
Deciphering the Polyglucosan Accumulation Present in Lafora Disease Using an Astrocytic Cellular Model
 , and
, and
Abstract
:1. Introduction
2. Results
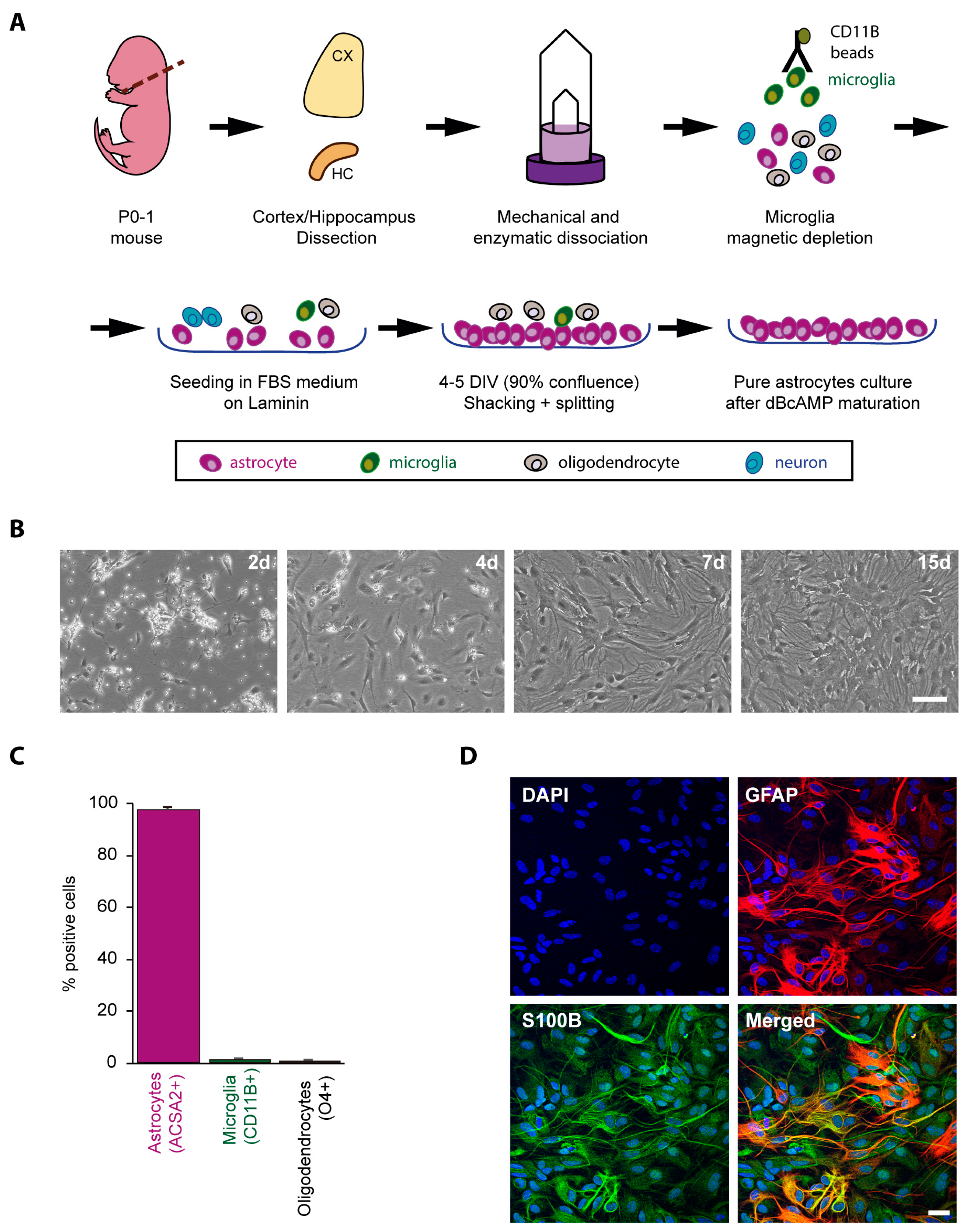
2.1. Obtaining a Primary Culture of Astrocytes from Control and LD Mouse Models
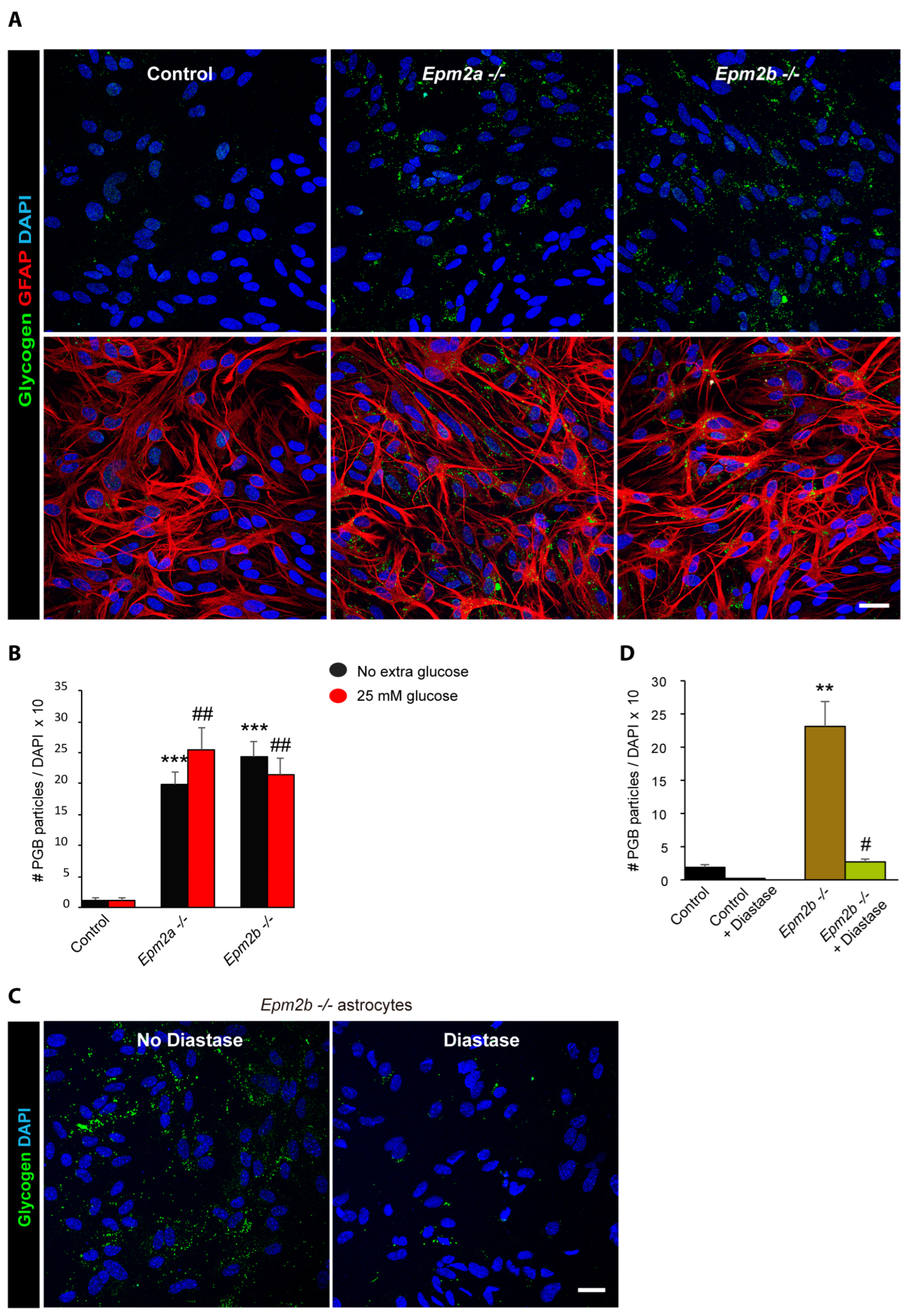
2.2. Astrocytes from LD Mice Accumulate More Polyglucosan Inclusions Than Controls
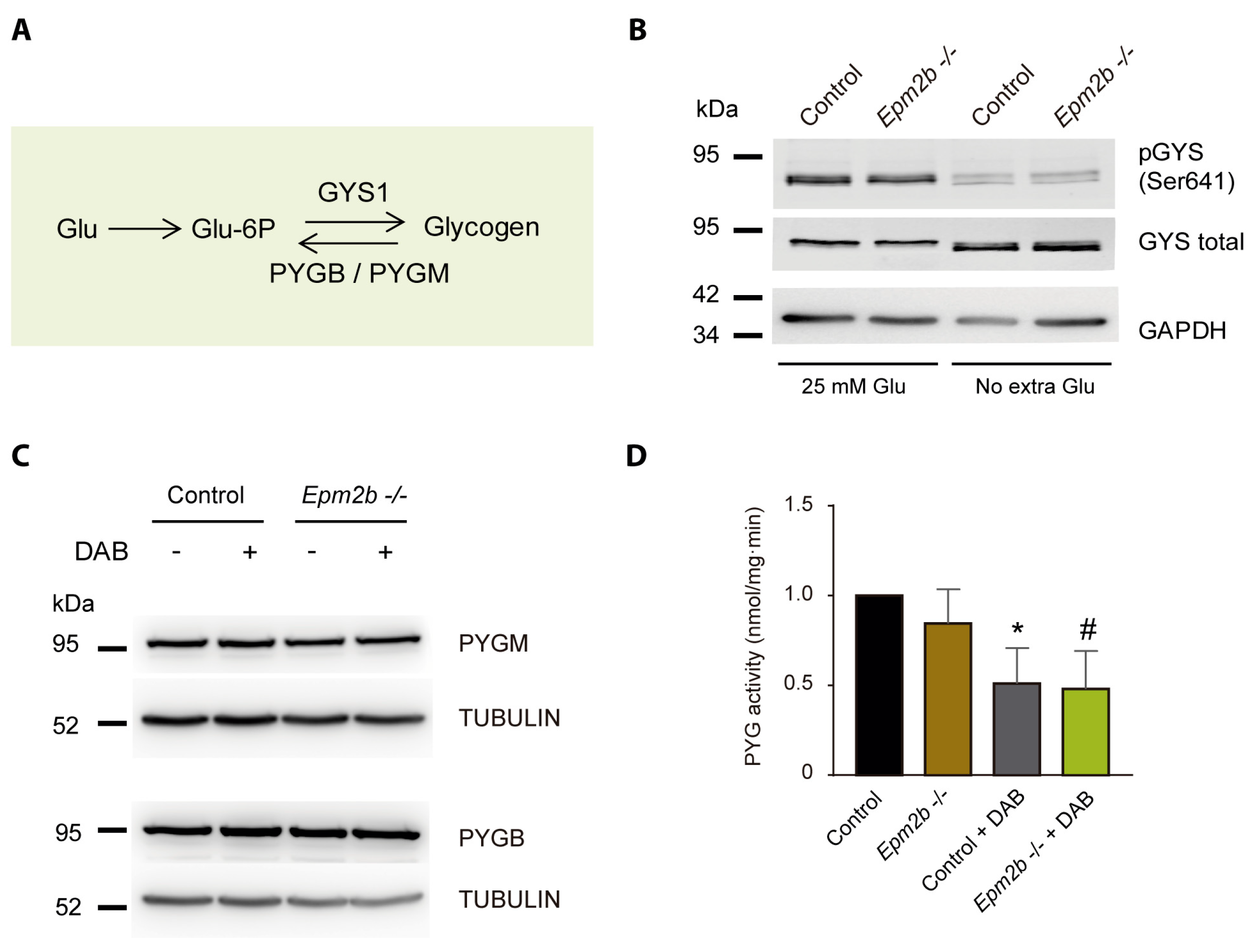
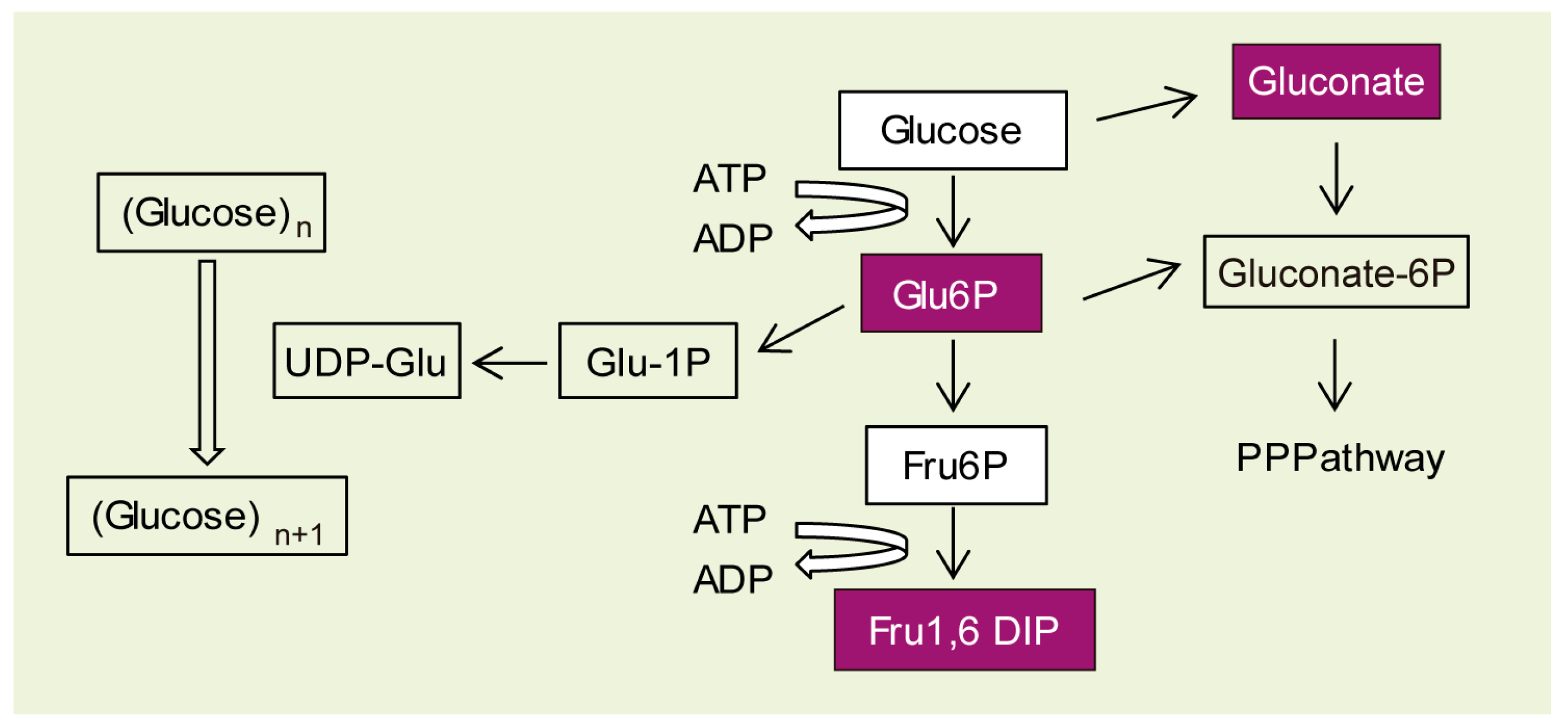
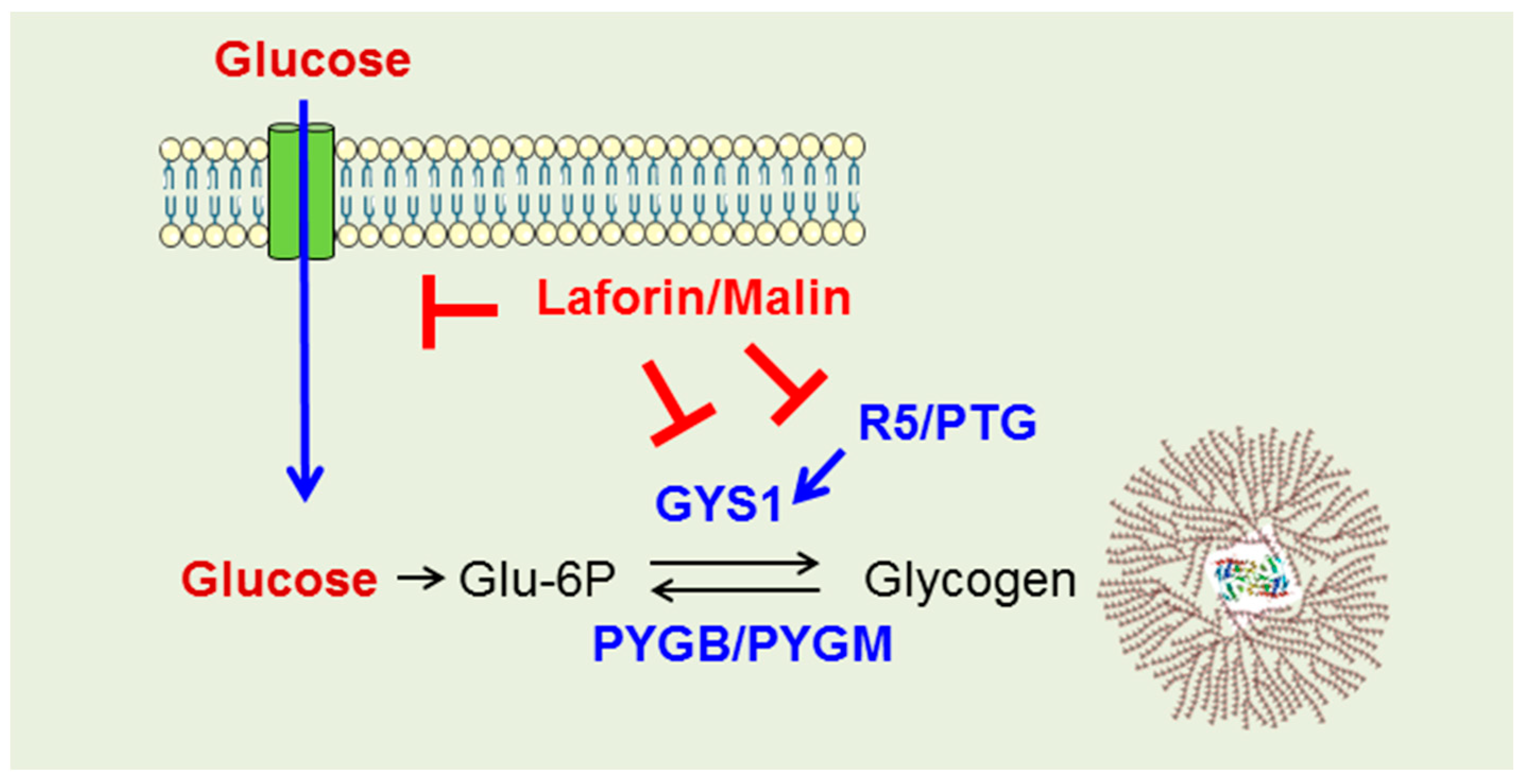
2.3. Why Do LD Astrocytes Accumulate More Polyglucosan Inclusions?
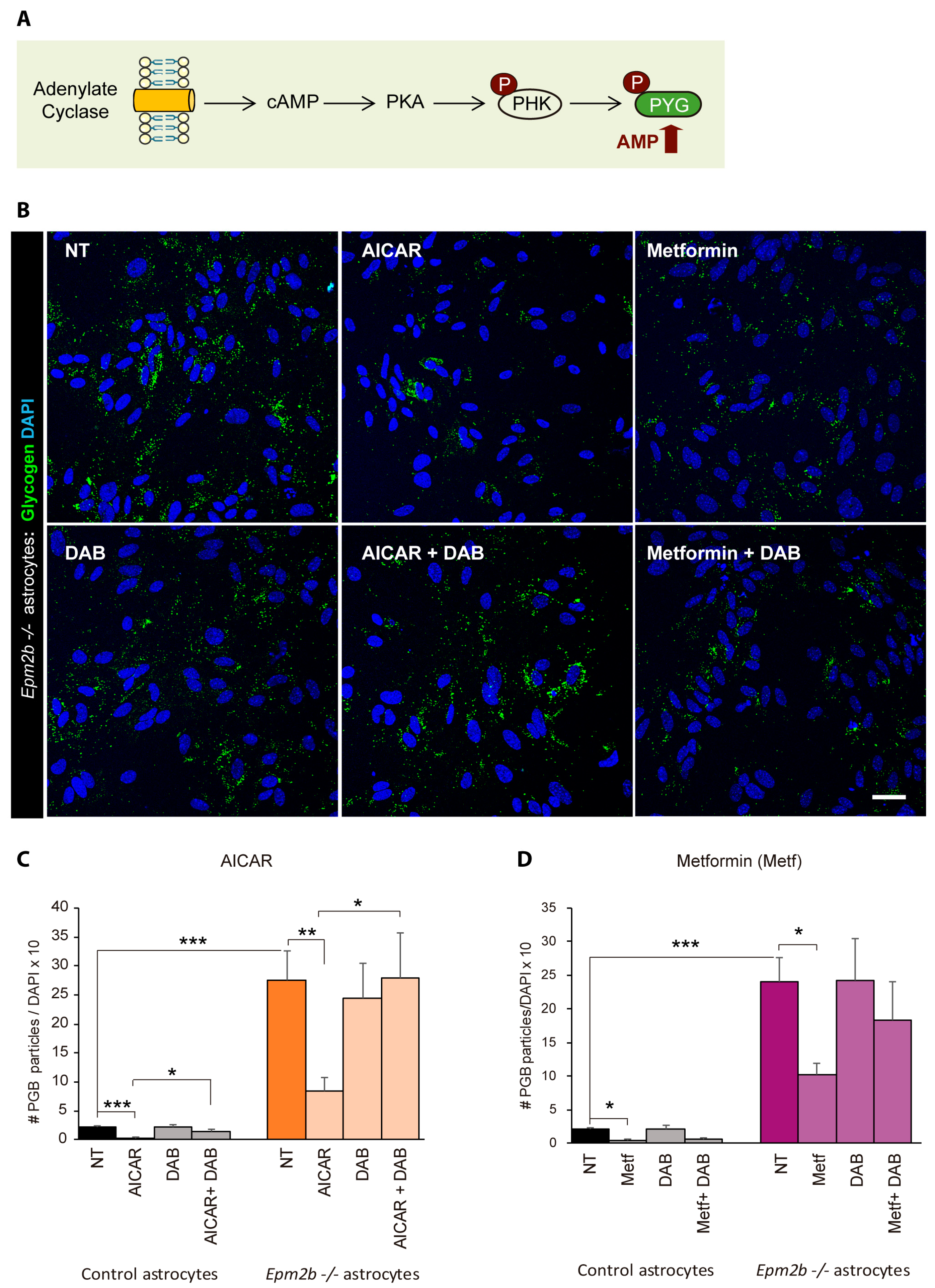
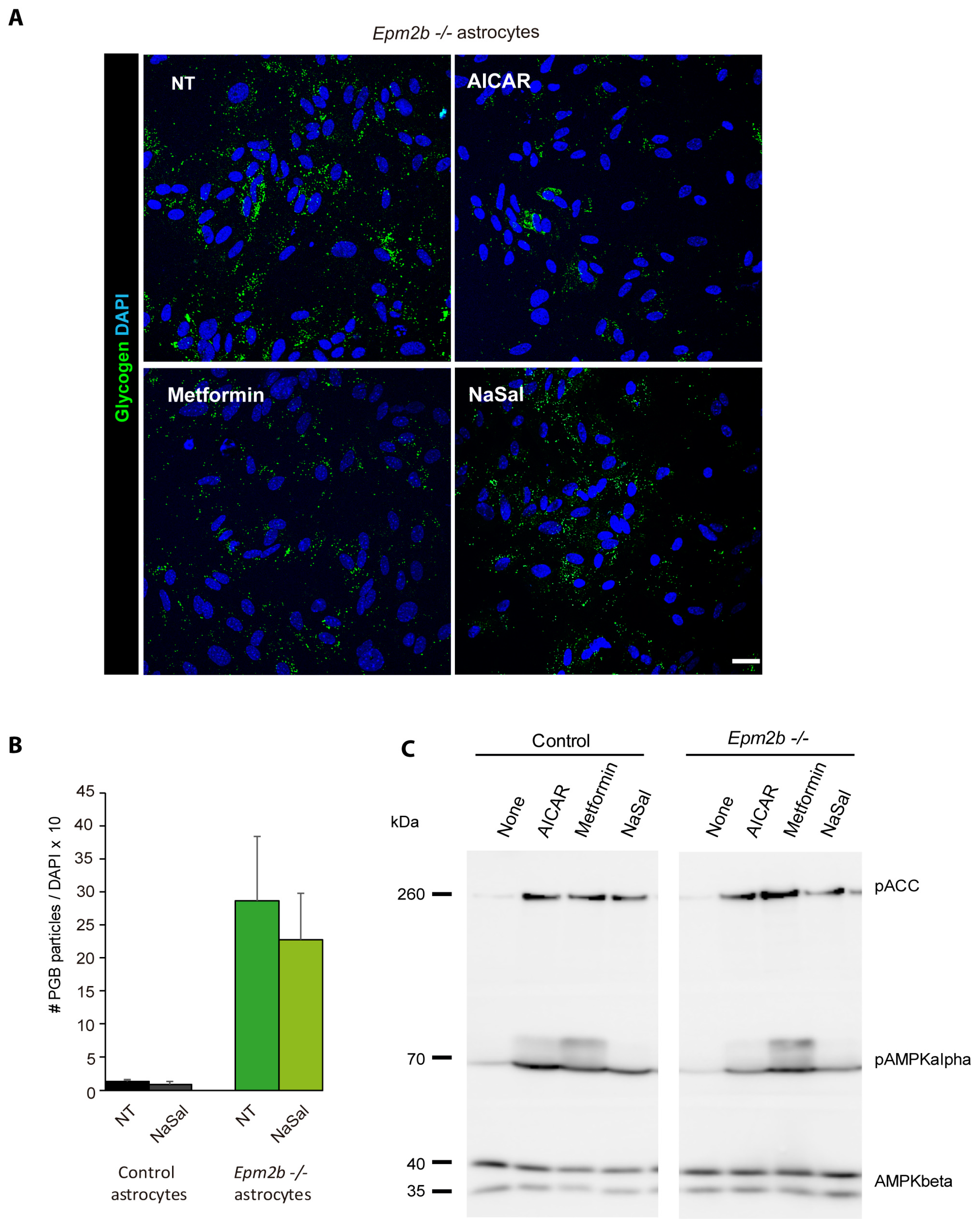
2.4. Can the Accumulation of Polyglucosan Inclusions in Epm2b−/− Astrocytes Be Prevented?
3. Discussion
4. Materials and Methods
4.1. Primary Mouse Astrocyte Isolation and Culture
4.2. Cytometry
4.3. Immunofluorescence (IF) Analyses
4.4. Western Blot Analyses
4.5. Glycogen Phosphorylase Enzymatic Assay
4.6. Metabolomics Analyses at Metabolon Co.
4.6.1. Sample collection
4.6.2. Sample Preparation and Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lafora, G.R.; Glueck, B. Beitrag zur histopathologie der myoklonischen epilepsie. Z. Für Die Gesamte Neurol. Und Psychiatr. 1911, 6, 1–14. [Google Scholar] [CrossRef]
- Sakai, M.; Austin, J.; Witmer, F.; Trueb, L. Studies in myoclonus epilepsy (lafora body form). Ii. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology 1970, 20, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Girard, J.-M.; Lohi, H.; Chan, E.M.; Wang, P.; Tiberia, E.; Omer, S.; Ahmed, M.; Bennett, C.; Chakrabarty, A.; et al. Early-onset lafora body disease. Brain 2012, 135, 2684–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, J.; Tiberia, E.; Striano, P.; Genton, P.; Carpenter, S.; Ackerley, C.A.; Minassian, B.A. Lafora disease. Epileptic Disord. 2016, 18, 38–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pondrelli, F.; Muccioli, L.; Licchetta, L.; Mostacci, B.; Zenesini, C.; Tinuper, P.; Vignatelli, L.; Bisulli, F. Natural history of lafora disease: A prognostic systematic review and individual participant data meta-analysis. Orphanet J. Rare Dis. 2021, 16, 362. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gimeno, M.A.; Knecht, E.; Sanz, P. Lafora disease: A ubiquitination-related pathology. Cells 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, S.; Delgado-Escueta, A.V.; Sakamoto, T.; Avila, M.R.; Machado-Salas, J.; Hoshii, Y.; Akagi, T.; Gomi, H.; Suzuki, T.; Amano, K.; et al. Targeted disruption of the epm2a gene causes formation of lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 2002, 11, 1251–1262. [Google Scholar] [CrossRef] [Green Version]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef]
- Garcia-Cabrero, A.M.; Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanchez, M.P. Enhanced sensitivity of laforin- and malin-deficient mice to the convulsant agent pentylenetetrazole. Front. Neurosci. 2014, 8, 291. [Google Scholar]
- Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado, O.; Vernia, S.; Boya, P.; Sanz, P.; de Cordoba, S.R.; Knecht, E.; Rubinsztein, D.C. Laforin, the most common protein mutated in lafora disease, regulates autophagy. Hum. Mol. Genet. 2010, 19, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Puri, R.; Ganesh, S. Laforin in autophagy: A possible link between carbohydrate and protein in lafora disease? Autophagy 2010, 6, 1229–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Cabrero, A.M.; Marinas, A.; Guerrero, R.; de Cordoba, S.R.; Serratosa, J.M.; Sanchez, M.P. Laforin and malin deletions in mice produce similar neurologic impairments. J. Neuropathol. Exp. Neurol. 2012, 71, 413–421. [Google Scholar] [CrossRef]
- Perez-Jimenez, E.; Viana, R.; Munoz-Ballester, C.; Vendrell-Tornero, C.; Moll-Diaz, R.; Garcia-Gimeno, M.A.; Sanz, P. Endocytosis of the glutamate transporter 1 is regulated by laforin and malin: Implications in lafora disease. Glia 2021, 69, 1170–1183. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, I.; Viana, R.; Sanz, P.; Ferrer, I. Inflammation in lafora disease: Evolution with disease progression in laforin and malin knock-out mouse models. Mol. Neurobiol. 2017, 54, 3119–3130. [Google Scholar] [CrossRef] [Green Version]
- Lahuerta, M.; Gonzalez, D.; Aguado, C.; Fathinajafabadi, A.; Garcia-Gimenez, J.L.; Moreno-Estelles, M.; Roma-Mateo, C.; Knecht, E.; Pallardo, F.V.; Sanz, P. Reactive glia-derived neuroinflammation: A novel hallmark in lafora progressive myoclonus epilepsy that progresses with age. Mol. Neurobiol. 2020, 57, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Rubio, T.; Viana, R.; Moreno-Estelles, M.; Campos-Rodriguez, A.; Sanz, P. Tnf and il6/jak2 signaling pathways are the main contributors of the glia-derived neuroinflammation present in lafora disease, a fatal form of progressive myoclonus epilepsy. Neurobiol. Dis. 2023, 176, 105964. [Google Scholar] [CrossRef]
- Gentry, M.S.; Afawi, Z.; Armstrong, D.D.; Delgado-Escueta, A.; Goldberg, Y.P.; Grossman, T.R.; Guinovart, J.J.; Harris, F.; Hurley, T.D.; Michelucci, R.; et al. The 5th international lafora epilepsy workshop: Basic science elucidating therapeutic options and preparing for therapies in the clinic. Epilepsy Behav. 2020, 103, 106839. [Google Scholar] [CrossRef] [PubMed]
- Pederson, B.A.; Turnbull, J.; Epp, J.R.; Weaver, S.A.; Zhao, X.; Pencea, N.; Roach, P.J.; Frankland, P.W.; Ackerley, C.A.; Minassian, B.A. Inhibiting glycogen synthesis prevents lafora disease in a mouse model. Ann. Neurol. 2013, 74, 297–300. [Google Scholar] [CrossRef]
- Turnbull, J.; DePaoli-Roach, A.A.; Zhao, X.; Cortez, M.A.; Pencea, N.; Tiberia, E.; Piliguian, M.; Roach, P.J.; Wang, P.; Ackerley, C.A.; et al. Ptg depletion removes lafora bodies and rescues the fatal epilepsy of lafora disease. PLoS Genet. 2011, 7, e1002037. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, J.; Epp, J.R.; Goldsmith, D.; Zhao, X.; Pencea, N.; Wang, P.; Frankland, P.W.; Ackerley, C.A.; Minassian, B.A. Ptg protein depletion rescues malin-deficient lafora disease in mouse. Ann. Neurol. 2014, 75, 442–446. [Google Scholar] [CrossRef]
- Tang, B.; Frasinyuk, M.S.; Chikwana, V.M.; Mahalingan, K.K.; Morgan, C.A.; Segvich, D.M.; Bondarenko, S.P.; Mrug, G.P.; Wyrebek, P.; Watt, D.S.; et al. Discovery and development of small-molecule inhibitors of glycogen synthase. J. Med. Chem. 2020, 63, 3538–3551. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, S.; Nitschke, S.; Grossman, T.R.; Kordasiewicz, H.; Wang, P.; Zhao, X.; Guisso, D.R.; Kasiri, S.; Nitschke, F.; Minassian, B.A. Gys1 antisense therapy rescues neuropathological bases of murine lafora disease. Brain 2021, 144, 2985–2993. [Google Scholar] [CrossRef]
- Gumusgoz, E.; Kasiri, S.; Guisso, D.R.; Wu, J.; Dear, M.; Verhalen, B.; Minassian, B.A. Aav-mediated artificial mirna reduces pathogenic polyglucosan bodies and neuroinflammation in adult polyglucosan body and lafora disease mouse models. Neurotherapeutics 2022, 19, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Brewer, M.K.; Uittenbogaard, A.; Austin, G.L.; Segvich, D.M.; DePaoli-Roach, A.; Roach, P.J.; McCarthy, J.J.; Simmons, Z.R.; Brandon, J.A.; Zhou, Z.; et al. Targeting pathogenic lafora bodies in lafora disease using an antibody-enzyme fusion. Cell Metab. 2019, 30, 689–705.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Austin, G.L.; Shaffer, R.; Armstrong, D.D.; Gentry, M.S. Antibody-mediated enzyme therapeutics and applications in glycogen storage diseases. Trends Mol. Med. 2019, 25, 1094–1109. [Google Scholar] [CrossRef]
- Rubio-Villena, C.; Viana, R.; Bonet, J.; Garcia-Gimeno, M.A.; Casado, M.; Heredia, M.; Sanz, P. Astrocytes: New players in progressive myoclonus epilepsy of lafora type. Hum. Mol. Genet. 2018, 27, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Ballester, C.; Santana, N.; Perez-Jimenez, E.; Viana, R.; Artigas, F.; Sanz, P. In vivo glutamate clearance defects in a mouse model of lafora disease. Exp. Neurol. 2019, 320, 112959. [Google Scholar] [CrossRef]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Khakh, B.S.; Deneen, B. The emerging nature of astrocyte diversity. Annu. Rev. Neurosci. 2019, 42, 187–207. [Google Scholar] [CrossRef]
- Davies, J.K.; Wells, D.J.; Liu, K.; Whitrow, H.R.; Daniel, T.D.; Grignani, R.; Lygate, C.A.; Schneider, J.E.; Noel, G.; Watkins, H.; et al. Characterization of the role of {gamma}2 r531g mutation in amp-activated protein kinase in cardiac hypertrophy and wolff-parkinson-white syndrome. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1942–H1951. [Google Scholar] [CrossRef] [Green Version]
- Auge, E.; Pelegri, C.; Manich, G.; Cabezon, I.; Guinovart, J.J.; Duran, J.; Vilaplana, J. Astrocytes and neurons produce distinct types of polyglucosan bodies in lafora disease. Glia 2018, 66, 2094–2107. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliabracci, V.S.; Turnbull, J.; Wang, W.; Girard, J.M.; Zhao, X.; Skurat, A.V.; Delgado-Escueta, A.V.; Minassian, B.A.; Depaoli-Roach, A.A.; Roach, P.J. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19262–19266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeau, O.W.; Fontes, J.D.; Carlson, G.M. The regulation of glycogenolysis in the brain. J. Biol. Chem. 2018, 293, 7099–7107. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, C.; Dupret, J.M.; Rodrigues-Lima, F. The structure and the regulation of glycogen phosphorylases in brain. Adv. Neurobiol. 2019, 23, 125–145. [Google Scholar]
- Walls, A.B.; Sickmann, H.M.; Brown, A.; Bouman, S.D.; Ransom, B.; Schousboe, A.; Waagepetersen, H.S. Characterization of 1,4-dideoxy-1,4-imino-d-arabinitol (dab) as an inhibitor of brain glycogen shunt activity. J. Neurochem. 2008, 105, 1462–1470. [Google Scholar] [CrossRef]
- Kumarasinghe, L.; Garcia-Gimeno, M.A.; Ramirez, J.; Mayor, U.; Zugaza, J.L.; Sanz, P. P-rex1 is a novel substrate of the e3 ubiquitin ligase malin associated with lafora disease. Neurobiol. Dis. 2023, 177, 105998. [Google Scholar] [CrossRef]
- Visnjic, D.; Lalic, H.; Dembitz, V.; Tomic, B.; Smoljo, T. Aicar, a widely used ampk activator with important ampk-independent effects: A systematic review. Cells 2021, 10, 1095. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and molecular mechanisms of metformin action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Sanz, P.; Serratosa, J.M.; Sanchez, M.P. Beneficial effects of metformin on the central nervous system, with a focus on epilepsy and lafora disease. Int. J. Mol. Sci. 2021, 22, 5351. [Google Scholar] [CrossRef]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates amp-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthier, A.; Paya, M.; Garcia-Cabrero, A.M.; Ballester, M.I.; Heredia, M.; Serratosa, J.M.; Sanchez, M.P.; Sanz, P. Pharmacological interventions to ameliorate neuropathological symptoms in a mouse model of lafora disease. Mol. Neurobiol. 2016, 53, 1296–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgos, D.F.; Machio-Castello, M.; Iglesias-Cabeza, N.; Giraldez, B.G.; Gonzalez-Fernandez, J.; Sanchez-Martin, G.; Sanchez, M.P.; Serratosa, J.M. Early treatment with metformin improves neurological outcomes in lafora disease. Neurotherapeutics, 2022; ahead of print. [Google Scholar] [CrossRef]
- Hertz, L.; Peng, L.; Lai, J.C. Functional studies in cultured astrocytes. Methods 1998, 16, 293–310. [Google Scholar] [CrossRef]
- Muller, M.S.; Fox, R.; Schousboe, A.; Waagepetersen, H.S.; Bak, L.K. Astrocyte glycogenolysis is triggered by store-operated calcium entry and provides metabolic energy for cellular calcium homeostasis. Glia 2014, 62, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Manthorpe, M.; Bloom, F.E.; Varon, S. Functional receptors for vasoactive intestinal polypeptide in cultured astroglia from neonatal rat brain. Regul. Pept. 1983, 6, 71–80. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epm2b−/− vs. Control | |||
|---|---|---|---|
| Biochemical Pathway | Metabolite | FC | p-Value |
| UP | |||
| Carbohydrate metabolism | Fru-1,6 diP | 5.19 | 0.023 |
| Glu-6P | 3.77 | 0.023 | |
| Gluconate | 2.27 | 0.063 | |
| Amino acid metabolism (His) | 1-ribosyl-5-imidazole acetate | 2.08 | 0.027 |
| Nucleotide metabolism (Purines) | 2-deoxyguanosine | 1.66 | 0.080 |
| DOWN | |||
| Carbohydrate metabolism | arabitol/xylitol | 0.58 | 0.053 |
| Amino acid metabolism (Glu) | N-methyl-GABA | 0.67 | 0.028 |
| Amino acid metabolism (Polyamines) | 4-Acetamido butanoate | 0.66 | 0.012 |
| Peptides (dipeptides) | Leucylalanine | 0.48 | 0.085 |
| Primary bile acid metabolism | Taurocholate | 0.60 | 0.087 |
| Benzoate metabolism | 3-hydroxy hippurate | 0.55 | 0.060 |
| Lipid metabolism (Monoacylglycerol) | 1-oleoylglycerol (18:1) | 0.50 | 0.037 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Estellés, M.; Campos-Rodríguez, Á.; Rubio-Villena, C.; Kumarasinghe, L.; Garcia-Gimeno, M.A.; Sanz, P. Deciphering the Polyglucosan Accumulation Present in Lafora Disease Using an Astrocytic Cellular Model. Int. J. Mol. Sci. 2023, 24, 6020. https://doi.org/10.3390/ijms24076020
Moreno-Estellés M, Campos-Rodríguez Á, Rubio-Villena C, Kumarasinghe L, Garcia-Gimeno MA, Sanz P. Deciphering the Polyglucosan Accumulation Present in Lafora Disease Using an Astrocytic Cellular Model. International Journal of Molecular Sciences. 2023; 24(7):6020. https://doi.org/10.3390/ijms24076020
Chicago/Turabian StyleMoreno-Estellés, Mireia, Ángela Campos-Rodríguez, Carla Rubio-Villena, Lorena Kumarasinghe, Maria Adelaida Garcia-Gimeno, and Pascual Sanz. 2023. "Deciphering the Polyglucosan Accumulation Present in Lafora Disease Using an Astrocytic Cellular Model" International Journal of Molecular Sciences 24, no. 7: 6020. https://doi.org/10.3390/ijms24076020