
Ion Channels in the Development and Remodeling of the Aortic Valve
Abstract
:1. Introduction
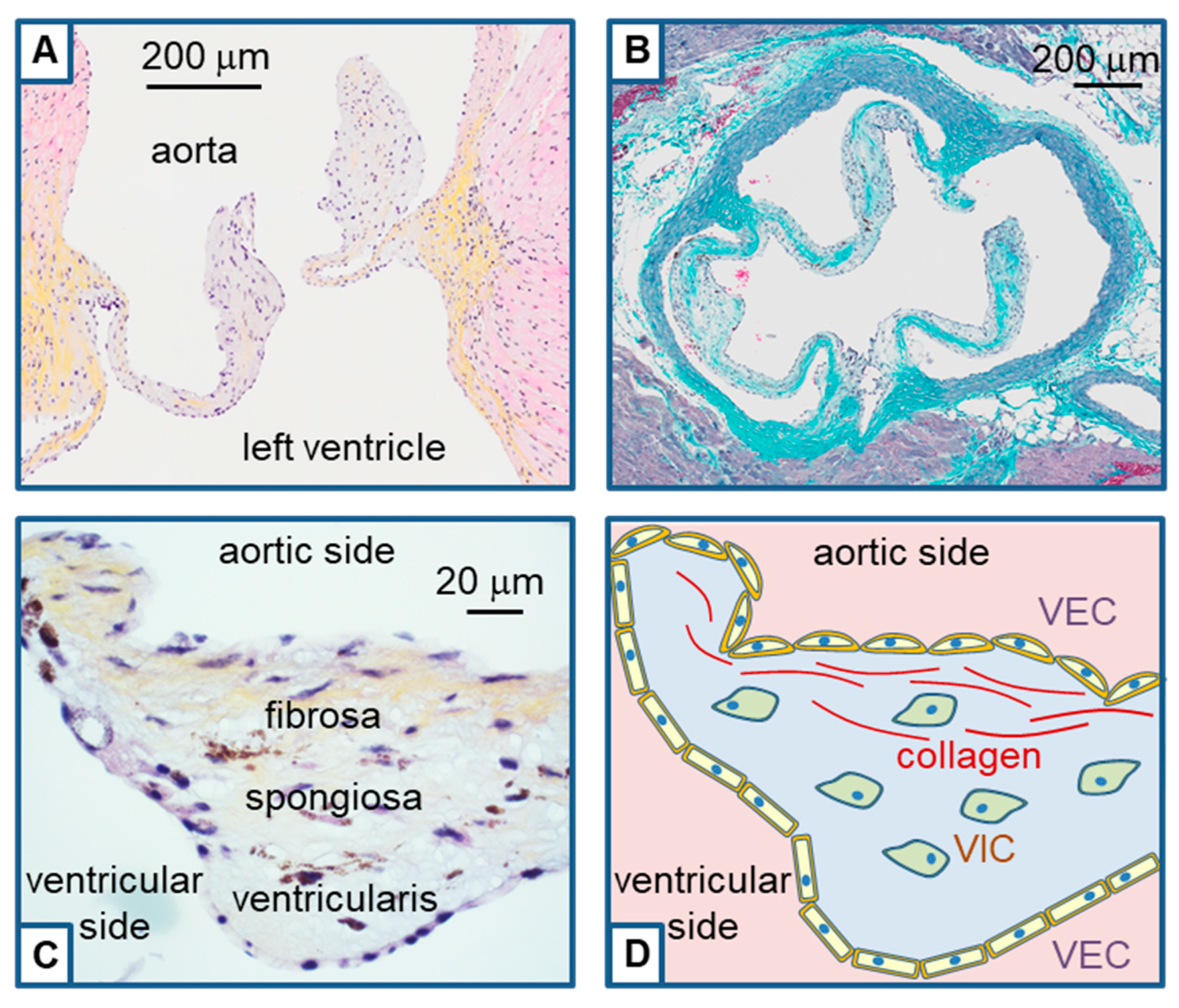
2. Histological Structure of the Aortic Valve and Remodeling
3. The Piezo and TRP Channel Families
4. Ion Channels in Aortic Valve Development
4.1. Piezo1 Channel in Aortic Valve Development
4.2. TRP Channels in Aortic Valve Development
4.3. Other Ion Channels in Aortic Valve Development
5. Ion Channels in Aortic Valve Remodeling
5.1. TRP Channels in VIC and Aortic Valve Remodeling
5.2. Voltage-Dependent Ca2+ Channels in VIC and Aortic Valve Remodeling
5.3. K+-Channels in VIC and Aortic Valve Remodeling
5.4. Ion Channels in VEC and Aortic Valve Remodeling
5.5. Ion Channels from Immune Cells and Their Implication in Valvular Remodeling
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, C. Clinical significance of the bicuspid aortic valve. Heart 2000, 83, 81–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iung, B.; Baron, G.; Butchart, E.G.; Delahaye, F.; Gohlke-Bärwolf, C.; Levang, O.W.; Tornos, P.; Vanoverschelde, J.L.; Vermeer, F.; Boersma, E.; et al. A prospective survey of patients with valvular heart disease in Europe: The Euro Heart Survey on Valvular Heart Disease. Eur. Heart J. 2003, 24, 1231–1243. [Google Scholar] [CrossRef] [Green Version]
- Goldbarg, S.H.; Elmariah, S.; Miller, M.A.; Fuster, V. Insights into degenerative aortic valve disease. J. Am. Coll. Cardiol. 2007, 50, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujral, D.M.; Lloyd, G.; Bhattacharyya, S. Radiation-induced valvular heart disease. Heart 2016, 102, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Sárközy, M.; Varga, Z.; Gáspár, R.; Szűcs, G.; Kovács, M.G.; Kovács, Z.Z.A.; Dux, L.; Kahán, Z.; Csont, T. Pathomechanisms and therapeutic opportunities in radiation-induced heart disease: From bench to bedside. Clin. Res. Cardiol. 2021, 110, 507–531. [Google Scholar] [CrossRef] [PubMed]
- Iung, B.; Delgado, V.; Rosenhek, R.; Price, S.; Prendergast, B.; Wendler, O.; De Bonis, M.; Tribouilloy, C.; Evangelista, A.; Bogachev-Prokophiev, A.; et al. Contemporary Presentation and Management of Valvular Heart Disease: The EURObservational Research Programme Valvular Heart Disease II Survey. Circulation 2019, 140, 1156–1169. [Google Scholar] [CrossRef]
- Nkomo, V.T.; Gardin, J.M.; Skelton, T.N.; Gottdiener, J.S.; Scott, C.G.; Enriquez-Sarano, M. Burden of valvular heart diseases: A population-based study. Lancet 2006, 368, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Yutzey, K.E. Heart valve structure and function in development and disease. Annu. Rev. Physiol. 2011, 73, 29–46. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.O.; Fiane, A.; Vaage, J. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 2017, 6, e006339. [Google Scholar] [CrossRef]
- Feulner, L.; van Vliet, P.P.; Puceat, M.; Andelfinger, G. Endocardial Regulation of Cardiac Development. J. Cardiovasc. Dev. Dis. 2022, 9, 122. [Google Scholar] [CrossRef]
- Dayawansa, N.H.; Baratchi, S.; Peter, K. Uncoupling the Vicious Cycle of Mechanical Stress and Inflammation in Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2022, 9, 783543. [Google Scholar] [CrossRef]
- Mathieu, P.; Bouchareb, R.; Boulanger, M.C. Innate and Adaptive Immunity in Calcific Aortic Valve Disease. J. Immunol. Res. 2015, 2015, 851945. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Choi, J.H. Involvement of inflammatory responses in the early development of calcific aortic valve disease: Lessons from statin therapy. Anim. Cells. Syst. (Seoul) 2018, 22, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Guauque-Olarte, S.; Messika-Zeitoun, D.; Droit, A.; Lamontagne, M.; Tremblay-Marchand, J.; Lavoie-Charland, E.; Gaudreault, N.; Arsenault, B.J.; Dubé, M.P.; Tardif, J.C.; et al. Calcium Signaling Pathway Genes RUNX2 and CACNA1C Are Associated With Calcific Aortic Valve Disease. Circ. Cardiovasc. Genet. 2015, 8, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Xie, S.; Huang, Y.; Zhou, T.; Liu, M.; Zhu, P.; Wang, C.; Shi, J.; Li, F.; Sellke, F.W.; et al. Cell-Type Transcriptome Atlas of Human Aortic Valves Reveal Cell Heterogeneity and Endothelial to Mesenchymal Transition Involved in Calcific Aortic Valve Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2910–2921. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; McCulloch, C.A. TRPV4 integrates matrix mechanosensing with Ca2+ signaling to regulate extracellular matrix remodeling. FEBS J. 2021, 288, 5867–5887. [Google Scholar] [CrossRef] [PubMed]
- LedFord, H.; Callaway, E. Medecine Nobel for duo who discovered biology of sense. Nature 2021, 598, 246. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Hof, T.; Chaigne, S.; Récalde, A.; Sallé, L.; Brette, F.; Guinamard, R. Transient receptor potential channels in cardiac health and disease. Nat. Rev. Cardiol. 2019, 16, 344–360. [Google Scholar] [CrossRef]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [Green Version]
- Parenti, A.; De Logu, F.; Geppetti, P.; Benemei, S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br. J. Pharmacol. 2016, 173, 953–969. [Google Scholar] [CrossRef]
- Nikolaev, Y.A.; Cox, C.D.; Ridone, P.; Rohde, P.R.; Cordero-Morales, J.F.; Vásquez, V.; Laver, D.R.; Martinac, B. Mammalian TRP ion channels are insensitive to membrane stretch. J. Cell. Sci. 2019, 132, jcs238360. [Google Scholar] [CrossRef] [Green Version]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coste, B.; Xiao, B.; Santos, J.S.; Syeda, R.; Grandl, J.; Spencer, K.S.; Kim, S.E.; Schmidt, M.; Mathur, J.; Dubin, A.E.; et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 2012, 483, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.Z.; Zhou, T.; Xu, J.Q.; Wang, Y.X.; Sun, M.M.; He, Y.J.; Pan, S.W.; Xiong, W.; Peng, Z.K.; Gao, X.H.; et al. Structure, kinetic properties and biological function of mechanosensitive Piezo channels. Cell Biosci. 2021, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Delmas, P.; Parpaite, T.; Coste, B. PIEZO channels and newcomers in the mammalian mechanosensitive ion channel family. Neuron 2022, 110, 2713–2727. [Google Scholar] [CrossRef]
- Saotome, K.; Murthy, S.E.; Kefauver, J.M.; Whitwam, T.; Patapoutian, A.; Ward, A.B. Structure of the mechanically activated ion channel Piezo1. Nature 2018, 554, 481–486. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhou, H.; Chi, S.; Wang, Y.; Wang, J.; Geng, J.; Wu, K.; Liu, W.; Zhang, T.; Dong, M.Q.; et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature 2018, 554, 487–492. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhou, H.; Li, X.; Xiao, B. The mechanosensitive Piezo1 channel: A three-bladed propeller-like structure and a lever-like mechanogating mechanism. FEBS J. 2019, 286, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faucherre, A.; Moha Ou Maati, H.; Nasr, N.; Pinard, A.; Theron, A.; Odelin, G.; Desvignes, J.P.; Salgado, D.; Collod-Béroud, G.; Avierinos, J.F.; et al. Piezo1 is required for outflow tract and aortic valve development. J. Mol. Cell. Cardiol. 2020, 143, 51–62. [Google Scholar] [CrossRef]
- Duchemin, A.L.; Vignes, H.; Vermot, J. Mechanically activated piezo channels modulate outflow tract valve development through the Yap1 and Klf2-Notch signaling axis. Elife 2019, 8, e44706. [Google Scholar] [CrossRef] [PubMed]
- Mpweme Bangando, H.; Simard, C.; Aize, M.; Lebrun, A.; Manrique, A.; Guinamard, R.; On Behalf Of The Stop-As Investigators. TRPM4 Participates in Irradiation-Induced Aortic Valve Remodeling in Mice. Cancers 2022, 14, 4477. [Google Scholar] [CrossRef]
- Heckel, E.; Boselli, F.; Roth, S.; Krudewig, A.; Belting, H.G.; Charvin, G.; Vermot, J. Oscillatory Flow Modulates Mechanosensitive klf2a Expression through trpv4 and trpp2 during Heart Valve Development. Curr. Biol. 2015, 25, 1354–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andelfinger, G.; Tapper, A.R.; Welch, R.C.; Vanoye, C.G.; George, A.L., Jr.; Benson, D.W. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am. J. Hum. Genet. 2002, 71, 663–668. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Yun, P.; Neuhaus, S.; Mohassel, P.; Dastgir, J.; Donkervoort, S.; Schindler, A.; Mankodi, A.; Foley, A.R.; Arai, A.E.; et al. Cardiac MRI identifies valvular and myocardial disease in a subset of ANO5-related muscular dystrophy patients. Neuromuscul. Disord. 2020, 30, 742–749. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, J.V.; Martinac, B.; Cox, C.D. Loss-of-Function Piezo1 Mutations Display Altered Stability Driven by Ubiquitination and Proteasomal Degradation. Front. Pharmacol. 2021, 12, 766416. [Google Scholar] [CrossRef]
- Nonomura, K.; Lukacs, V.; Sweet, D.T.; Goddard, L.M.; Kanie, A.; Whitwam, T.; Ranade, S.S.; Fujimori, T.; Kahn, M.L.; Patapoutian, A. Mechanically activated ion channel PIEZO1 is required for lymphatic valve formation. Proc. Natl. Acad. Sci. USA 2018, 115, 12817–12822. [Google Scholar] [CrossRef] [Green Version]
- Luxán, G.; D’Amato, G.; MacGrogan, D.; de la Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- Shah, V.; Patel, S.; Shah, J. Emerging role of Piezo ion channels in cardiovascular development. Dev. Dyn. 2022, 251, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, L.; Chi, C.; Wu, X.; Yang, X.; Han, M.; Xu, T.; Zhuang, Y.; Deng, K. Adam10 is essential for early embryonic cardiovascular development. Dev. Dyn. 2010, 239, 2594–2602. [Google Scholar] [CrossRef]
- Alabi, R.O.; Glomski, K.; Haxaire, C.; Weskam, G.; Monette, S.; Blobel, C.P. ADAM10-Dependent Signaling Through Notch1 and Notch4 Controls Development of Organ-Specific Vascular Beds. Circ. Res. 2016, 119, 519–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabi, R.O.; Farber, G.; Blobel, C.P. Intriguing Roles for Endothelial ADAM10/Notch Signaling in the Development of Organ-Specific Vascular Beds. Physiol. Rev. 2018, 98, 2025–2061. [Google Scholar] [CrossRef] [PubMed]
- Caolo, V.; Debant, M.; Endesh, N.; Futers, T.S.; Lichtenstein, L.; Bartoli, F.; Parsonage, G.; Jones, E.A.; Beech, D.J. Shear stress activates ADAM10 sheddase to regulate Notch1 via the Piezo1 force sensor in endothelial cells. Elife 2020, 9, e50684. [Google Scholar] [CrossRef]
- Bornhorst, D.; Abdelilah-Seyfried, S. Strong as a Hippo’s Heart: Biomechanical Hippo Signaling During Zebrafish Cardiac Development. Front. Cell Dev. Biol. 2021, 9, 731101. [Google Scholar] [CrossRef]
- Loukin, S.; Zhou, X.; Su, Z.; Saimi, Y.; Kung, C. Wild-type and brachyolmia-causing mutant TRPV4 channels respond directly to stretch force. J. Biol. Chem. 2010, 285, 27176–27181. [Google Scholar] [CrossRef] [Green Version]
- Demion, M.; Thireau, J.; Gueffier, M.; Finan, A.; Khoueiry, Z.; Cassan, C.; Serafini, N.; Aimond, F.; Granier, M.; Pasquié, J.L.; et al. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS ONE 2014, 9, e115256. [Google Scholar] [CrossRef] [Green Version]
- Simard, C.; Hof, T.; Keddache, Z.; Launay, P.; Guinamard, R. The TRPM4 non-selective cation channel contributes to the mammalian atrial action potential. J. Mol. Cell. Cardiol. 2013, 59, 11–19. [Google Scholar] [CrossRef]
- Fernández, B.; Soto-Navarrete, M.T.; López-García, A.; López-Unzu, M.Á.; Durán, A.C.; Fernández, M.C. Bicuspid Aortic Valve in 2 Model Species and Review of the Literature. Vet. Pathol. 2020, 57, 321–331. [Google Scholar] [CrossRef]
- Kruse, M.; Schulze-Bahr, E.; Corfield, V.; Beckmann, A.; Stallmeyer, B.; Kurtbay, G.; Ohmert, I.; Schulze-Bahr, E.; Brink, P.; Pongs, O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Investig. 2009, 119, 2737–2744. [Google Scholar] [CrossRef]
- Liu, H.; El Zein, L.; Kruse, M.; Guinamard, R.; Beckmann, A.; Bozio, A.; Kurtbay, G.; Mégarbané, A.; Ohmert, I.; Blaysat, G.; et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 2010, 3, 374–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Chatel, S.; Simard, C.; Syam, N.; Salle, L.; Probst, V.; Morel, J.; Millat, G.; Lopez, M.; Abriel, H.; et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS ONE 2013, 8, e54131. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, J.; Güttsches, A.K.; Schara-Schmidt, U.; Vorgerd, M.; Heute, C.; Preusse, C.; Stenzel, W.; Roos, A. ANO5-related muscle diseases: From clinics and genetics to pathology and research strategies. Genes Dis. 2022, 9, 1506–1520. [Google Scholar] [CrossRef]
- Al-Shammari, H.; Latif, N.; Sarathchandra, P.; McCormack, A.; Rog-Zielinska, E.A.; Raja, S.; Kohl, P.; Yacoub, M.H.; Peyronnet, R.; Chester, A.H. Expression and function of mechanosensitive ion channels in human valve interstitial cells. PLoS ONE 2020, 15, e0240532. [Google Scholar] [CrossRef]
- Batan, D.; Peters, D.K.; Schroeder, M.E.; Aguado, B.A.; Young, M.W.; Weiss, R.M.; Anseth, K.S. Hydrogel cultures reveal Transient Receptor Potential Vanilloid 4 regulation of myofibroblast activation and proliferation in valvular interstitial cells. FASEB J. 2022, 36, e22306. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Macdougall, L.J.; GonzalezRodriguez, A.; Schroeder, M.E.; Batan, D.; Weiss, R.M.; Anseth, K.S. Calcium Signaling Regulates Valvular Interstitial Cell Alignment and Myofibroblast Activation in Fast-Relaxing Boronate Hydrogels. Macromol. Biosci. 2020, 20, e2000268. [Google Scholar] [CrossRef]
- Matsui, M.; Bouchareb, R.; Storto, M.; Hussain, Y.; Gregg, A.; Marx, S.O.; Pitt, G.S. Increased Ca2+ influx through CaV1.2 drives aortic valve calcification. JCI Insight. 2022, 7, e155569. [Google Scholar] [CrossRef]
- Wang, H.; Tibbitt, M.W.; Langer, S.J.; Leinwand, L.A.; Anseth, K.S. Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 19336–19341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simard, C.; Magaud, C.; Adjlane, R.; Dupas, Q.; Sallé, L.; Manrique, A.; Bois, P.; Faivre, J.F.; Guinamard, R. TRPM4 non-selective cation channel in human atrial fibroblast growth. Pflugers Arch. 2020, 472, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adapala, R.K.; Katari, V.; Teegala, L.R.; Thodeti, S.; Paruchuri, S.; Thodeti, C.K. TRPV4 Mechanotransduction in Fibrosis. Cells 2021, 10, 3053. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, Z.Y.; Wu, J.; Gong, H.; Kesteven, S.; Iismaa, S.E.; Chan, A.Y.; Holman, S.; Pinto, S.; Pironet, A.; et al. The Ca2+-activated cation channel TRPM4 is a positive regulator of pressure overload-induced cardiac hypertrophy. Elife 2021, 10, e66582. [Google Scholar] [CrossRef]
- Yu, Z.Y.; Gong, H.; Kesteven, S.; Guo, Y.; Wu, J.; Vero Li, J.; Cheng, D.; Zhou, Z.; Lismaa, S.E.; Kaidonis, X.; et al. Piezo1 is the cardiac mechanosensor that initiates the cardiomyocyte hypertrophic response to pressure overload in adult mice. Nat. Cardiovasc. Res. 2022, 1, 577–591. [Google Scholar] [CrossRef]
- Matthews, B.D.; Thodeti, C.K.; Tytell, J.D.; Mammoto, A.; Overby, D.R.; Ingber, D.E. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr. Biol. 2010, 2, 435–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ali, M.S.; Lacerda, C.M.R. A Three-Dimensional Collagen-Elastin Scaffold for Heart Valve Tissue Engineering. Bioengineering 2018, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Gwanyanya, A.; Mubagwa, K. Emerging role of transient receptor potential (TRP) ion channels in cardiac fibroblast pathophysiology. Front. Physiol. 2022, 13, 968393. [Google Scholar] [CrossRef] [PubMed]
- Guauque-Olarte, S.; Droit, A.; Tremblay-Marchand, J.; Gaudreault, N.; Kalavrouziotis, D.; Dagenais, F.; Seidman, J.G.; Body, S.C.; Pibarot, P.; Mathieu, P.; et al. RNA expression profile of calcified bicuspid, tricuspid, and normal human aortic valves by RNA sequencing. Physiol. Genom. 2016, 48, 749–761. [Google Scholar] [CrossRef] [Green Version]
- Diszházi, G.; Magyar, Z.É.; Lisztes, E.; Tóth-Molnár, E.; Nánási, P.P.; Vennekens, R.; Tóth, B.I.; Almássy, J. TRPM4 links calcium signaling to membrane potential in pancreatic acinar cells. J. Biol. Chem. 2021, 297, 101015. [Google Scholar] [CrossRef]
- Gerhold, K.A.; Schwartz, M.A. Ion Channels in Endothelial Responses to Fluid Shear Stress. Physiology 2016, 31, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, J.T.; Tressel, S.; Johnson, T.; Turner, D.; Sorescu, G.; Jo, H.; Nerem, R.M. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: Influence of shear stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.D.; Tchir, A.; Mirza, A.; Chaparro, D.; Herrera, R.E.; Hutcheson, J.D.; Ramaswamy, S. Valve Endothelial Cell Exposure to High Levels of Flow Oscillations Exacerbates Valve Interstitial Cell Calcification. Bioengineering 2022, 9, 393. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Yacoub, M.H.; El-Hamamsy, I.; Sanchez-Alonso, J.L.; Moshkov, A.; Mongkoldhumrongkul, N.; Padala, M.; Paramagurunathan, S.; Sarathchandra, P.; Korchev, Y.E.; et al. Side-specific mechanical properties of valve endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H15–H24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, J.; El-Hamamsy, I.; Chen, S.; Sarang, Z.; Sarathchandra, P.; Yacoub, M.H.; Chester, A.H.; Butcher, J.T. Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am. J. Pathol. 2013, 182, 1922–1931. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef]
- Aziz, Q.; Li, Y.; Anderson, N.; Ojake, L.; Tsisanova, E.; Tinker, A. Molecular and functional characterization of the endothelial ATP-sensitive potassium channel. J. Biol. Chem. 2017, 292, 17587–17597. [Google Scholar] [CrossRef] [Green Version]
- Bian, W.; Wang, Z.; Sun, C.; Zhang, D.M. Pathogenesis and Molecular Immune Mechanism of Calcified Aortic Valve Disease. Front. Cardiovasc. Med. 2021, 8, 765419. [Google Scholar] [CrossRef]
- Launay, P.; Cheng, H.; Srivatsan, S.; Penner, R.; Fleig, A.; Kinet, J.P. TRPM4 regulates calcium oscillations after T cell activation. Science 2004, 306, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Barbet, G.; Demion, M.; Moura, I.C.; Serafini, N.; Léger, T.; Vrtovsnik, F.; Monteiro, R.C.; Guinamard, R.; Kinet, J.P.; Launay, P. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat. Immunol. 2008, 9, 1148–1156. [Google Scholar] [CrossRef] [Green Version]
- Serafini, N.; Dahdah, A.; Barbet, G.; Demion, M.; Attout, T.; Gautier, G.; Arcos-Fajardo, M.; Souchet, H.; Jouvin, M.H.; Vrtovsnik, F.; et al. The TRPM4 channel controls monocyte and macrophage, but not neutrophil, function for survival in sepsis. J. Immunol. 2012, 189, 3689–3699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.N.; Siddiqui, G.; Veldhuis, N.A.; Poole, D.P. Diverse Roles of TRPV4 in Macrophages: A Need for Unbiased Profiling. Front. Immunol. 2022, 12, 828115. [Google Scholar] [CrossRef]
- Atcha, H.; Jairaman, A.; Holt, J.R.; Meli, V.S.; Nagalla, R.R.; Veerasubramanian, P.K.; Brumm, K.T.; Lim, H.E.; Othy, S.; Cahalan, M.D.; et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat. Commun. 2021, 12, 3256. [Google Scholar] [CrossRef] [PubMed]
- Baratchi, S.; Zaldivia, M.T.K.; Wallert, M.; Loseff-Silver, J.; Al-Aryahi, S.; Zamani, J.; Thurgood, P.; Salim, A.; Htun, N.M.; Stub, D.; et al. Transcatheter Aortic Valve Implantation Represents an Anti-Inflammatory Therapy Via Reduction of Shear Stress-Induced, Piezo-1-Mediated Monocyte Activation. Circulation 2020, 142, 1092–1105. [Google Scholar] [CrossRef]
- Zeng, Y.; Riquelme, M.A.; Hua, R.; Zhang, J.; Acosta, F.M.; Gu, S.; Jiang, J.X. Mechanosensitive piezo1 calcium channel activates connexin 43 hemichannels through PI3K signaling pathway in bone. Cell Biosci. 2022, 12, 191. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, G.; Oliveira, A.P.; Noleto, I.R.S.G.; Araújo, A.K.; Lopes, A.L.F.; Sousa, F.B.M.; Chaves, L.S.; Alves, E.H.P.; Vasconcelos, D.F.P.; Araujo, A.R.; et al. Activation of transient receptor potential vanilloid channel 4 contributes to the development of ethanol-induced gastric injury in mice. Eur. J. Pharmacol. 2021, 902, 174113. [Google Scholar] [CrossRef]
- Csípő, T.; Czikora, Á.; Fülöp, G.Á.; Gulyás, H.; Rutkai, I.; Tóth, E.P.; Pórszász, R.; Szalai, A.; Bölcskei, K.; Helyes, Z.; et al. A Central Role for TRPM4 in Ca2+-Signal Amplification and Vasoconstriction. Int. J. Mol. Sci. 2022, 23, 1465. [Google Scholar] [CrossRef]
- Imbrici, P.; Liantonio, A.; Camerino, G.M.; De Bellis, M.; Camerino, C.; Mele, A.; Giustino, A.; Pierno, S.; De Luca, A.; Tricarico, D.; et al. Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery. Front. Pharmacol. 2016, 7, 121. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Channel | Selectivity | Regulation | Effect on Aortic Valve Development | Reference |
|---|---|---|---|---|
| Piezo1 | Ca2+ > Na+, K+ | Mechano-sensitive |
| [30] [30,31] |
| TRPM4 | Na+, K+ | Ca2+-activated | Knock-out in mice: increase in bicuspid valve occurrence? | [32] |
| TRPP2 | Ca2+ > Na+, K+ | Mechano-sensitive? | Knock-out in zebrafish: delayed valve formation | [31,33] |
| TRPV4 | Ca2+ > Na+, K+ | Mechano-sensitive? | Knock-out in zebrafish: thickening of the valve | [31] |
| Kir2.1 (KCNJ2 gene) | K+ | Inward rectifier | R67W loss of function mutation in human: increase in bicuspid valve occurrence | [34] |
| TMEM16E (ANO5 gene) | Cl− | Ca2+-activated | N64Kfs mutations in human: thickened aortic valve | [35] |
| Channel | Selectivity | Regulation | Effect on Aortic Valve Remodeling | Reference |
|---|---|---|---|---|
| TRPC6 | Ca2+ > Na+, K+ | DAG-activated, SOCE | High expression in human osteoblastic VIC (in vivo) | [55] |
| TRPM4 | Na+, K+ | Ca2+-activated | Promotes radiation-induced valvular fibrosis and thickening in mice (in vivo) | [32] |
| TRPV4 | Ca2+ > Na+, K+ | Mechano-sensitive? |
| [55] [55] [56,57] |
| CaV1.2 (CACNA1 gene) | Ca2+ | Voltage-gated(VDCa) |
| [14,58] [58] [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simard, C.; Aize, M.; Chaigne, S.; Mpweme Bangando, H.; Guinamard, R. Ion Channels in the Development and Remodeling of the Aortic Valve. Int. J. Mol. Sci. 2023, 24, 5860. https://doi.org/10.3390/ijms24065860
Simard C, Aize M, Chaigne S, Mpweme Bangando H, Guinamard R. Ion Channels in the Development and Remodeling of the Aortic Valve. International Journal of Molecular Sciences. 2023; 24(6):5860. https://doi.org/10.3390/ijms24065860
Chicago/Turabian StyleSimard, Christophe, Margaux Aize, Sébastien Chaigne, Harlyne Mpweme Bangando, and Romain Guinamard. 2023. "Ion Channels in the Development and Remodeling of the Aortic Valve" International Journal of Molecular Sciences 24, no. 6: 5860. https://doi.org/10.3390/ijms24065860