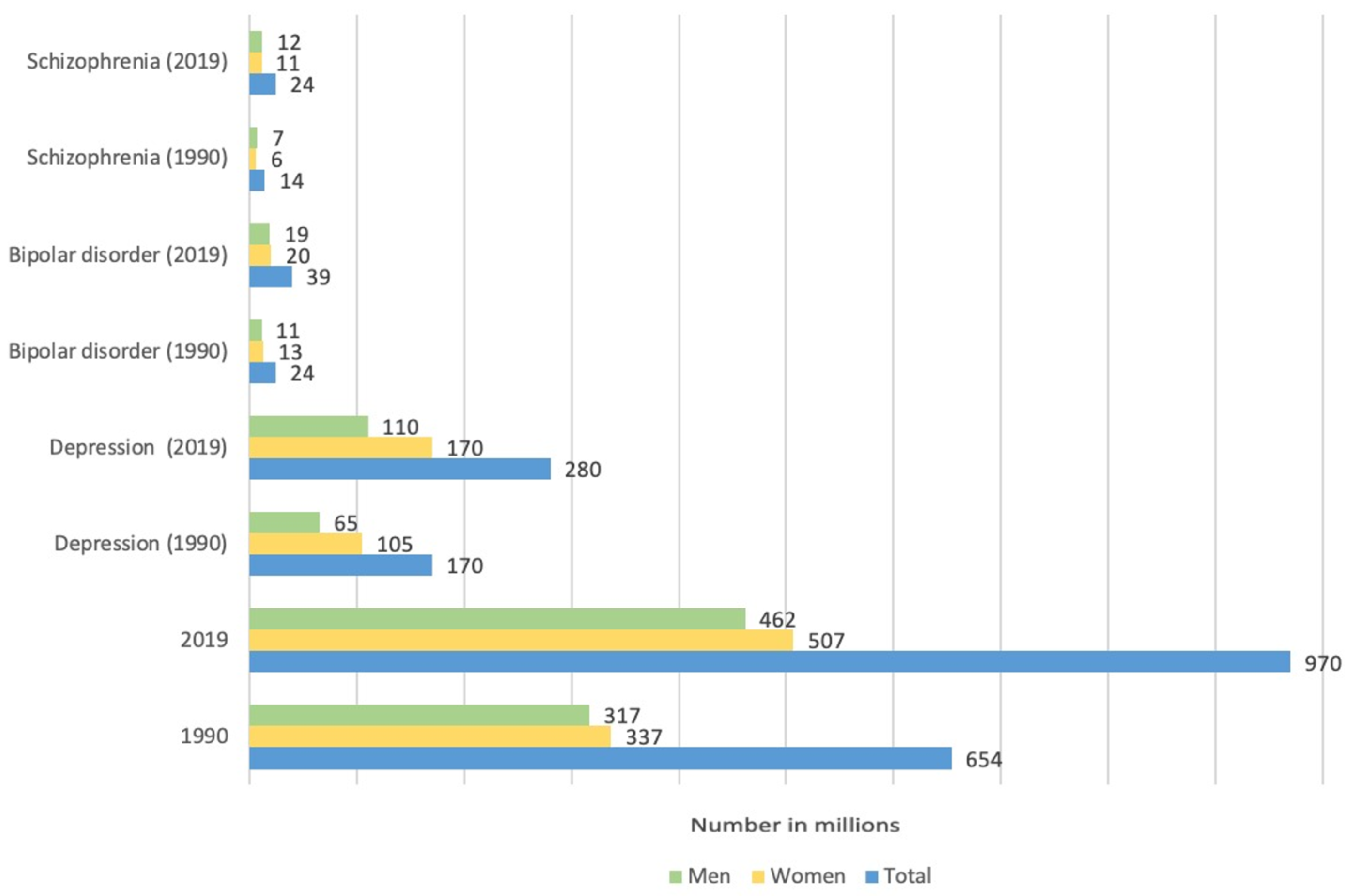
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.F.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.J.; Whiteford, H.A. Global Epidemiology and Burden of Schizophrenia: Findings From the Global Burden of Disease Study 2016. Schizophr. Bull. 2018, 44, 1195–1203. [Google Scholar] [CrossRef]
- Velligan, D.I.; Rao, S. The Epidemiology and Global Burden of Schizophrenia. J. Clin. Psychiatry 2023, 84, 45094. [Google Scholar] [CrossRef]
- Laszlovszky, I.; Barabássy, Á.; Németh, G. Cariprazine, A Broad-Spectrum Antipsychotic for the Treatment of Schizophrenia: Pharmacology, Efficacy, and Safety. Adv. Ther. 2021, 38, 3652–3673. [Google Scholar] [CrossRef] [PubMed]
- Mauri, M.C.; Paletta, S.; Maffini, M.; Moliterno, D.; Altamura, A.C. Suicide attempts in schizophrenic patients: Clinical variables. Asian J. Psychiatr. 2013, 6, 421–427. [Google Scholar] [CrossRef]
- Palmer, B.A.; Pankratz, V.S.; Bostwick, J.M. The Lifetime Risk of Suicide in Schizophrenia. Arch. Gen. Psychiatry 2005, 62, 247. [Google Scholar] [CrossRef] [PubMed]
- Jarskog, L.F.; Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Batalla, A.; Bargalló, N.; Gassó, P.; Molina, O.; Pareto, D.; Mas, S.; Roca, J.M.; Bernardo, M.; Lafuente, A.; Parellada, E. Apoptotic markers in cultured fibroblasts correlate with brain metabolites and regional brain volume in antipsychotic-naive first-episode schizophrenia and healthy controls. Transl. Psychiatry 2015, 5, e626. [Google Scholar] [CrossRef] [PubMed]
- Aram, L.; Yacobi-Sharon, K.; Arama, E. CDPs: Caspase-dependent non-lethal cellular processes. Cell Death Differ. 2017, 24, 1307–1310. [Google Scholar] [CrossRef]
- Hyman, B.T.; Yuan, J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 2012, 13, 395–406. [Google Scholar] [CrossRef]
- Takayanagi, Y.; Sasabayashi, D.; Takahashi, T.; Furuichi, A.; Kido, M.; Nishikawa, Y.; Nakamura, M.; Noguchi, K.; Suzuki, M. Reduced Cortical Thickness in Schizophrenia and Schizotypal Disorder. Schizophr. Bull. 2019, 46, 387–394. [Google Scholar] [CrossRef]
- Maas, D.A.; Vallès, A.; Martens, G.J.M. Oxidative stress, prefrontal cortex hypomyelination and cognitive symptoms in schizophrenia. Transl. Psychiatry 2017, 7, e1171. [Google Scholar] [CrossRef]
- Flores, G.; Morales-Medina, J.C.; Diaz, A. Neuronal and brain morphological changes in animal models of schizophrenia. Behav. Brain Res. 2016, 301, 190–203. [Google Scholar] [CrossRef]
- Xing, G. Decreased calcium-dependent constitutive nitric oxide synthase (cNOS) activity in prefrontal cortex in schizophrenia and depression. Schizophr. Res. 2002, 58, 21–30. [Google Scholar] [CrossRef]
- Zoupa, E.; Pitsikas, N. The Nitric Oxide (NO) Donor Sodium Nitroprusside (SNP) and Its Potential for the Schizophrenia Therapy: Lights and Shadows. Molecules 2021, 26, 3196. [Google Scholar] [CrossRef] [PubMed]
- Ermakov, E.A.; Dmitrieva, E.M.; Parshukova, D.A.; Kazantseva, D.V.; Vasilieva, A.R.; Smirnova, L.P. Oxidative Stress-Related Mechanisms in Schizophrenia Pathogenesis and New Treatment Perspectives. Oxid. Med. Cell. Longev. 2021, 2021, 8881770. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Zhou, Y. NMDAR Hypofunction Animal Models of Schizophrenia. Front. Mol. Neurosci. 2019, 12, 185. [Google Scholar] [CrossRef]
- Callicott, J.H.; Straub, R.E.; Pezawas, L.; Egan, M.F.; Mattay, V.S.; Hariri, A.R.; Verchinski, B.A.; Meyer-Lindenberg, A.; Balkissoon, R.; Kolachana, B.; et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 8627–8632. [Google Scholar] [CrossRef]
- Zoubovsky, S.P.; Pogorelov, V.M.; Taniguchi, Y.; Kim, S.-H.; Yoon, P.; Nwulia, E.; Sawa, A.; Pletnikov, M.V.; Kamiya, A. Working memory deficits in neuronal nitric oxide synthase knockout mice: Potential impairments in prefrontal cortex mediated cognitive function. Biochem. Biophys. Res. Commun. 2011, 408, 707–712. [Google Scholar] [CrossRef]
- Fossier, P.; Blanchard, B.; Ducrocq, C.; Leprince, C.; Tauc, L.; Baux, G. Nitric oxide transforms serotonin into an inactive form and this affects neuromodulation. Neuroscience 1999, 93, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.-J.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef]
- Jarskog, L.F. Apoptosis in schizophrenia: Pathophysiologic and therapeutic considerations. Curr. Opin. Psychiatry 2006, 19, 307–312. [Google Scholar] [CrossRef]
- Ide, M.; Ohnishi, T.; Toyoshima, M.; Balan, S.; Maekawa, M.; Shimamoto-Mitsuyama, C.; Iwayama, Y.; Ohba, H.; Watanabe, A.; Ishii, T.; et al. Excess hydrogen sulfide and polysulfides production underlies a schizophrenia pathophysiology. EMBO Mol. Med. 2019, 11, e10695. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Wei, B.; Li, Y.; Zhan, J.; Jiang, S.; Chen, H.; Yan, K.; Yu, B.; Yang, Y. Decreased plasma levels of gasotransmitter hydrogen sulfide in patients with schizophrenia: Correlation with psychopathology and cognition. Psychopharmacology 2018, 235, 2267–2274. [Google Scholar] [CrossRef]
- Sommer, O.; Aug, R.L.; Schmidt, A.J.; Heiser, P.; Schulz, E.; Vedder, H.; Clement, H.-W. Hydrogen Sulfide Affects Radical Formation in the Hippocampus of LPS Treated Rats and the Effect of Antipsychotics on Hydrogen Sulfide Forming Enzymes in Human Cell Lines. Front. Psychiatry 2018, 9, 501. [Google Scholar] [CrossRef]
- Pearce, M.; Garcia, L.; Abbas, A.; Strain, T.; Schuch, F.B.; Golubic, R.; Kelly, P.; Khan, S.; Utukuri, M.; Laird, Y.; et al. Association Between Physical Activity and Risk of Depression. JAMA Psychiatry 2022, 79, 550. [Google Scholar] [CrossRef]
- Papp, M.; Cubała, W.J.; Swiecicki, L.; Newman-Tancredi, A.; Willner, P. Perspectives for therapy of treatment-resistant depression. Br. J. Pharmacol. 2022, 179, 4181–4200. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, Y. Animal models for the study of depressive disorder. CNS Neurosci. Ther. 2021, 27, 633–642. [Google Scholar] [CrossRef]
- McKernan, D.P.; Dinan, T.G.; Cryan, J.F. “Killing the Blues”: A role for cellular suicide (apoptosis) in depression and the antidepressant response? Prog. Neurobiol. 2009, 88, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi M Nitric oxide: Antidepressant mechanisms and inflammation. Adv Pharmacol. 2019, 86, 121–152.
- Chrapko, W.; Jurasz, P.; Radomski, M.W.; Archer, S.L.; Newman, S.C.; Baker, G.; Lara, N.; Le Mellédo, J.-M. Alteration of Decreased Plasma NO Metabolites and Platelet NO Synthase Activity by Paroxetine in Depressed Patients. Neuropsychopharmacology 2006, 31, 1286–1293. [Google Scholar] [CrossRef]
- Erkan Ozcan, M.; Gulec, M.; Ozerol, E.; Polat, R.; Akyol, O. Antioxidant enzyme activities and oxidative stress in affective disorders. Int. Clin. Psychopharmacol. 2004, 19, 89–95. [Google Scholar] [CrossRef]
- Talarowska, M.; Gałecki, P.; Maes, M.; Orzechowska, A.; Chamielec, M.; Bartosz, G.; Kowalczyk, E. Nitric oxide plasma concentration associated with cognitive impairment in patients with recurrent depressive disorder. Neurosci. Lett. 2012, 510, 127–131. [Google Scholar] [CrossRef]
- Moreno, J.; Gaspar, E.; López-Bello, G.; Juárez, E.; Alcázar-Leyva, S.; González-Trujano, E.; Pavón, L.; Alvarado-Vásquez, N. Increase in nitric oxide levels and mitochondrial membrane potential in platelets of untreated patients with major depression. Psychiatry Res. 2013, 209, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Kong, F.-Z.; Hong, X.-H.; Zhang, L.; Zhao, W.-H.; Yang, J.-C.; Zhang, H. Neuronal Nitric Oxide Synthase Regulates Depression-like Behaviors in Shortening-Induced Obese Mice. Nutrients 2022, 14, 4302. [Google Scholar] [CrossRef] [PubMed]
- McLeod, T.M.; López-Figueroa, A.L.; López-Figueroa, M.O. Nitric oxide, stress, and depression. Psychopharmacol. Bull. 2001, 35, 24–41. [Google Scholar]
- Dhir, A.; Kulkarni, S.K. Nitric oxide and major depression. Nitric Oxide 2011, 24, 125–131. [Google Scholar] [CrossRef]
- Kulikova, E.A.; Kulikov, V.A.; Sinyakova, N.A.; Kulikov, A.V.; Popova, N. The effect of long-term hindlimb unloading on the expression of risk neurogenes encoding elements of serotonin-, dopaminergic systems and apoptosis; comparison with the effect of actual spaceflight on mouse brain. Neurosci. Lett. 2017, 640, 88–92. [Google Scholar] [CrossRef]
- Bhatt, S.; Nagappa, A.N.; Patil, C.R. Role of oxidative stress in depression. Drug Discov. Today 2020, 25, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Herken, H.; Gurel, A.; Selek, S.; Armutcu, F.; Ozen, M.E.; Bulut, M.; Kap, O.; Yumru, M.; Savas, H.A.; Akyol, O. Adenosine Deaminase, Nitric Oxide, Superoxide Dismutase, and Xanthine Oxidase in Patients with Major Depression: Impact of Antidepressant Treatment. Arch. Med. Res. 2007, 38, 247–252. [Google Scholar] [CrossRef]
- Yang, D.; Liu, X.; Zhang, R.; Cheng, K.; Mu, J.; Fang, L.; Xie, P. Increased apoptosis and different regulation of pro-apoptosis protein bax and anti-apoptosis protein bcl-2 in the olfactory bulb of a rat model of depression. Neurosci. Lett. 2011, 504, 18–22. [Google Scholar] [CrossRef]
- Moreno-Lopez, B. Nitric Oxide Is a Physiological Inhibitor of Neurogenesis in the Adult Mouse Subventricular Zone and Olfactory Bulb. J. Neurosci. 2004, 24, 85–95. [Google Scholar] [CrossRef]
- Scaini, G.; Mason, B.L.; Diaz, A.P.; Jha, M.K.; Soares, J.C.; Trivedi, M.H.; Quevedo, J. Dysregulation of mitochondrial dynamics, mitophagy and apoptosis in major depressive disorder: Does inflammation play a role? Mol. Psychiatry 2022, 27, 1095–1102. [Google Scholar] [CrossRef]
- Yang, Y.-J.; Chen, C.-N.; Zhan, J.-Q.; Liu, Q.-S.; Liu, Y.; Jiang, S.-Z.; Wei, B. Decreased Plasma Hydrogen Sulfide Level Is Associated With the Severity of Depression in Patients With Depressive Disorder. Front. Psychiatry 2021, 12, 765664. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Jiang, L.; Lan, F.; Tang, Y.-Y.; Zhang, P.; Zou, W.; Chen, Y.-J.; Tang, X.-Q. Hydrogen sulfide antagonizes sleep deprivation-induced depression- and anxiety-like behaviors by inhibiting neuroinflammation in a hippocampal Sirt1-dependent manner. Brain Res. Bull. 2021, 177, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Roch, G.; Batallé, G.; Bai, X.; Pouso-Vázquez, E.; Rodríguez, L.; Pol, O. The Beneficial Effects of Heme Oxygenase 1 and Hydrogen Sulfide Activation in the Management of Neuropathic Pain, Anxiety- and Depressive-like Effects of Paclitaxel in Mice. Antioxidants 2022, 11, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.; Xin, Y.; Zhang, J.; Wang, S.; Yang, Z.; Liu, C. Hydrogen sulfide alleviates the anxiety-like and depressive-like behaviors of type 1 diabetic mice via inhibiting inflammation and ferroptosis. Life Sci. 2021, 278, 119551. [Google Scholar] [CrossRef]
- Luo, Y.; Ullah, R.; Wang, J.; Du, Y.; Huang, S.; Meng, L.; Gao, Y.; Gong, M.; Galaj, E.; Yin, X.; et al. Exogenous Carbon Monoxide Produces Rapid Antidepressant- and Anxiolytic-Like Effects. Front. Pharmacol. 2021, 12, 757417. [Google Scholar] [CrossRef]
- Ueno, K.; Morstein, J.; Ofusa, K.; Naganos, S.; Suzuki-Sawano, E.; Minegishi, S.; Rezgui, S.P.; Kitagishi, H.; Michel, B.W.; Chang, C.J.; et al. Carbon Monoxide, a Retrograde Messenger Generated in Postsynaptic Mushroom Body Neurons, Evokes Noncanonical Dopamine Release. J. Neurosci. 2020, 40, 3533–3548. [Google Scholar] [CrossRef]
- Latalova, K.; Kamaradova, D.; Prasko, J. Suicide in bipolar disorder: A review. Psychiatr Danub 2014, 26, 108–114. [Google Scholar]
- Arnold, I.; Dehning, J.; Grunze, A.; Hausmann, A. Old Age Bipolar Disorder—Epidemiology, Aetiology and Treatment. Medicina 2021, 57, 587. [Google Scholar] [CrossRef]
- Gigante, A.D.; Young, L.T.; Yatham, L.N.; Andreazza, A.C.; Nery, F.G.; Grinberg, L.T.; Heinsen, H.; Lafer, B. Morphometric post-mortem studies in bipolar disorder: Possible association with oxidative stress and apoptosis. Int. J. Neuropsychopharmacol. 2011, 14, 1075–1089. [Google Scholar] [CrossRef]
- Uranova, N.; Orlovskaya, D.; Vikhreva, O.; Zimina, I.; Kolomeets, N.; Vostrikov, V.; Rachmanova, V. Electron microscopy of oligodendroglia in severe mental illness. Brain Res. Bull. 2001, 55, 597–610. [Google Scholar] [CrossRef]
- Kim, H.-W.; Rapoport, S.I.; Rao, J.S. Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol. Dis. 2010, 37, 596–603. [Google Scholar] [CrossRef]
- Kauer-Sant’Anna, M.; Kapczinski, F.; Andreazza, A.C.; Bond, D.J.; Lam, R.W.; Young, L.T.; Yatham, L.N. Brain-derived neurotrophic factor and inflammatory markers in patients with early- vs. late-stage bipolar disorder. Int. J. Neuropsychopharmacol. 2009, 12, 447. [Google Scholar] [CrossRef]
- Selek, S.; Savas, H.A.; Gergerlioglu, H.S.; Bulbul, F.; Uz, E.; Yumru, M. The course of nitric oxide and superoxide dismutase during treatment of bipolar depressive episode. J. Affect. Disord. 2008, 107, 89–94. [Google Scholar] [CrossRef]
- Bielau, H.; Brisch, R.; Bernard-Mittelstaedt, J.; Dobrowolny, H.; Gos, T.; Baumann, B.; Mawrin, C.; Bernstein, H.-G.; Bogerts, B.; Steiner, J. Immunohistochemical evidence for impaired nitric oxide signaling of the locus coeruleus in bipolar disorder. Brain Res. 2012, 1459, 91–99. [Google Scholar] [CrossRef]
- Penninx, B.W.; Pine, D.S.; Holmes, E.A.; Reif, A. Anxiety disorders. Lancet 2021, 397, 914–927. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Y.; Wang, J.; Liu, Y.; Xiao, M.; Song, C.; Bai, Y.; Yinuo Han, N.; Han, F. Synapse impairment associated with enhanced apoptosis in post-traumatic stress disorder. Synapse 2020, 74, e22134. [Google Scholar] [CrossRef]
- Li, X.M.; Han, F.; Liu, D.J.; Shi, Y.X. Single-prolonged stress induced mitochondrial-dependent apoptosis in hippocampus in the rat model of post-traumatic stress disorder. J. Chem. Neuroanat. 2010, 40, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Han, Y.; Wang, X.; Han, F. Role of apoptosis in the Post-traumatic stress disorder model-single prolonged stressed rats. Psychoneuroendocrinology 2018, 95, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Gulati, K.; Rai, N.; Ray, A. Nitric Oxide and Anxiety. Vitam. Horm. 2017, 103, 169–192. [Google Scholar] [PubMed]
- Pałasz, A.; Menezes, I.C.; Worthington, J.J. The role of brain gaseous neurotransmitters in anxiety. Pharmacol. Rep. 2021, 73, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chanana, P. Role of Nitric Oxide in Stress-Induced Anxiety: From pathophysiology to therapeutic target. Vitam. Horm. 2017, 103, 147–167. [Google Scholar] [PubMed]
- Spiga, F.; Lightman, S.L.; Shekhar, A.; Lowry, C.A. Injections of urocortin 1 into the basolateral amygdala induce anxiety-like behavior and c-Fos expression in brainstem serotonergic neurons. Neuroscience 2006, 138, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Gong, S.; Luo, J.; Zheng, Z.; Song, B.; Ma, S.; Guo, J.; Hu, C.; Thiel, G.; Vinson, C.; et al. Opposing Roles for ATF2 and c-Fos in c-Jun-Mediated Neuronal Apoptosis. Mol. Cell. Biol. 2009, 29, 2431–2442. [Google Scholar] [CrossRef] [PubMed]
- Sugama, S.; Kakinuma, Y. Stress and brain immunity: Microglial homeostasis through hypothalamus-pituitary-adrenal gland axis and sympathetic nervous system. Brain, Behav. Immun.-Health 2020, 7, 100111. [Google Scholar] [CrossRef]
- Orlando, G.F.; Langnaese, K.; Schulz, C.; Wolf, G.; Engelmann, M. Neuronal nitric oxide synthase gene inactivation reduces the expression of vasopressin in the hypothalamic paraventricular nucleus and of catecholamine biosynthetic enzymes in the adrenal gland of the mouse. Stress 2008, 11, 42–51. [Google Scholar] [CrossRef]
- Bi, X.L.; Yang, J.Y.; Dong, Y.X.; Wang, J.M.; Cui, Y.H.; Ikeshima, T.; Zhao, Y.Q.; Wu, C.F. Resveratrol inhibits nitric oxide and TNF-α production by lipopolysaccharide-activated microglia. Int. Immunopharmacol. 2005, 5, 185–193. [Google Scholar] [CrossRef]
- Beheshti, F.; Hashemzehi, M.; Hosseini, M.; Marefati, N.; Memarpour, S. Inducible nitric oxide synthase plays a role in depression- and anxiety-like behaviors chronically induced by lipopolysaccharide in rats: Evidence from inflammation and oxidative stress. Behav. Brain Res. 2020, 392, 112720. [Google Scholar] [CrossRef]
- Carvalho-Costa, P.G.; Branco, L.G.S.; Leite-Panissi, C.R.A. Activation of locus coeruleus heme oxygenase-carbon monoxide pathway promoted an anxiolytic-like effect in rats. Braz. J. Med. Biol. Res. 2016, 49, e5135. [Google Scholar] [CrossRef]
- Cazuza, R.A.; Pol, O.; Leite-Panissi, C.R.A. Enhanced expression of heme oxygenase-1 in the locus coeruleus can be associated with anxiolytic-like effects. Behav. Brain Res. 2018, 336, 204–210. [Google Scholar] [CrossRef]
- Habibitabar, E.; Moridi, H.; Shateri, H.; Karimi, S.A.; Salehi, I.; Komaki, A.; Sarihi, A. Chronic NaHS treatment improves spatial and passive avoidance learning and memory and anxiety-like behavior and decreases oxidative stress in rats fed with a high-fat diet. Brain Res. Bull. 2020, 164, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, O.; Bogatova, K.; Mukhtarova, R.; Yakovlev, A.; Shakhmatova, V.; Gerasimova, E.; Ziyatdinova, G.; Hermann, A.; Sitdikova, G. Hydrogen Sulfide Alleviates Anxiety, Motor, and Cognitive Dysfunctions in Rats with Maternal Hyperhomocysteinemia via Mitigation of Oxidative Stress. Biomolecules 2020, 10, 995. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Pritchard, C.; Fortune, D.; Kodi, P.; Grados, M. Hydrogen sulfide: A target to modulate oxidative stress and neuroplasticity for the treatment of pathological anxiety. Expert Rev. Neurother. 2020, 20, 109–121. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodkin, S.; Nwosu, C.; Sannikov, A.; Tyurin, A.; Chulkov, V.S.; Raevskaya, M.; Ermakov, A.; Kirichenko, E.; Gasanov, M. The Role of Gasotransmitter-Dependent Signaling Mechanisms in Apoptotic Cell Death in Cardiovascular, Rheumatic, Kidney, and Neurodegenerative Diseases and Mental Disorders. Int. J. Mol. Sci. 2023, 24, 6014. https://doi.org/10.3390/ijms24076014
Rodkin S, Nwosu C, Sannikov A, Tyurin A, Chulkov VS, Raevskaya M, Ermakov A, Kirichenko E, Gasanov M. The Role of Gasotransmitter-Dependent Signaling Mechanisms in Apoptotic Cell Death in Cardiovascular, Rheumatic, Kidney, and Neurodegenerative Diseases and Mental Disorders. International Journal of Molecular Sciences. 2023; 24(7):6014. https://doi.org/10.3390/ijms24076014
Chicago/Turabian StyleRodkin, Stanislav, Chizaram Nwosu, Alexander Sannikov, Anton Tyurin, Vasilii Sergeevich Chulkov, Margarita Raevskaya, Alexey Ermakov, Evgeniya Kirichenko, and Mitkhat Gasanov. 2023. "The Role of Gasotransmitter-Dependent Signaling Mechanisms in Apoptotic Cell Death in Cardiovascular, Rheumatic, Kidney, and Neurodegenerative Diseases and Mental Disorders" International Journal of Molecular Sciences 24, no. 7: 6014. https://doi.org/10.3390/ijms24076014