
Inhibitory Effects of 3-Cyclopropylmethoxy-4-(difluoromethoxy) Benzoic Acid on TGF-β1-Induced Epithelial–Mesenchymal Transformation of In Vitro and Bleomycin-Induced Pulmonary Fibrosis In Vivo
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
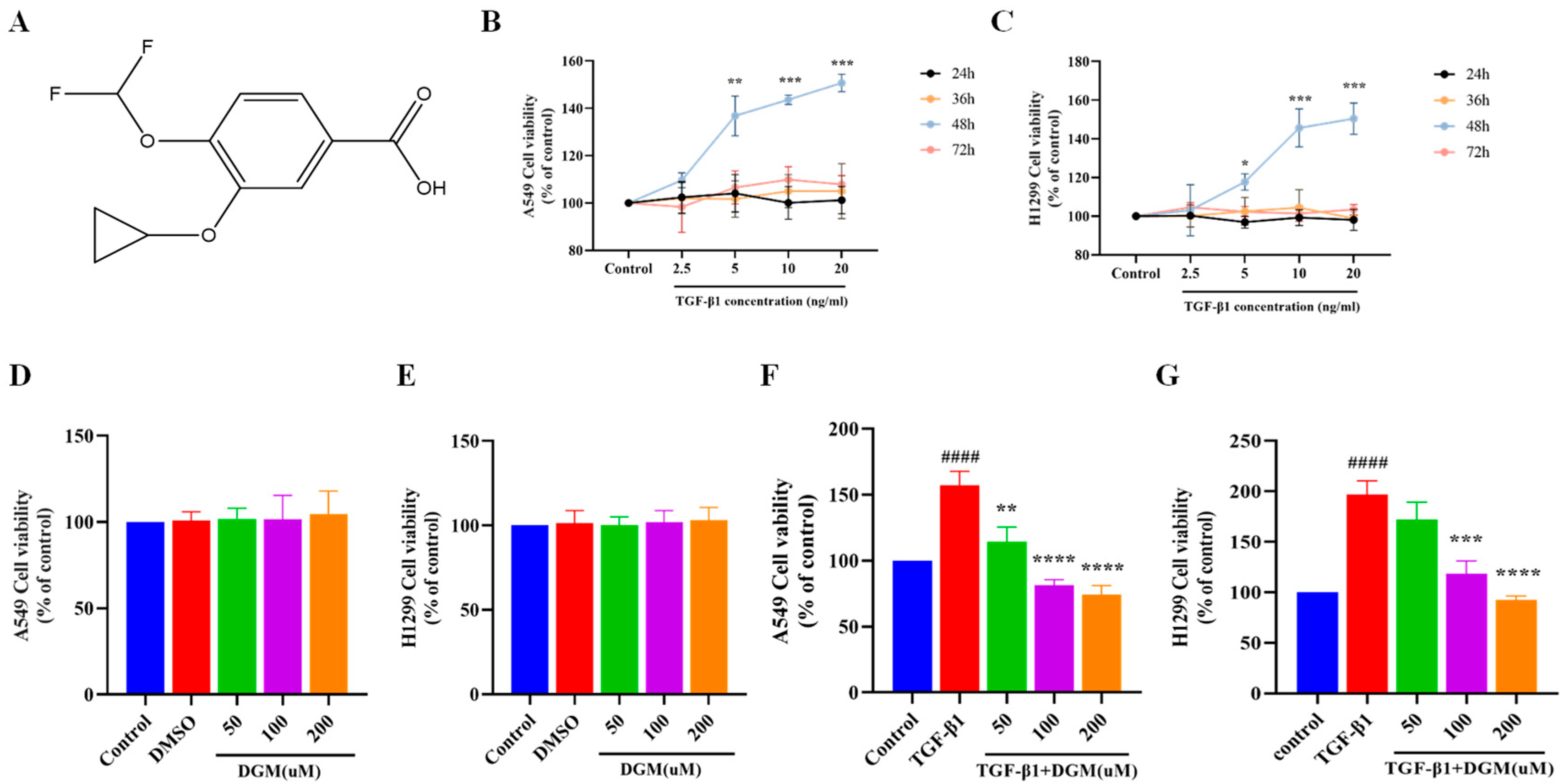
2.1. Effects of DGM on TGF-β1 Induced Viability of A549 and H1299 Cells
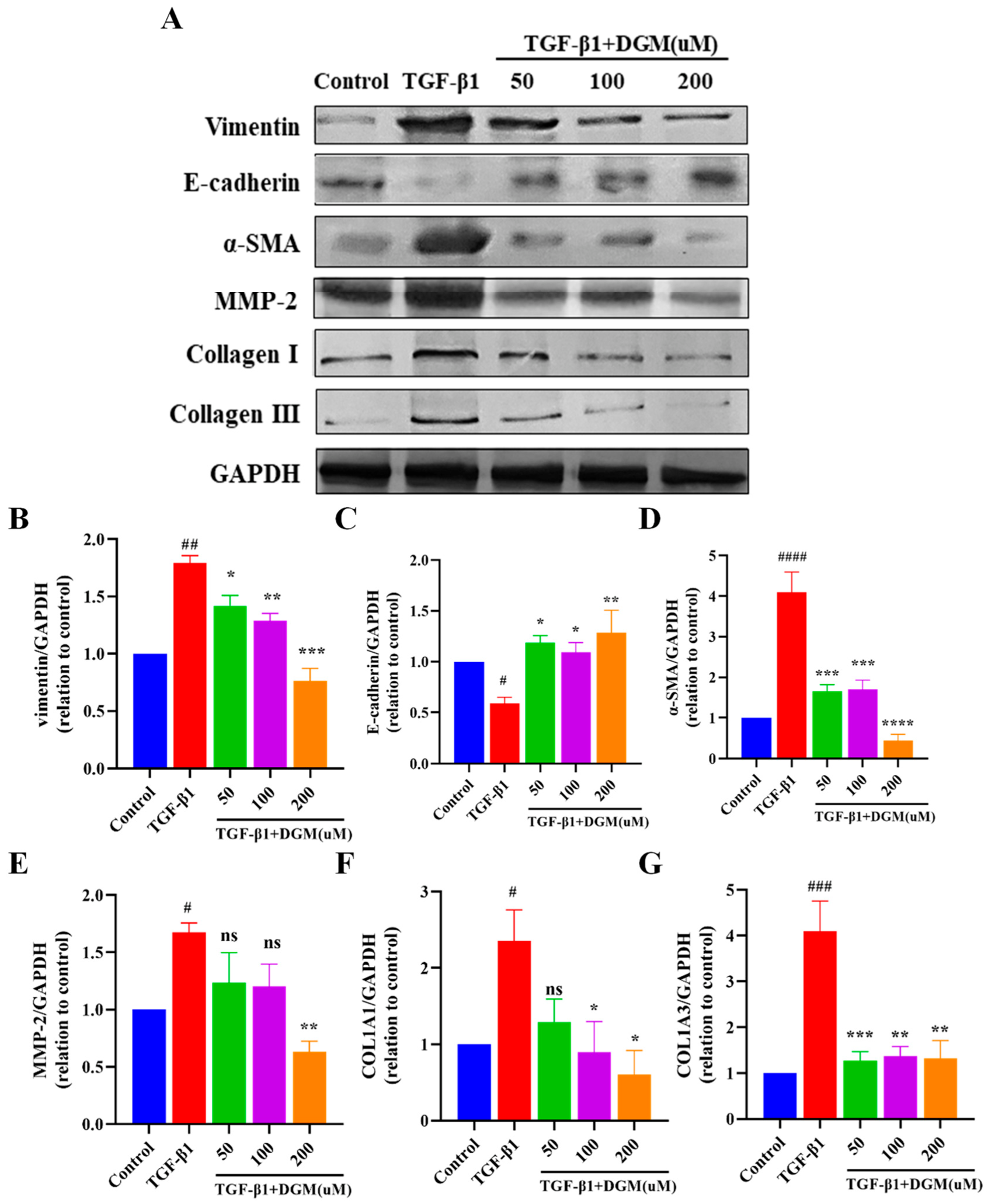
2.2. DGM Alleviated the EMT Process Induced by TGF-β1 in A549 Cells
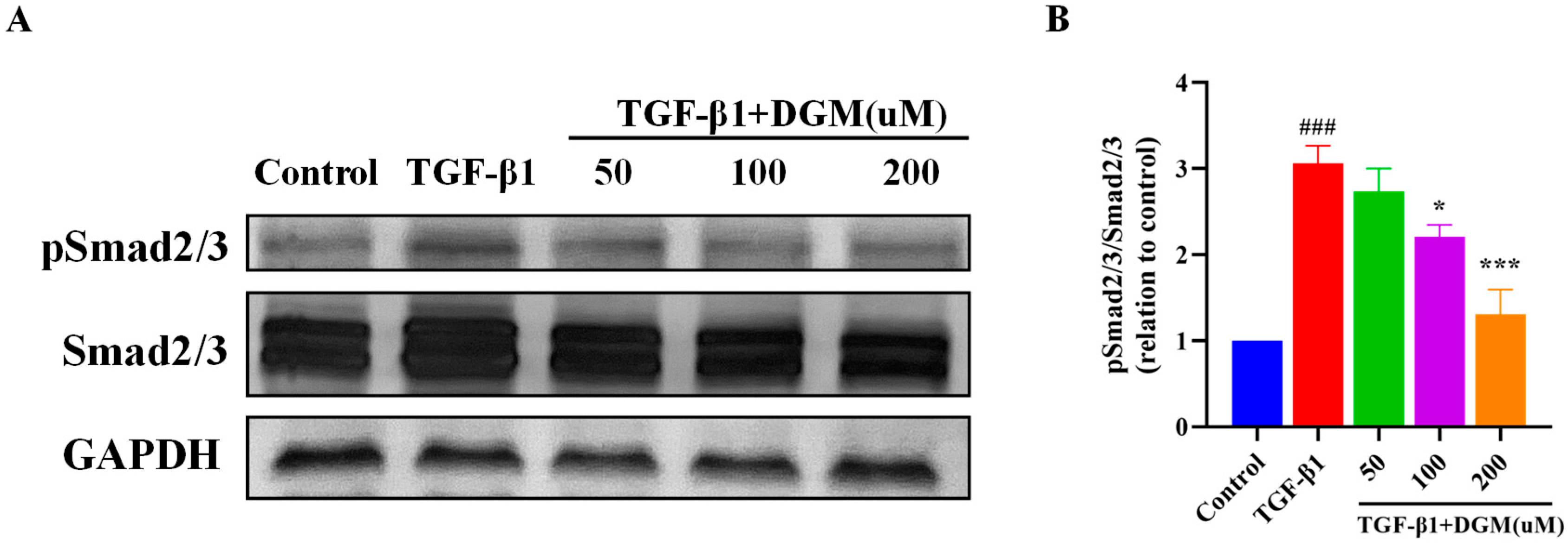
2.3. DGM Inhibited the Activation of the Smad Signaling Pathway Stimulated by TGF-β1
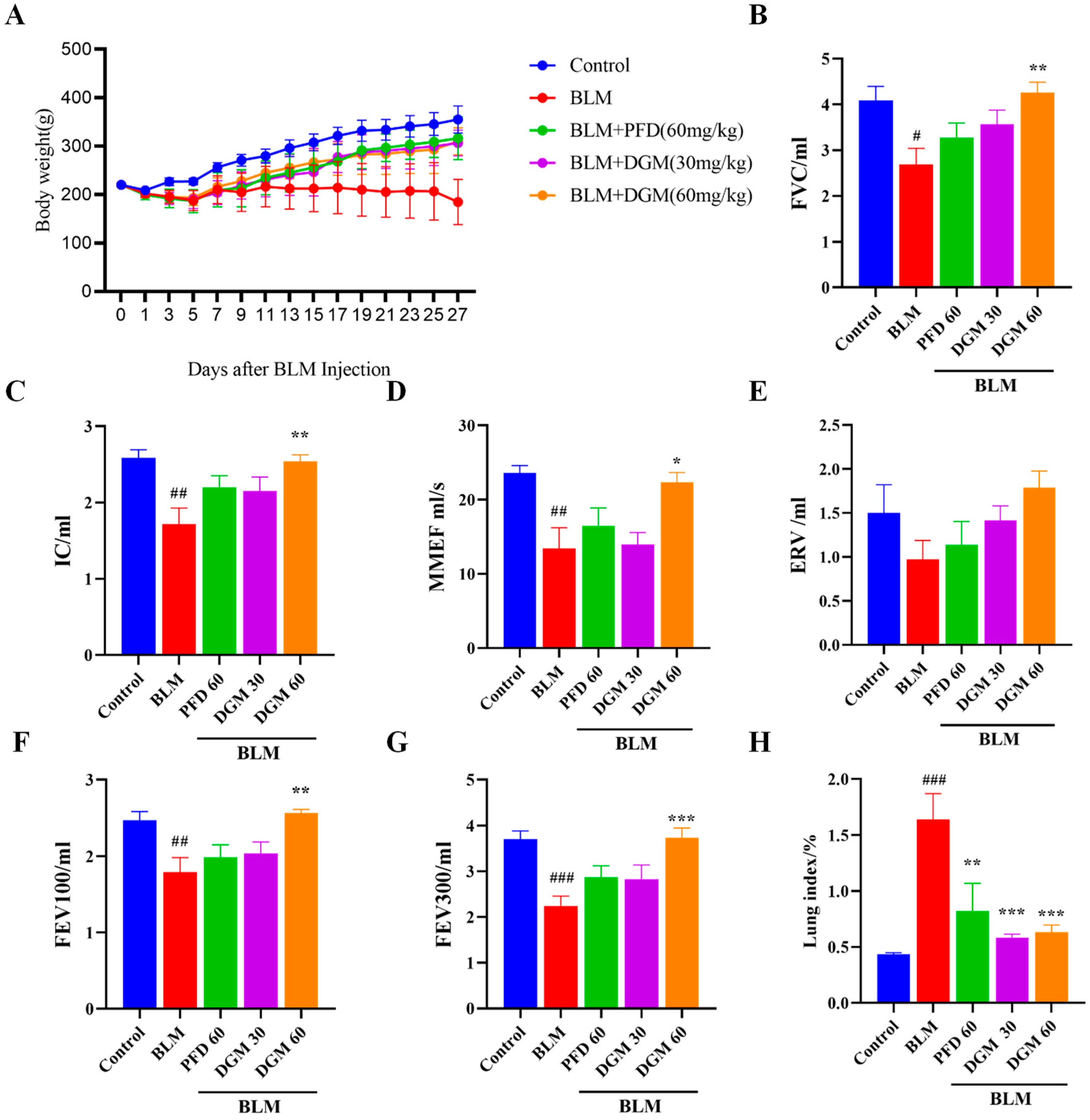
2.4. DGM Improved Bleomycin-Induced Weight Loss and Decreased Lung Function in Rats
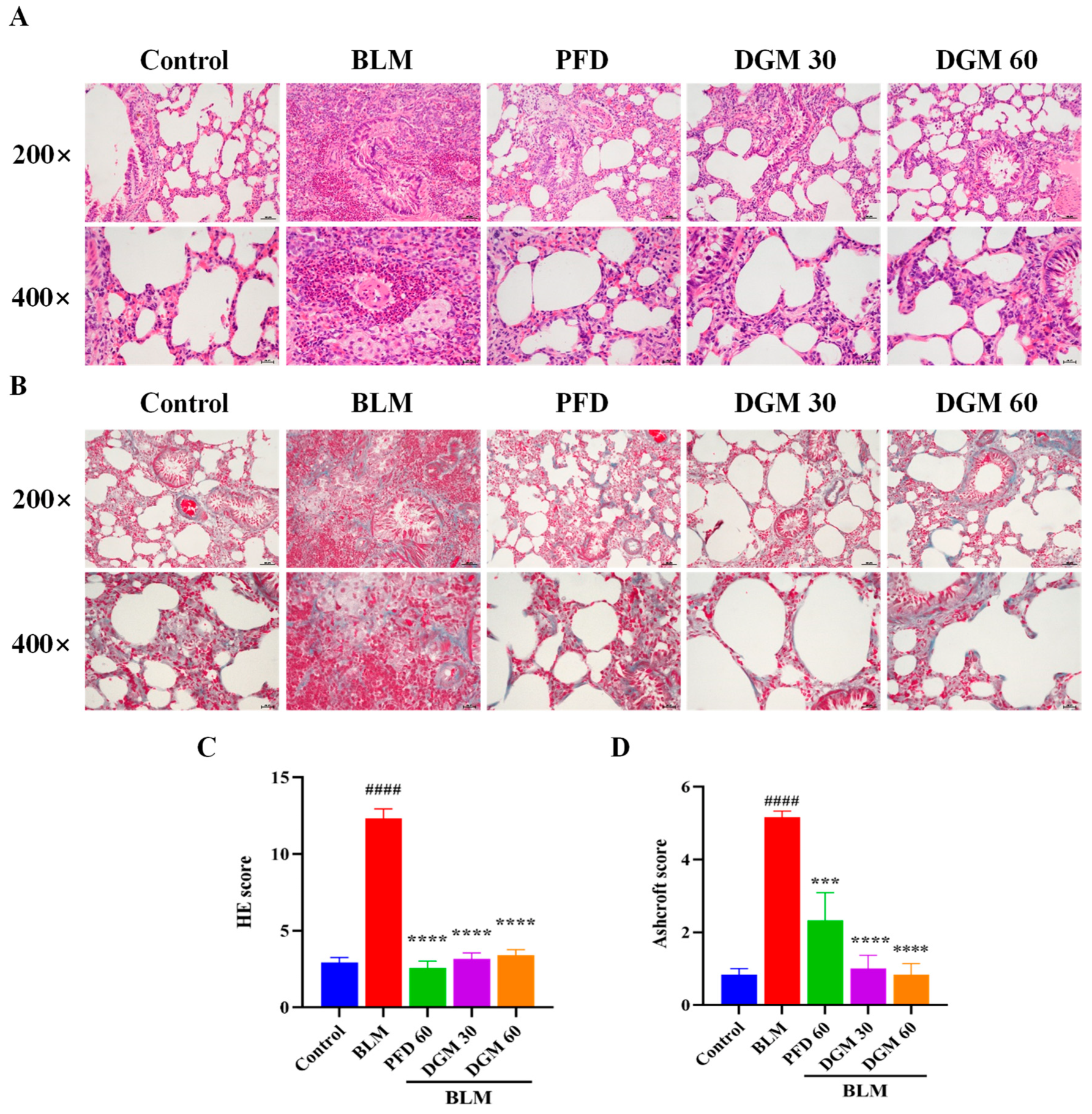
2.5. DGM Reduced BLM-Induced Lung Inflammation and Fibrosis
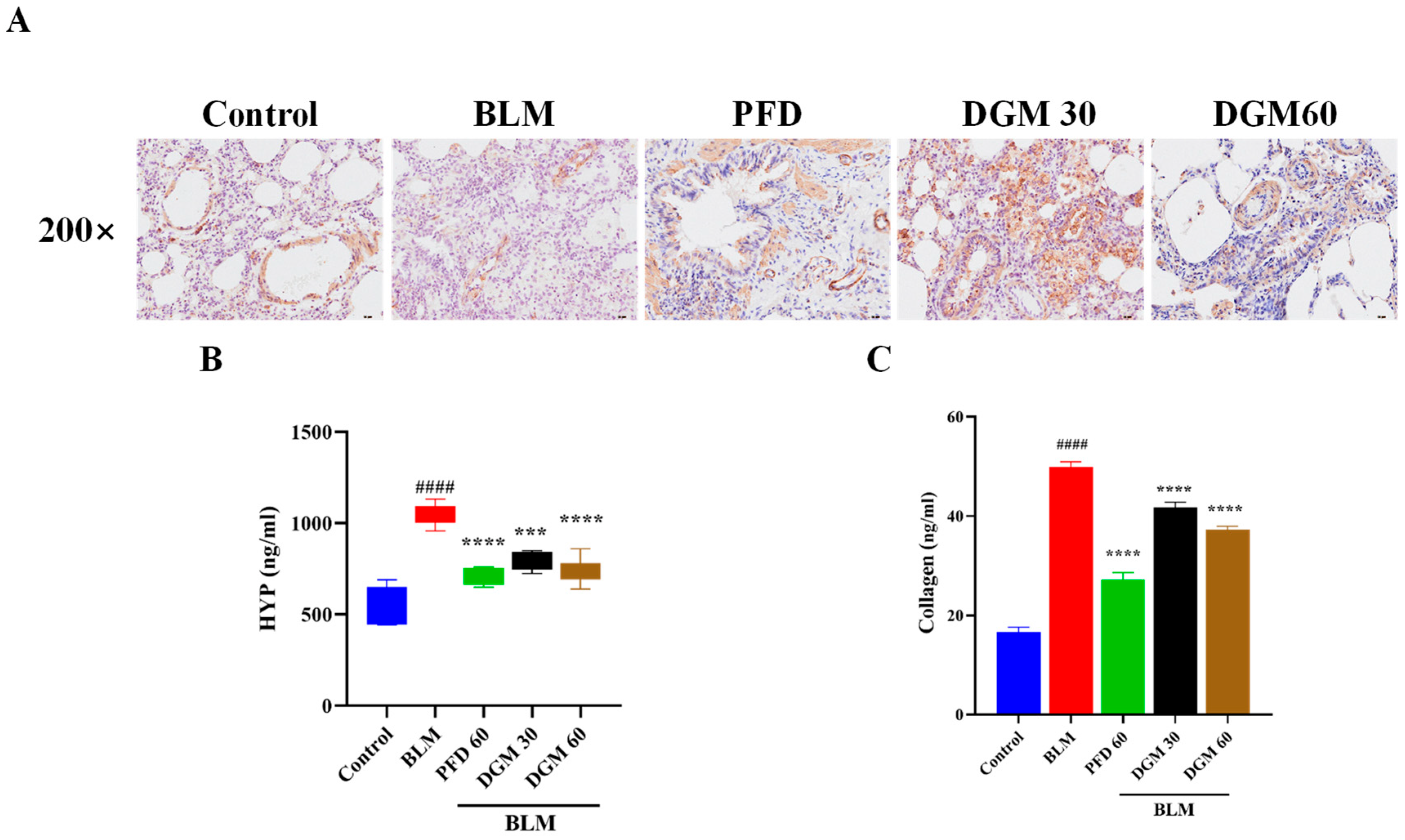
2.6. DGM Reduced the Expression of α-SMA, Hydroxyproline, and Total Collagen in Lung Tissue
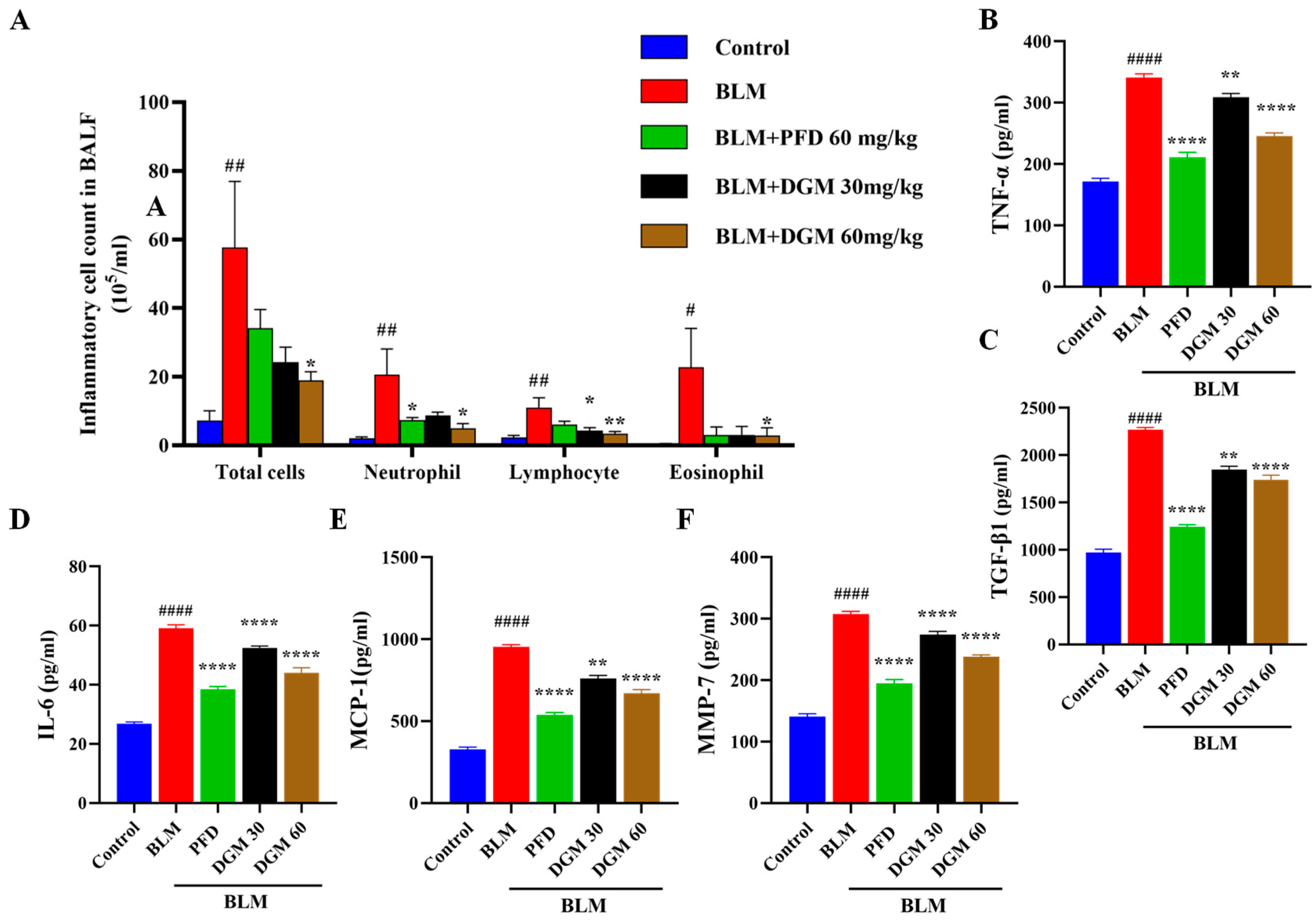
2.7. DGM Reduced the Content of Inflammatory Cells and Inflammatory Cytokines in Rat Bronchoalveolar Lavage Fluid
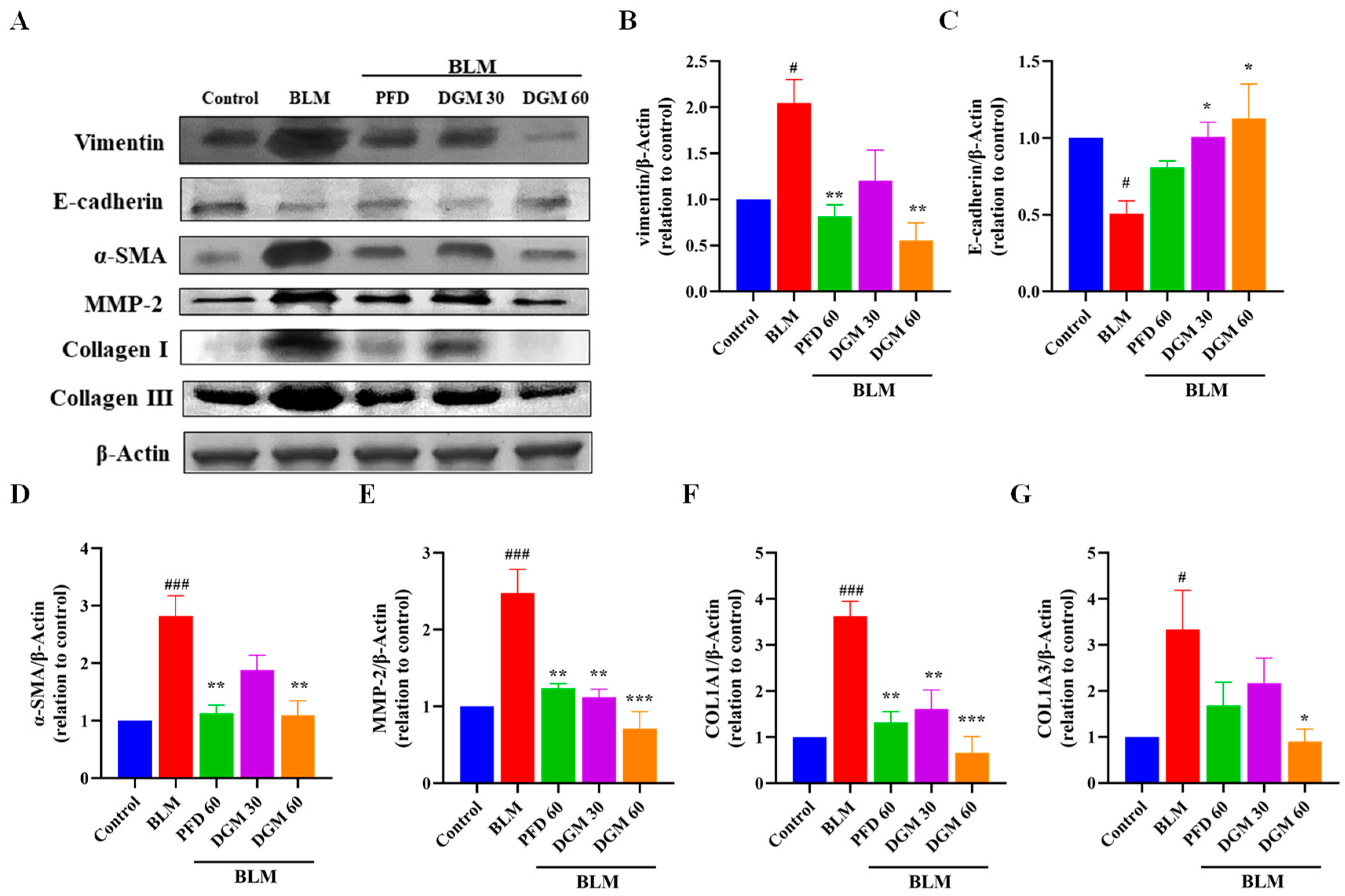
2.8. DGM Repressed the BLM-Induced EMT and Fibroblast Activation in Rat Lung Tissue
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Cell Culture and Treatment
4.3. MTT Assay
4.4. Western Blotting Analysis
4.5. Animal Models
4.6. Mold Making and Treatment Scheme
4.7. Pulmonary Function Tests
4.8. Lung Coefficient Detection
4.9. H&E and Masson’s Staining
4.10. Immunohistochemical Staining
4.11. Determination of Hydroproline and Collagen
4.12. BALF Collection and Inflammatory Cell Count
4.13. ELISA Method for the Detection of Cytokines in BALF Supernatant
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| FBS | fetal bovine serum |
| BLM | bleomycin |
| PFD | pirfenidone |
| α-SMA | α-smooth muscle actin |
| HYP | hydroxyproline |
| BALF | bronchoalveolar lavage fluid |
| MMP-7 | matrix metallopeptidase 7 |
References
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, J.T.; Hunninghake, W.G. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2001, 345, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [Green Version]
- Ley, B.; Collard, H.R. Epidemiology of idiopathic pulmonary fibrosis. Clin. Epidemiol. 2013, 5, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Shah, A.B.; Chowdhury, A.B. Forced Vital Capacity in Idiopathic Pulmonary Fibrosis—FDA Review of Pirfenidone and Nintedanib. N. Engl. J. Med. 2015, 372, 1187–1191. [Google Scholar] [CrossRef]
- Kalafatis, D.; Löfdahl, A.; Näsman, P.; Dellgren, G.; Wheelock, Å.M.; Elowsson Rendin, L.; Sköld, M.; Westergren-Thorsson, G. Distal Lung Microenvironment Triggers Release of Mediators Recognized as Potential Systemic Biomarkers for Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 13421. [Google Scholar] [CrossRef]
- Salton, F.; Ruaro, B.; Confalonieri, P.; Confalonieri, M. Epithelial-Mesenchymal Transition: A Major Pathogenic Driver in Idiopathic Pulmonary Fibrosis? Medicina 2020, 56, 608. [Google Scholar] [CrossRef]
- Engelmann, T.A.; Knudsen, L.; Leitz, D.H.W.; Duerr, J.; Beers, M.F.; Mall, M.A.; Ochs, M. Linking Fibrotic Remodeling and Ultrastructural Alterations of Alveolar Epithelial Cells after Deletion of Nedd4-2. Int. J. Mol. Sci. 2021, 22, 7607. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Ward, C.; Eapen, M.S.; Myers, S.; Hallgren, O.; Levine, H.; Sohal, S.S. Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev. Dyn. 2018, 247, 346–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, G.N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef] [PubMed]
- Gauldie, J.; Kolb, M.; Sime, J.P. A new direction in the pathogenesis of idiopathic pulmonary fibrosis? Respir. Res. 2002, 3, 1. [Google Scholar] [CrossRef]
- Ruaro, B.; Soldano, S.; Smith, V.; Paolino, S.; Contini, P.; Montagna, P.; Pizzorni, C.; Casabella, A.; Tardito, S.; Sulli, A.; et al. Correlation between circulating fibrocytes and dermal thickness in limited cutaneous systemic sclerosis patients: A pilot study. Rheumatol. Int. 2019, 39, 1369–1376. [Google Scholar] [CrossRef]
- Kelly, M.; Kolb, M.; Bonniaud, P.; Gauldie, J. Re-evaluation of Fibrogenic Cytokines in Lung Fibrosis. Curr. Pharm. Des. 2003, 9, 39–49. [Google Scholar] [CrossRef]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.L.; Zhang, X.; Bai, J.; Gai, L.; Ye, X.L.; Zhang, L.; Xu, Q.; Zhang, Y.X.; Xu, L.; Li, H.P.; et al. Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: Potential roles in the inhibition of epithelial-mesenchymal transition and fibroblast activation. Cell Death Dis. 2013, 4, e665. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Desmouliere, A.; Darby, I.A.; Gabbiani, G. Normal and pathologic soft tissue remodeling: Role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab. Investig. 2003, 83, 1689–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmouliere, A. Factors influencing myofibroblast differentiation during wound healing and fibrosis. Cell Biol. Int. 1995, 19, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, J.; Patel, S.; Suzuki, N.; Buxton, R.; Takagi, K.I.; Suzuki, J.; Sumi, Y.; Imaizumi, A.; Mason, R.M.; Zhang, Z. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J. Am. Soc. Nephrol. 2004, 15, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ma, L.; Huang, K.; Wei, Y.; Long, S.; Liu, Q.; Zhang, D.; Wu, S.; Wang, W.; Yang, G.; et al. Regorafenib-Attenuated, Bleomycin-Induced Pulmonary Fibrosis by Inhibiting the TGF-β1 Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 1985. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. TGF-beta signaling by Smad proteins. Cytokine Growth Factor Rev. 2000, 11, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.J.; Ten Dijke, P. TGF-beta-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef] [Green Version]
- Heldin, H.C.; Miyazono, K.; Dijke, T.P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Noble, P.W. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123–129. [Google Scholar] [CrossRef]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-beta: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Guillot, L.; Nathan, N.; Tabary, O.; Thouvenin, G.; Le Rouzic, P.; Corvol, H.; Amselem, S.; Clement, A. Alveolar epithelial cells: Master regulators of lung homeostasis. Int. J. Biochem. Cell Biol. 2013, 45, 2568–2573. [Google Scholar] [CrossRef]
- Winters, N.I.; Burman, A.; Kropski, J.A.; Blackwell, T.S. Epithelial Injury and Dysfunction in the Pathogenesis of Idiopathic PulmonaryFibrosis. Am. J. Med. Sci. 2019, 357, 374–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Shen, Y.; Hong, J.; Xia, Q.; Zhou, F.; Liu, X. The contribution of TGF-beta in Epithelial-Mesenchymal Transition (EMT): Down-regulation of E-cadherin via snail. Neoplasma 2015, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.; Fu, Y.; Wei, Z.; Yu, Y.; Zhang, X.; Yang, Z. Thymol attenuates allergic airway inflammation in ovalbumin (OVA)-induced mouse asthma. Fitoterapia 2014, 96, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.M.; Qin, B.Y.; Tiwari, A.; Shi, G.; Lam, S.; Hayward, L.J.; de Caestecker, M.; Lin, K. Structural Basis of Heteromeric Smad Protein Assembly in TGF-β Signaling. Mol. Cell 2004, 15, 813–823. [Google Scholar] [CrossRef] [PubMed]
- DeMaio, L.; Buckley, S.T.; Krishnaveni, M.S.; Flodby, P.; Dubourd, M.; Banfalvi, A.; Xing, Y.; Ehrhardt, C.; Minoo, P.; Zhou, B.; et al. Ligand-independent transforming growth factor-β type I receptor signalling mediates type I collagen-induced epithelial-mesenchymal transition. J. Pathol. 2012, 226, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; De Los Santos, F.G.; Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar]
- Shimbori, C.; Upagupta, C.; Bellaye, P.S.; Ayaub, E.A.; Sato, S.; Yanagihara, T.; Zhou, Q.; Ognjanovic, A.; Ask, K.; Gauldie, J.; et al. Mechanical stress-induced mast cell degranulation activates TGF-beta1 signalling pathway in pulmonary fibrosis. Thorax 2019, 74, 455–465. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, T.; Shan, S.; Wang, S.; Bian, W.; Ren, T.; Yang, D. MiR-124 regulates transforming growth factor-beta1 induced differentiation of lung resident mesenchymal stem cells to myofibroblast by repressing Wnt/beta-catenin signaling. Dev. Biol. 2019, 449, 115–121. [Google Scholar] [CrossRef]
- Hou, J.; Ma, T.; Cao, H.; Chen, Y.; Wang, C.; Chen, X.; Xiang, Z.; Han, X. TNF-alpha-induced NF-kappaB activation promotes myofibroblast differentiation of LR-MSCs and exacerbates bleomycin-induced pulmonary fibrosis. J. Cell Physiol. 2018, 233, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Mouratis, M.A.; Aidinis, V. Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm. Med. 2011, 17, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Upagupta, C.; Vierhout, M.; Ayaub, E.; Bellaye, P.S.; Gauldie, J.; Shimbori, C.; Inman, M.; Ask, K.; Kolb, M.R.J. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur. Respir. J. 2020, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Qu, J.; Huang, X.; Kurundkar, A.; Zhu, L.; Yang, N.; Venado, A.; Ding, Q.; Liu, G.; Antony, V.B.; et al. Mechanosensing by the alpha6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat. Commun. 2016, 7, 12564. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, T.; Li, H.; Zhang, Y.; Xiong, G.; Liang, Y.; Lu, F.; Zheng, R.; Zou, Q.; Hao, J. Inhibitory Effects of 3-Cyclopropylmethoxy-4-(difluoromethoxy) Benzoic Acid on TGF-β1-Induced Epithelial–Mesenchymal Transformation of In Vitro and Bleomycin-Induced Pulmonary Fibrosis In Vivo. Int. J. Mol. Sci. 2023, 24, 6172. https://doi.org/10.3390/ijms24076172
Sun T, Li H, Zhang Y, Xiong G, Liang Y, Lu F, Zheng R, Zou Q, Hao J. Inhibitory Effects of 3-Cyclopropylmethoxy-4-(difluoromethoxy) Benzoic Acid on TGF-β1-Induced Epithelial–Mesenchymal Transformation of In Vitro and Bleomycin-Induced Pulmonary Fibrosis In Vivo. International Journal of Molecular Sciences. 2023; 24(7):6172. https://doi.org/10.3390/ijms24076172
Chicago/Turabian StyleSun, Tianxiao, Haihua Li, Yan Zhang, Guixin Xiong, Yuerun Liang, Fang Lu, Rong Zheng, Qi Zou, and Jiejie Hao. 2023. "Inhibitory Effects of 3-Cyclopropylmethoxy-4-(difluoromethoxy) Benzoic Acid on TGF-β1-Induced Epithelial–Mesenchymal Transformation of In Vitro and Bleomycin-Induced Pulmonary Fibrosis In Vivo" International Journal of Molecular Sciences 24, no. 7: 6172. https://doi.org/10.3390/ijms24076172