
Molecular Mechanisms and Pathophysiological Significance of Eryptosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
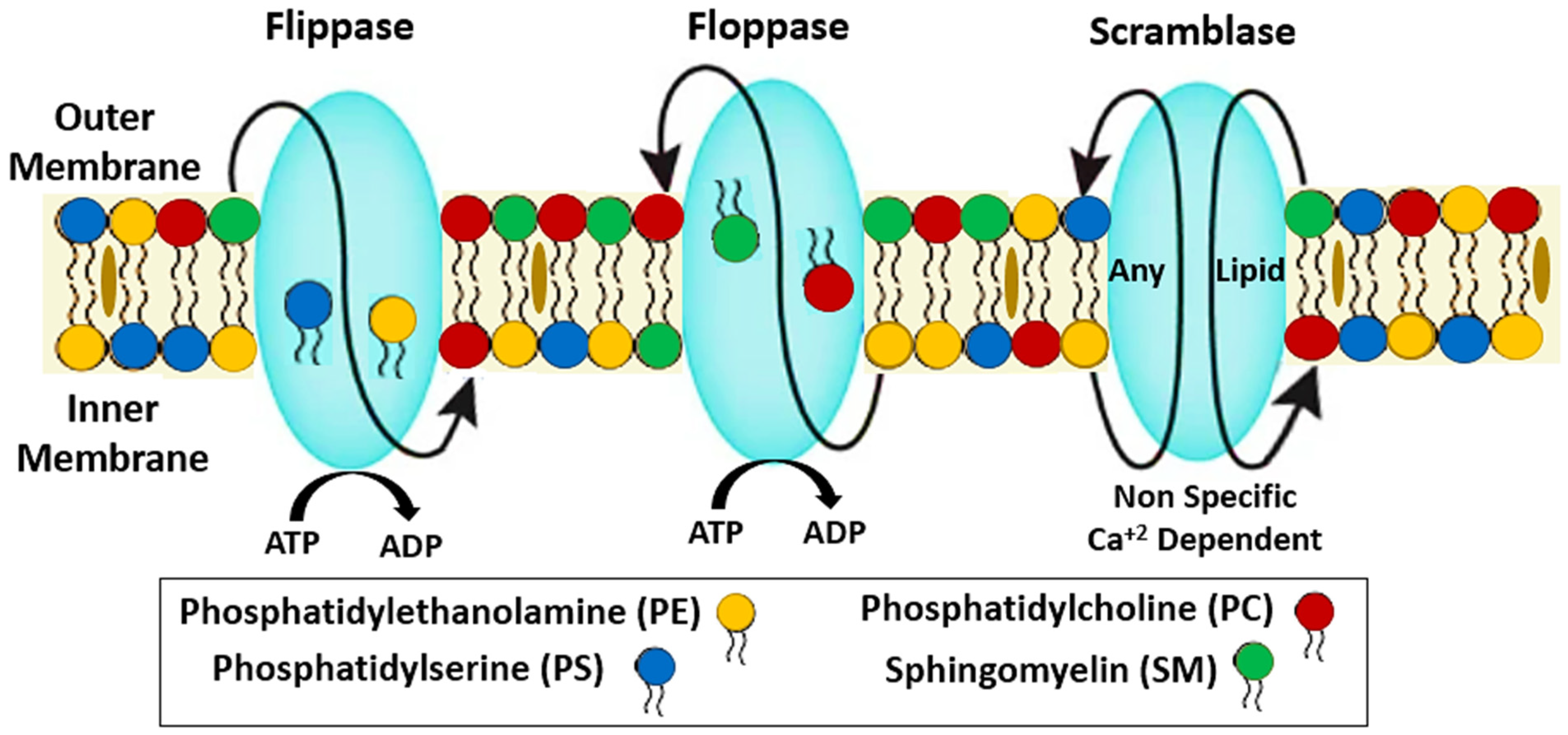
2. Structural Stability of RBCs
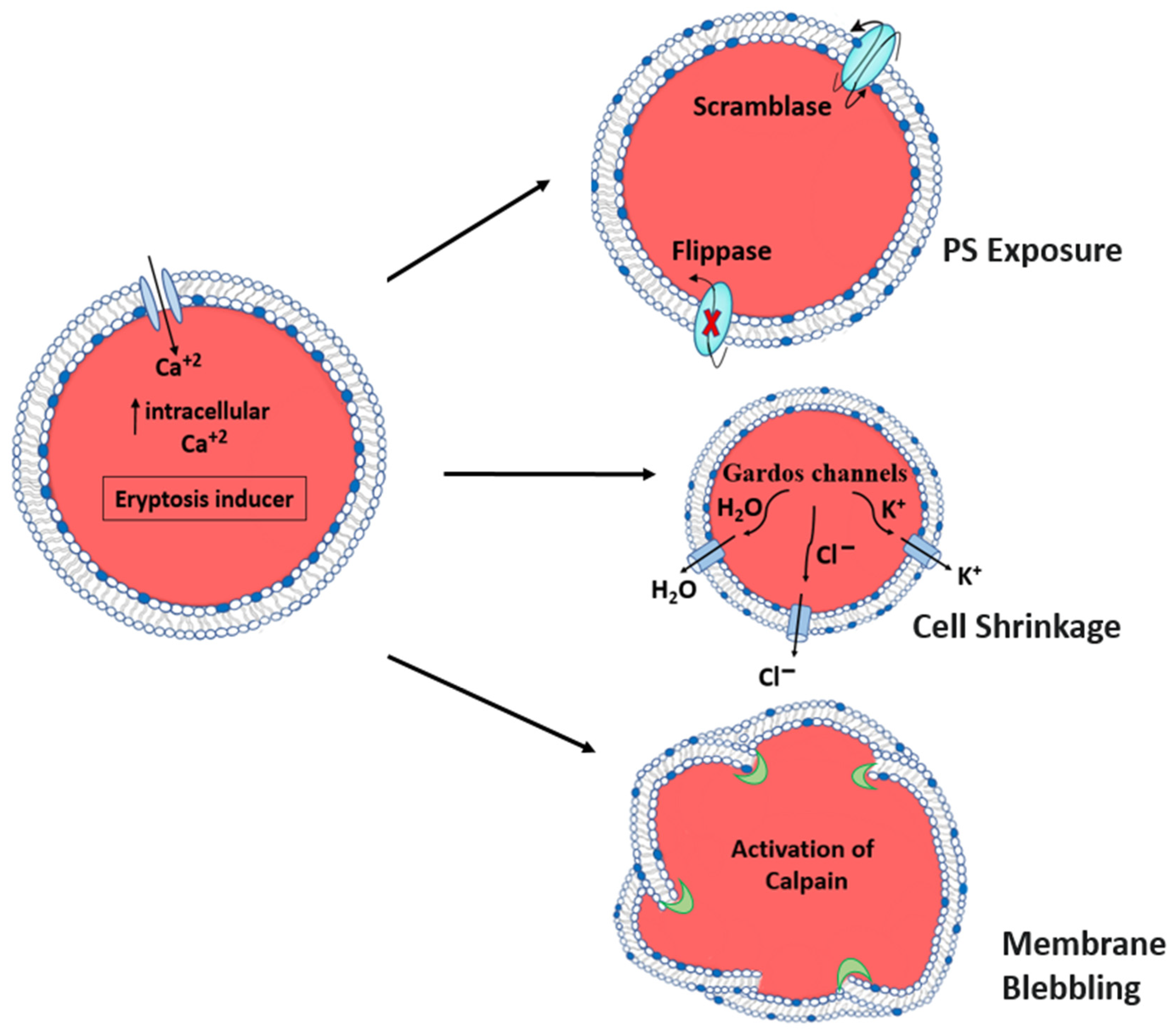
3. Biochemical and Molecular Mechanisms of Eryptosis
4. Physiological Roles of Eryptosis
5. Modulation of Eryptosis
- (a)
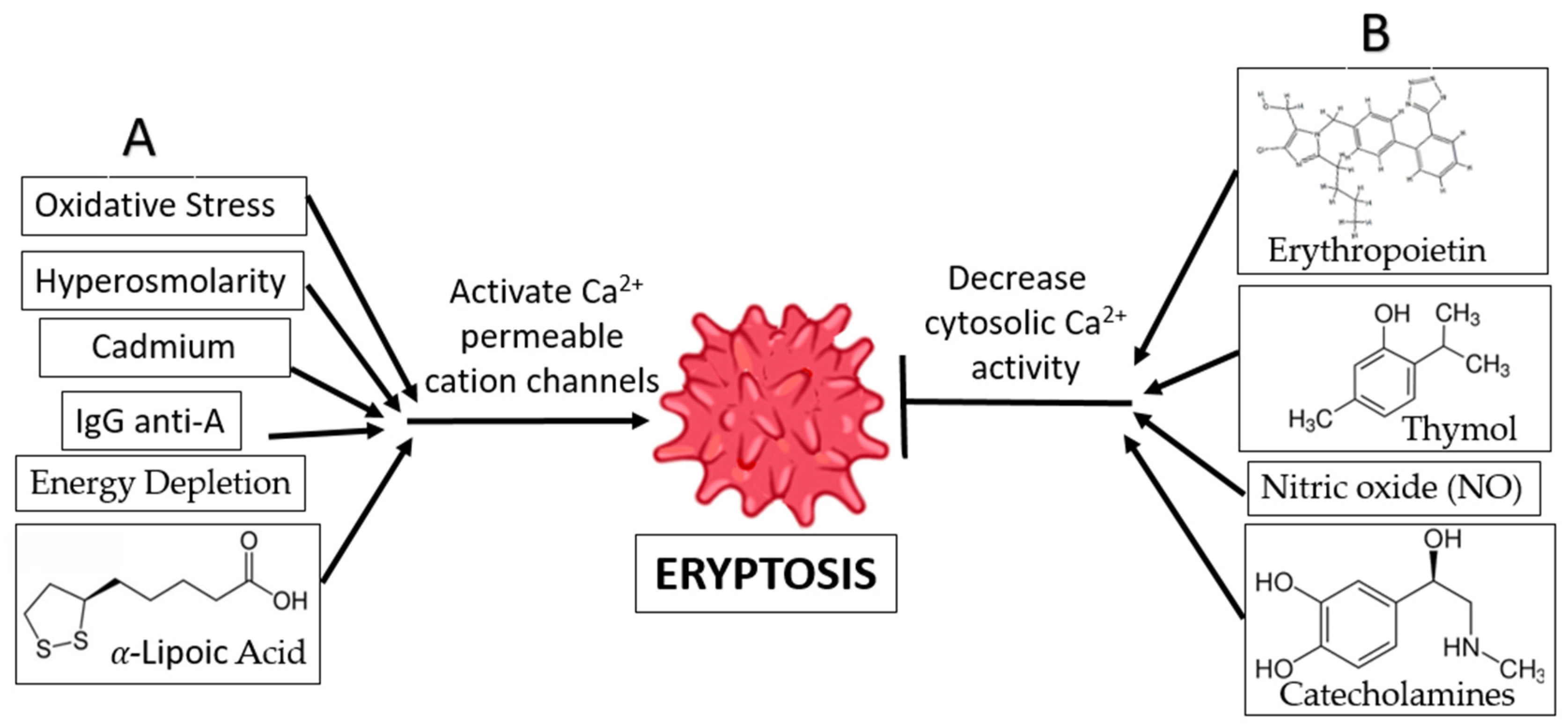
- Oxidative Stress and Hyperosmolarity: Oxidative stress and hyperosmolarity activate Cl− and Ca2+-permeable cation channels, as well as cysteinyl, aspartyl, and proteases [38]. Eryptosis is induced by a rise in intracellular Ca2+ levels, which is the result of PGE2 production in the absence of Cl− ions. Furthermore, oxidative stress activates erythrocyte-produced caspases, resulting in enhanced PS exposure and recognition of the erythrocyte by circulating macrophages. The existence of hyperosmolarity does not require the activation of caspase [38].
- (b)
- Energy Depletion: Inadequate glutathione (GSH) replenishment during calorie restriction has been associated with decreases in erythrocyte antioxidant activity. Energy deprivation also activates Ca2+-permeable cation channels in erythrocyte cell membranes, which in turn triggers eryptosis and the production of cyclooxygenase-2 (COX2) and (PGE2) [38]. Energy deprivation may also impact the phosphorylation of membrane proteins and the activity of the protein kinase C, resulting in the release of PS and the subsequent shrinking of the cell. Direct activation of eryptosis and an increase in intracellular Ca2+ ion concentration are the results of PKC activation [38].
- (c)
- Hyperthermia: When subjected to hyperthermia, erythrocytes are incapable of upregulating the production of protective proteins, and increased Ca2+ entry results in uniform cell suicide. The increase in cytosolic Ca2+ activity affects the architecture of the cytoskeleton through a decrease in erythrocyte volume, membrane scrambling, and cell shrinkage, resulting in membrane blebbing, another characteristic of eryptosis [50].
- (d)
- α-Lipoic Acid: Ca2+-sensitive K+ channels are activated in response to exposure to α-lipoic acid, leading to erythrocyte shrinking. This phenomenon may be partially or entirely explained by the increase in cytosolic Ca2+ concentration. As a bonus, α-lipoic acid is known to cause eryptosis by decreasing ATP levels and increasing ceramide synthesis. In addition, α-lipoic acid stimulates eryptosis by triggering caspases and generating oxidative stress. Because of its antioxidant qualities, α-lipoic acid is used for the treatment and prevention of several diseases. [51].
- (e)
- Xenobiotics: Eryptosis can be triggered by several different types of xenobiotics. One such cause of eryptosis is cadmium poisoning, which increases the Ca2+ ion concentration in erythrocytes while decreasing the K+ ion concentration. This clarifies why some people who have been exposed to cadmium develop anemia [52]. In addition, exposure of erythrocytes to aluminum ions [53], hexavalent chromium [4], lithium [54], and the drug Azathioprine [55] have been reported to induce suicidal erythrocyte death by decreasing cytosolic ATP, increasing the intracellular Ca2+ ion concentration, activating cell membrane scrambling, and cell shrinkage.
- (f)
- IgG Anti-A: It is also known that anti-A IgG antibodies enhance Ca2+ ion influx into erythrocytes, resulting in erythrocyte clearance. This is consistent with the immune system’s response to antigen A in autoimmune disorders and following an ABO blood transfusion [56].
- (a)
- (b)
- Xenobiotics: Many xenobiotics have been shown to suppress eryptosis. The antimicrobial agent thymol is a naturally occurring substance found in plants that prevents eryptosis by reducing cytosolic Ca2+ activity and preventing oxidative damage. However, it does not prevent the occurrence of cell shrinkage [58]. Flufenamic acid, a nonsteroidal anti-inflammatory medication, has been demonstrated to suppress eryptosis via altering Ca2+-permeable cation channels [59].
- (c)
- Catecholamines: Certain catecholamines, including dopamine, epinephrine, and isoproterenol, are considered to inhibit eryptosis by impairing the Ca2+ cation channels’ ability to enhance the entry of Ca2+ ions [36]. The literature indicates that the amounts of catecholamines required to exert an anti-eryptotic impact are lower in the body than those required to produce these effects [60] Contrary to this, research has demonstrated that dopamine can be used to treat some disorders associated with erythrocyte toxicity by preventing the erythrocytes from entering eryptosis [36].
- (d)
- (e)
6. Pathophysiological Significance of Eryptosis
7. Role of Eryptosis in Disease
7.1. Nervous System
7.1.1. Parkinson’s Disease
7.1.2. Alzheimer’s Disease
7.2. Cardiovascular System
7.3. Immune System
COVID-19
7.4. Kidney Disease
7.5. Digestive System
7.6. Diabetes Mellitus
7.7. Malignancy
7.8. Chronic Inflammatory Disease
7.9. Aging
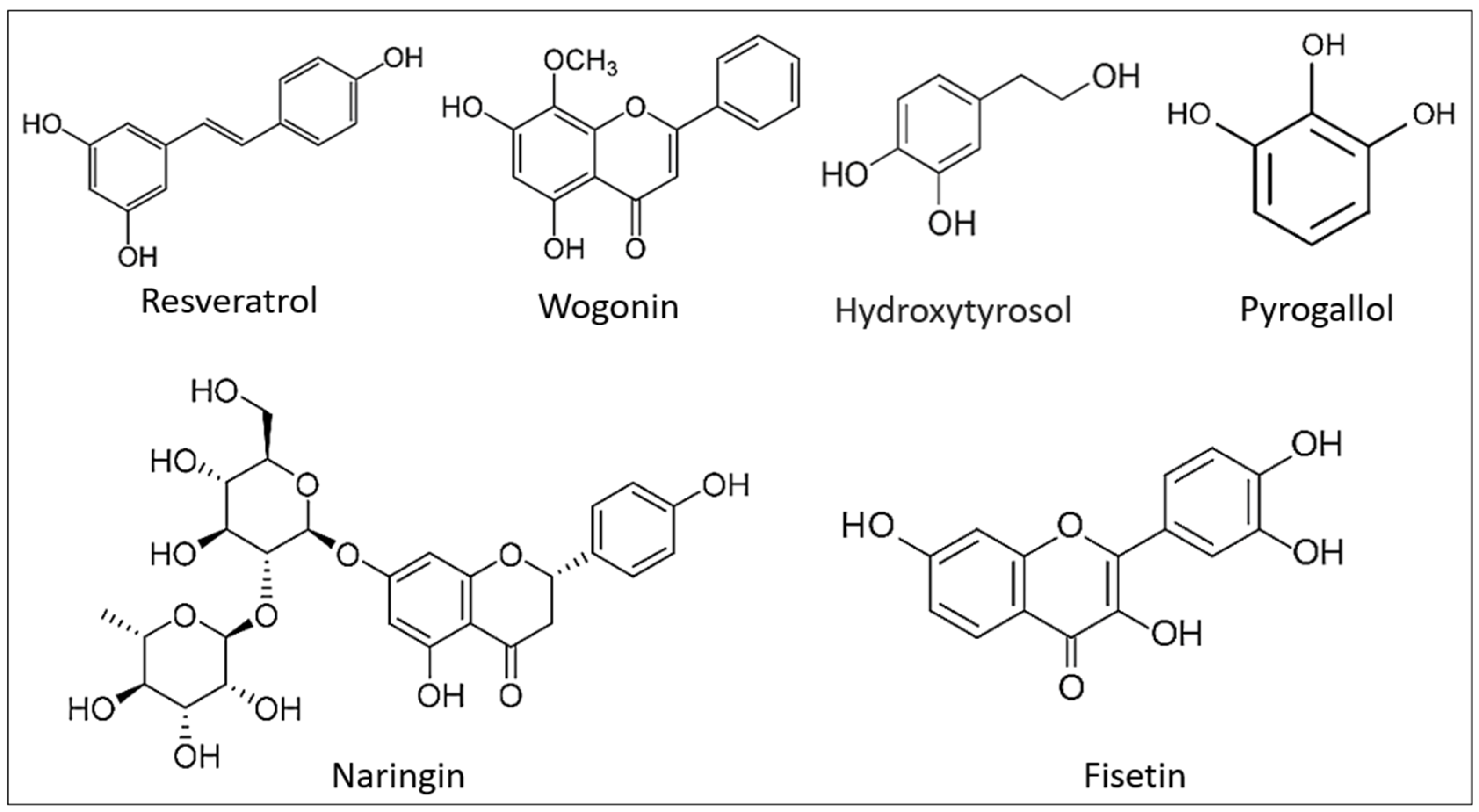
8. Prevention of Eryptosis by Natural Compounds
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vahedi, A.; Bigdelou, P.; Farnoud, A.M. Quantitative analysis of red blood cell membrane phospholipids and modulation of cell-macrophage interactions using cyclodextrins. Sci. Rep. 2020, 10, 15111. [Google Scholar] [CrossRef]
- Repsold, L.; Joubert, A.M. Eryptosis: An Erythrocyte’s Suicidal Type of Cell Death. Biomed. Res. Int. 2018, 2018, 9405617. [Google Scholar] [CrossRef] [PubMed]
- Sultan, S.A.; Khawaji, M.H.; Alsughayyir, J.; Alfhili, M.A.; Alamri, H.S.; Alrfaei, B.M. Antileukemic activity of sulfoxide nutraceutical allicin against THP-1 cells is associated with premature phosphatidylserine exposure in human erythrocytes. Saudi J. Biol. Sci. 2020, 27, 3376–3384. [Google Scholar] [CrossRef]
- Lupescu, A.; Shaik, N.; Jilani, K.; Zelenak, C.; Lang, E.; Pasham, V.; Zbidah, M.; Plate, A.; Bitzer, M.; Foller, M.; et al. Enhanced erythrocyte membrane exposure of phosphatidylserine following sorafenib treatment: An in vivo and in vitro study. Cell Physiol. Biochem. 2012, 30, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; du Plooy, J.N.; Bester, J. A Comprehensive Review on Eryptosis. Cell Physiol. Biochem. 2016, 39, 1977–2000. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Alamri, H.S.; Alsughayyir, J.; Basudan, A.M. Induction of hemolysis and eryptosis by occupational pollutant nickel chloride is mediated through calcium influx and p38 MAP kinase signaling. Int. J. Occup. Med. Environ. Health 2022, 35, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gatidis, S.; Zelenak, C.; Fajol, A.; Lang, E.; Jilani, K.; Michael, D.; Qadri, S.M.; Lang, F. p38 MAPK activation and function following osmotic shock of erythrocytes. Cell Physiol. Biochem. 2011, 28, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Lang, E.; Foller, M. Physiology and pathophysiology of eryptosis. Transfus. Med. Hemother. 2012, 39, 308–314. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Lee, M.H. Flow Cytofluorometric Analysis of Molecular Mechanisms of Premature Red Blood Cell Death. Methods Mol. Biol. 2021, 2326, 155–165. [Google Scholar] [CrossRef]
- Al Mamun Bhuyan, A.; Lang, F. Stimulation of Eryptosis by Afatinib. Cell Physiol. Biochem. 2018, 47, 1259–1273. [Google Scholar] [CrossRef] [Green Version]
- Gbotosho, G.O.; Okuboyejo, T.; Happi, C.T.; Sowunmi, A. Fall in hematocrit per 1000 parasites cleared from peripheral blood: A simple method for estimating drug-related fall in hematocrit after treatment of malaria infections. Am. J. Ther. 2014, 21, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Alfhili, M.A.; Basudan, A.M.; Alsughayyir, J. Antiproliferative Wnt inhibitor wogonin prevents eryptosis following ionophoric challenge, hyperosmotic shock, oxidative stress, and metabolic deprivation. J. Food Biochem. 2021, 45, e13977. [Google Scholar] [CrossRef]
- Foller, M.; Lang, F. Ion Transport in Eryptosis, the Suicidal Death of Erythrocytes. Front. Cell Dev. Biol. 2020, 8, 597. [Google Scholar] [CrossRef]
- Sathi, A.; Viswanad, V.; Aneesh, T.P.; Kumar, B.A. Pros and cons of phospholipid asymmetry in erythrocytes. J. Pharm. Bioallied Sci. 2014, 6, 81–85. [Google Scholar] [CrossRef]
- Bissinger, R.; Bhuyan, A.A.M.; Qadri, S.M.; Lang, F. Oxidative stress, eryptosis and anemia: A pivotal mechanistic nexus in systemic diseases. FEBS J. 2019, 286, 826–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.; Bissinger, R.; Abed, M.; Artunc, F. Eryptosis-the Neglected Cause of Anemia in End Stage Renal Disease. Kidney Blood Press Res. 2017, 42, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Barodka, V.M.; Nagababu, E.; Mohanty, J.G.; Nyhan, D.; Berkowitz, D.E.; Rifkind, J.M.; Strouse, J.J. New insights provided by a comparison of impaired deformability with erythrocyte oxidative stress for sickle cell disease. Blood Cells Mol. Dis. 2014, 52, 230–235. [Google Scholar] [CrossRef]
- Dreischer, P.; Duszenko, M.; Stein, J.; Wieder, T. Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells. Cells 2022, 11, 503. [Google Scholar] [CrossRef]
- Akiel, M.; Alsughayyir, J.; Basudan, A.M.; Alamri, H.S.; Dera, A.; Barhoumi, T.; Al Subayyil, A.M.; Basmaeil, Y.S.; Aldakheel, F.M.; Alakeel, R.; et al. Physcion Induces Hemolysis and Premature Phosphatidylserine Externalization in Human Erythrocytes. Biol. Pharm. Bull. 2021, 44, 372–378. [Google Scholar] [CrossRef]
- Restivo, I.; Attanzio, A.; Tesoriere, L.; Allegra, M. Suicidal Erythrocyte Death in Metabolic Syndrome. Antioxidants 2021, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Pretini, V.; Koenen, M.H.; Kaestner, L.; Fens, M.; Schiffelers, R.M.; Bartels, M.; Van Wijk, R. Red Blood Cells: Chasing Interactions. Front. Physiol. 2019, 10, 945. [Google Scholar] [CrossRef] [Green Version]
- Vahedi-Mazdabadi, Y.; Karimpour-Razkenari, E.; Akbarzadeh, T.; Lotfian, H.; Toushih, M.; Roshanravan, N.; Saeedi, M.; Ostadrahimi, A. Anti-cholinesterase and Neuroprotective Activities of Sweet and Bitter Apricot Kernels (Prunus armeniaca L.). Iran J. Pharm. Res. 2020, 19, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Xiang, Y.; Liu, Y.; Xiang, L.; Li, F.; Deng, X.; Xiao, Y.; Chen, L.; Chen, L.; Li, Z. Eryptosis Indices as a Novel Predictive Parameter for Biocompatibility of Fe3O4 Magnetic Nanoparticles on Erythrocytes. Sci. Rep. 2015, 5, 16209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohvo-Rekila, H.; Ramstedt, B.; Leppimaki, P.; Slotte, J.P. Cholesterol interactions with phospholipids in membranes. Prog. Lipid. Res. 2002, 41, 66–97. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef] [Green Version]
- Salzer, U.; Prohaska, R. Stomatin, flotillin-1, and flotillin-2 are major integral proteins of erythrocyte lipid rafts. Blood 2001, 97, 1141–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Sirachainan, N.; Thongsad, J.; Pakakasama, S.; Hongeng, S.; Chuansumrit, A.; Kadegasem, P.; Tirakanjana, A.; Archararit, N.; Sirireung, S. Normalized coagulation markers and anticoagulation proteins in children with severe beta-thalassemia disease after stem cell transplantation. Thromb. Res. 2012, 129, 765–770. [Google Scholar] [CrossRef]
- Weiss, E.; Cytlak, U.M.; Rees, D.C.; Osei, A.; Gibson, J.S. Deoxygenation-induced and Ca(2+) dependent phosphatidylserine externalisation in red blood cells from normal individuals and sickle cell patients. Cell Calcium 2012, 51, 51–56. [Google Scholar] [CrossRef]
- Pomorski, T.; Menon, A.K. Lipid flippases and their biological functions. Cell Mol. Life Sci. 2006, 63, 2908–2921. [Google Scholar] [CrossRef]
- Lux, S.E.t. Anatomy of the red cell membrane skeleton: Unanswered questions. Blood 2016, 127, 187–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbarino, F.; Waschenbach, L.; Cavalho-Lemos, V.; Dillenberger, M.; Becker, K.; Gohlke, H.; Cortese-Krott, M.M. Targeting spectrin redox switches to regulate the mechanoproperties of red blood cells. Biol. Chem. 2021, 402, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Akel, A.; Dreischer, P.; Toulany, M.; Koberle, M.; Skabytska, Y.; Saki, M.; Biedermann, T.; Duszenko, M.; et al. The impact of erythrocyte age on eryptosis. Br. J. Haematol. 2012, 157, 606–614. [Google Scholar] [CrossRef]
- Lang, K.S.; Lang, P.A.; Bauer, C.; Duranton, C.; Wieder, T.; Huber, S.M.; Lang, F. Mechanisms of suicidal erythrocyte death. Cell Physiol. Biochem. 2005, 15, 195–202. [Google Scholar] [CrossRef]
- Berg, C.P.; Engels, I.H.; Rothbart, A.; Lauber, K.; Renz, A.; Schlosser, S.F.; Schulze-Osthoff, K.; Wesselborg, S. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 2001, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.M.; Mahmud, H.; Lang, E.; Gu, S.; Bobbala, D.; Zelenak, C.; Jilani, K.; Siegfried, A.; Foller, M.; Lang, F. Enhanced suicidal erythrocyte death in mice carrying a loss-of-function mutation of the adenomatous polyposis coli gene. J. Cell Mol. Med. 2012, 16, 1085–1093. [Google Scholar] [CrossRef]
- Boas, F.E.; Forman, L.; Beutler, E. Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc. Natl. Acad. Sci. USA 1998, 95, 3077–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolay, J.P.; Schneider, J.; Niemoeller, O.M.; Artunc, F.; Portero-Otin, M.; Haik, G., Jr.; Thornalley, P.J.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell Physiol. Biochem. 2006, 18, 223–232. [Google Scholar] [CrossRef]
- Clarke, R.J.; Hossain, K.R.; Cao, K. Physiological roles of transverse lipid asymmetry of animal membranes. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183382. [Google Scholar] [CrossRef]
- Kuhn, V.; Diederich, L.; Keller, T.C.S.t.; Kramer, C.M.; Luckstadt, W.; Panknin, C.; Suvorava, T.; Isakson, B.E.; Kelm, M.; Cortese-Krott, M.M. Red Blood Cell Function and Dysfunction: Redox Regulation, Nitric Oxide Metabolism, Anemia. Antioxid. Redox Signal. 2017, 26, 718–742. [Google Scholar] [CrossRef] [Green Version]
- Boulet, C.; Doerig, C.D.; Carvalho, T.G. Manipulating Eryptosis of Human Red Blood Cells: A Novel Antimalarial Strategy? Front. Cell Infect. Microbiol. 2018, 8, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryer, E.; Henry, D. Isolated hypoglossal nerve palsy as a presenting symptom of metastatic peripheral T-cell lymphoma-not otherwise specified (PTCL-NOS): A unique case & a review of the literature. Int. J. Hematol. Oncol. 2018, 7, IJH03. [Google Scholar] [CrossRef] [Green Version]
- Lang, E.; Lang, F. Mechanisms and pathophysiological significance of eryptosis, the suicidal erythrocyte death. Semin. Cell Dev. Biol. 2015, 39, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Alfhili, M.A.; Aljuraiban, G.S. Lauric Acid, a Dietary Saturated Medium-Chain Fatty Acid, Elicits Calcium-Dependent Eryptosis. Cells 2021, 10, 3388. [Google Scholar] [CrossRef]
- Alsughayyir, J.; Alshaiddi, W.; Alsubki, R.; Alshammary, A.; Basudan, A.M.; Alfhili, M.A. Geraniin inhibits whole blood IFN-gamma and IL-6 and promotes IL-1beta and IL-8, and stimulates calcium-dependent and sucrose-sensitive erythrocyte death. Toxicol. Appl. Pharmacol. 2022, 436, 115881. [Google Scholar] [CrossRef]
- Kim-Shapiro, D.B.; Gladwin, M.T. Mechanisms of nitrite bioactivation. Nitric Oxide 2014, 38, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, G.; Roberts, A.I.; Shi, Y. Adhesion molecules: Key players in Mesenchymal stem cell-mediated immunosuppression. Cell Adh. Migr. 2011, 5, 20–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foller, M.; Huber, S.M.; Lang, F. Erythrocyte programmed cell death. IUBMB Life 2008, 60, 661–668. [Google Scholar] [CrossRef]
- Lang, E.; Bissinger, R.; Qadri, S.M.; Lang, F. Suicidal death of erythrocytes in cancer and its chemotherapy: A potential target in the treatment of tumor-associated anemia. Int. J. Cancer 2017, 141, 1522–1528. [Google Scholar] [CrossRef] [Green Version]
- Foller, M.; Braun, M.; Qadri, S.M.; Lang, E.; Mahmud, H.; Lang, F. Temperature sensitivity of suicidal erythrocyte death. Eur. J. Clin. Investig. 2010, 40, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Kucherenko, Y.V.; Bhavsar, S.K.; Grischenko, V.I.; Fischer, U.R.; Huber, S.M.; Lang, F. Increased cation conductance in human erythrocytes artificially aged by glycation. J. Membr. Biol. 2010, 235, 177–189. [Google Scholar] [CrossRef]
- Sopjani, M.; Foller, M.; Dreischer, P.; Lang, F. Stimulation of eryptosis by cadmium ions. Cell Physiol. Biochem. 2008, 22, 245–252. [Google Scholar] [CrossRef]
- Niemoeller, O.M.; Kiedaisch, V.; Dreischer, P.; Wieder, T.; Lang, F. Stimulation of eryptosis by aluminium ions. Toxicol. Appl. Pharmacol 2006, 217, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, J.P.; Gatz, S.; Lang, F.; Lang, U.E. Lithium-induced suicidal erythrocyte death. J. Psychopharmacol. 2010, 24, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Geiger, C.; Foller, M.; Herrlinger, K.R.; Lang, F. Azathioprine-induced suicidal erythrocyte death. Inflamm. Bowel. Dis. 2008, 14, 1027–1032. [Google Scholar] [CrossRef]
- Attanasio, P.; Shumilina, E.; Hermle, T.; Kiedaisch, V.; Lang, P.A.; Huber, S.M.; Wieder, T.; Lang, F. Stimulation of eryptosis by anti-A IgG antibodies. Cell Physiol. Biochem. 2007, 20, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, C.R.; Snyder, L.M. Oxidation and erythrocyte senescence. Curr. Opin. Hematol. 2000, 7, 113–116. [Google Scholar] [CrossRef]
- Mahmud, H.; Mauro, D.; Foller, M.; Lang, F. Inhibitory effect of thymol on suicidal erythrocyte death. Cell Physiol. Biochem. 2009, 24, 407–414. [Google Scholar] [CrossRef]
- Kasinathan, R.S.; Foller, M.; Koka, S.; Huber, S.M.; Lang, F. Inhibition of eryptosis and intraerythrocytic growth of Plasmodium falciparum by flufenamic acid. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 374, 255–264. [Google Scholar] [CrossRef]
- Lang, P.A.; Kempe, D.S.; Akel, A.; Klarl, B.A.; Eisele, K.; Podolski, M.; Hermle, T.; Niemoeller, O.M.; Attanasio, P.; Huber, S.M.; et al. Inhibition of erythrocyte “apoptosis” by catecholamines. Naunyn Schmiedebergs Arch. Pharmacol. 2005, 372, 228–235. [Google Scholar] [CrossRef]
- Floride, E.; Foller, M.; Ritter, M.; Lang, F. Caffeine inhibits suicidal erythrocyte death. Cell Physiol. Biochem. 2008, 22, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Gulbins, E.; Lerche, H.; Huber, S.M.; Kempe, D.S.; Foller, M. Eryptosis, a window to systemic disease. Cell Physiol. Biochem. 2008, 22, 373–380. [Google Scholar] [CrossRef]
- Setty, B.N.; Betal, S.G. Microvascular endothelial cells express a phosphatidylserine receptor: A functionally active receptor for phosphatidylserine-positive erythrocytes. Blood 2008, 111, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Wautier, M.P.; Heron, E.; Picot, J.; Colin, Y.; Hermine, O.; Wautier, J.L. Red blood cell phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in central retinal vein occlusion. J. Thromb. Haemost. 2011, 9, 1049–1055. [Google Scholar] [CrossRef]
- Pretorius, E. Erythrocyte deformability and eryptosis during inflammation, and impaired blood rheology. Clin. Hemorheol. Microcirc. 2018, 69, 545–550. [Google Scholar] [CrossRef]
- Foller, M.; Bobbala, D.; Koka, S.; Huber, S.M.; Gulbins, E.; Lang, F. Suicide for survival--death of infected erythrocytes as a host mechanism to survive malaria. Cell Physiol. Biochem. 2009, 24, 133–140. [Google Scholar] [CrossRef]
- Lang, F.; Abed, M.; Lang, E.; Foller, M. Oxidative stress and suicidal erythrocyte death. Antioxid. Redox Signal. 2014, 21, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Lau, P.M.; Kong, S.K. Mitochondrial toxin betulinic acid induces in vitro eryptosis in human red blood cells through membrane permeabilization. Arch. Toxicol. 2014, 88, 755–768. [Google Scholar] [CrossRef]
- Funke, C.; Schneider, S.A.; Berg, D.; Kell, D.B. Genetics and iron in the systems biology of Parkinson’s disease and some related disorders. Neurochem. Int. 2013, 62, 637–652. [Google Scholar] [CrossRef]
- Pretorius, E.; Swanepoel, A.C.; Buys, A.V.; Vermeulen, N.; Duim, W.; Kell, D.B. Eryptosis as a marker of Parkinson’s disease. Aging 2014, 6, 788–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samantaray, S.; Knaryan, V.H.; Shields, D.C.; Banik, N.L. Critical role of calpain in spinal cord degeneration in Parkinson’s disease. J. Neurochem. 2013, 127, 880–890. [Google Scholar] [CrossRef] [Green Version]
- Diepenbroek, M.; Casadei, N.; Esmer, H.; Saido, T.C.; Takano, J.; Kahle, P.J.; Nixon, R.A.; Rao, M.V.; Melki, R.; Pieri, L.; et al. Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P]alphaSyn transgenic mice. Hum. Mol. Genet. 2014, 23, 3975–3989. [Google Scholar] [CrossRef]
- Arshad, A.; Chen, X.; Cong, Z.; Qing, H.; Deng, Y. TRPC1 protects dopaminergic SH-SY5Y cells from MPP+, salsolinol, and N-methyl-(R)-salsolinol-induced cytotoxicity. Acta Biochim. Biophys. Sin 2014, 46, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Esteves, A.R.; Cardoso, S.M.; Oliveira, C.R. Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: Relevance to Parkinson’s disease. Neurochem. Int. 2009, 55, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Qadri, S.M. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif. 2012, 33, 125–130. [Google Scholar] [CrossRef]
- Carelli-Alinovi, C.; Misiti, F. Erythrocytes as Potential Link between Diabetes and Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Uscinska, E.; Idzkowska, E.; Sobkowicz, B.; Musial, W.J.; Tycinska, A.M. Anemia in Intensive Cardiac Care Unit patients—An underestimated problem. Adv. Med. Sci. 2015, 60, 307–314. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S.; Jankowska, E.A.; Ponikowski, P.; Anker, S.D. Anemia in heart failure: An overview of current concepts. Future Cardiol. 2011, 7, 119–129. [Google Scholar] [CrossRef]
- Attanasio, P.; Bissinger, R.; Haverkamp, W.; Pieske, B.; Wutzler, A.; Lang, F. Enhanced suicidal erythrocyte death in acute cardiac failure. Eur. J. Clin. Investig. 2015, 45, 1316–1324. [Google Scholar] [CrossRef]
- Blokhin, I.O.; Lentz, S.R. Mechanisms of thrombosis in obesity. Curr. Opin. Hematol. 2013, 20, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, E.; Vaya, A.; Martinez, M.; Moscardo, A.; Corella, D.; Santaolaria, M.L.; Espana, F.; Hernandez-Mijares, A. Erythrocyte membrane phosphatidylserine exposure in obesity. Obesity 2009, 17, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Wiewiora, M.; Piecuch, J.; Sedek, L.; Mazur, B.; Sosada, K. The effects of obesity on CD47 expression in erythrocytes. Cytom. B Clin. Cytom. 2017, 92, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Unruh, D.; Srinivasan, R.; Benson, T.; Haigh, S.; Coyle, D.; Batra, N.; Keil, R.; Sturm, R.; Blanco, V.; Palascak, M.; et al. Red Blood Cell Dysfunction Induced by High-Fat Diet: Potential Implications for Obesity-Related Atherosclerosis. Circulation 2015, 132, 1898–1908. [Google Scholar] [CrossRef] [Green Version]
- Pinzon-Diaz, C.E.; Calderon-Salinas, J.V.; Rosas-Flores, M.M.; Hernandez, G.; Lopez-Betancourt, A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in hypertensive and dyslipidemic patients. Mol. Cell Biochem. 2018, 440, 105–113. [Google Scholar] [CrossRef]
- Arca, M.; Natoli, S.; Micheletta, F.; Riggi, S.; Di Angelantonio, E.; Montali, A.; Antonini, T.M.; Antonini, R.; Diczfalusy, U.; Iuliano, L. Increased plasma levels of oxysterols, in vivo markers of oxidative stress, in patients with familial combined hyperlipidemia: Reduction during atorvastatin and fenofibrate therapy. Free Radic. Biol. Med. 2007, 42, 698–705. [Google Scholar] [CrossRef]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, M.H. Rates of cholesterol exchange between human erythrocytes and plasma lipoproteins. Biochim. Biophys. Acta 1980, 600, 530–541. [Google Scholar] [CrossRef]
- van Zwieten, R.; Bochem, A.E.; Hilarius, P.M.; van Bruggen, R.; Bergkamp, F.; Hovingh, G.K.; Verhoeven, A.J. The cholesterol content of the erythrocyte membrane is an important determinant of phosphatidylserine exposure. Biochim. Biophys. Acta 2012, 1821, 1493–1500. [Google Scholar] [CrossRef]
- Barcellini, W. New Insights in the Pathogenesis of Autoimmune Hemolytic Anemia. Transfus. Med. Hemother. 2015, 42, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Packman, C.H. The Clinical Pictures of Autoimmune Hemolytic Anemia. Transfus. Med. Hemother. 2015, 42, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, D.; Zeerleder, S. Complement inhibitors to treat IgM-mediated autoimmune hemolysis. Haematologica 2015, 100, 1388–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Suzuki, J.; Segawa, K.; Fujii, T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016, 23, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, A.; Hartnack, D.; Lindemann, H.W.; Lange, H.J.; Rummel, M.; Loew, A. The effect of erythropoiesis-stimulating agents in patients with therapy-refractory autoimmune hemolytic anemia. Transfus. Med. Hemother. 2014, 41, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Bartolmas, T.; Mayer, B.; Balola, A.H.; Salama, A. Eryptosis in autoimmune haemolytic anaemia. Eur. J. Haematol. 2018, 100, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Bouchla, A.; Kriebardis, A.G.; Georgatzakou, H.T.; Fortis, S.P.; Thomopoulos, T.P.; Lekkakou, L.; Markakis, K.; Gkotzias, D.; Panagiotou, A.; Papageorgiou, E.G.; et al. Red Blood Cell Abnormalities as the Mirror of SARS-CoV-2 Disease Severity: A Pilot Study. Front. Physiol. 2021, 12, 825055. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Dreischer, P.; Wieder, T.; Koberle, M. Coronavirus disease 2019 (COVID-19), human erythrocytes and the PKC-alpha/-beta inhibitor chelerythrine -possible therapeutic implication. Cell Cycle 2020, 19, 3399–3405. [Google Scholar] [CrossRef]
- Clementi, A.; Virzi, G.M.; Milan Manani, S.; Battaglia, G.G.; Ronco, C.; Zanella, M. Eryptosis in Patients with Chronic Kidney Disease: A Possible Relationship with Oxidative Stress and Inflammatory Markers. J. Clin. Med. 2022, 11, 7167. [Google Scholar] [CrossRef]
- Li, D.; Zheng, X.; Zhang, Y.; Li, X.; Chen, X.; Yin, Y.; Hu, J.; Li, J.; Guo, M.; Wang, X. What Should Be Responsible for Eryptosis in Chronic Kidney Disease? Kidney Blood Press Res. 2022, 47, 375–390. [Google Scholar] [CrossRef]
- Alzoubi, K.; Honisch, S.; Abed, M.; Lang, F. Triggering of suicidal erythrocyte death by penta-O-galloyl-beta-D-glucose. Toxins 2013, 6, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press Res. 2013, 37, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Vodosek Hojs, N.; Bevc, S.; Ekart, R.; Hojs, R. Oxidative Stress Markers in Chronic Kidney Disease with Emphasis on Diabetic Nephropathy. Antioxidants 2020, 9, 925. [Google Scholar] [CrossRef]
- Sudnitsyna, J.; Skverchinskaya, E.; Dobrylko, I.; Nikitina, E.; Gambaryan, S.; Mindukshev, I. Microvesicle Formation Induced by Oxidative Stress in Human Erythrocytes. Antioxidants 2020, 9, 929. [Google Scholar] [CrossRef]
- Maheshwari, A.; Mishra, R.; Thuluvath, P.J. Post-liver-transplant anemia: Etiology and management. Liver Transpl. 2004, 10, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.W. Hematologic manifestations of liver disease. Semin. Hematol. 2013, 50, 216–221. [Google Scholar] [CrossRef]
- Lang, E.; Gatidis, S.; Freise, N.F.; Bock, H.; Kubitz, R.; Lauermann, C.; Orth, H.M.; Klindt, C.; Schuier, M.; Keitel, V.; et al. Conjugated bilirubin triggers anemia by inducing erythrocyte death. Hepatology 2015, 61, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.; Huber, S.M.; Szabo, I.; Gulbins, E. Plasma membrane ion channels in suicidal cell death. Arch. Biochem. Biophys. 2007, 462, 189–194. [Google Scholar] [CrossRef]
- Soma, P.; Pretorius, E. Interplay between ultrastructural findings and atherothrombotic complications in type 2 diabetes mellitus. Cardiovasc. Diabetol. 2015, 14, 96. [Google Scholar] [CrossRef] [Green Version]
- Mohammedi, K.; Bellili-Munoz, N.; Marklund, S.L.; Driss, F.; Le Nagard, H.; Patente, T.A.; Fumeron, F.; Roussel, R.; Hadjadj, S.; Marre, M.; et al. Plasma extracellular superoxide dismutase concentration, allelic variations in the SOD3 gene and risk of myocardial infarction and all-cause mortality in people with type 1 and type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 845. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, B.L.; Sharma, P.; Das, R. Anemia in malignancies: Pathogenetic and diagnostic considerations. Hematology 2015, 20, 18–25. [Google Scholar] [CrossRef]
- Gilreath, J.A.; Stenehjem, D.D.; Rodgers, G.M. Diagnosis and treatment of cancer-related anemia. Am. J. Hematol. 2014, 89, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.M.; Bissinger, R.; Solh, Z.; Oldenborg, P.A. Eryptosis in health and disease: A paradigm shift towards understanding the (patho)physiological implications of programmed cell death of erythrocytes. Blood Rev. 2017, 31, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Bissinger, R.; Lang, F. Mitoxantrone-induced suicidal erythrocyte death. Cell Physiol. Biochem. 2014, 34, 1756–1767. [Google Scholar] [CrossRef]
- Jacobi, J.; Lang, E.; Bissinger, R.; Frauenfeld, L.; Modicano, P.; Faggio, C.; Abed, M.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by mitotane. Cell Physiol. Biochem. 2014, 33, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhang, X.; Feng, X.; Fan, X.; Jin, Z. The crosstalk between microRNAs and the Wnt/beta-catenin signaling pathway in cancer. Oncotarget 2017, 8, 14089–14106. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, A.; Wier, E.M.; Fu, K.; Sun, X.; Wan, F. Ultrasound imaging of splenomegaly as a proxy to monitor colon tumor development in Apc(min716/+) mice. Cancer Med. 2016, 5, 2469–2476. [Google Scholar] [CrossRef]
- Stein, R.S. The role of erythropoietin in the anemia of myelodysplastic syndrome. Clin. Lymphoma 2003, 4 (Suppl. 1), S36–S40. [Google Scholar] [CrossRef]
- Basu, S.; Banerjee, D.; Ghosh, M.; Chakrabarti, A. Erythrocyte membrane defects and asymmetry in paroxysmal nocturnal hemoglobinuria and myelodysplastic syndrome. Hematology 2010, 15, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Abioye, A.I.; Park, S.; Ripp, K.; McDonald, E.A.; Kurtis, J.D.; Wu, H.; Pond-Tor, S.; Sharma, S.; Ernerudh, J.; Baltazar, P.; et al. Anemia of Inflammation during Human Pregnancy Does Not Affect Newborn Iron Endowment. J. Nutr. 2018, 148, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Bester, J.; Pretorius, E. Effects of IL-1beta, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci. Rep. 2016, 6, 32188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissinger, R.; Kempe-Teufel, D.S.; Honisch, S.; Qadri, S.M.; Randrianarisoa, E.; Haring, H.U.; Henes, J.; Lang, F. Stimulated Suicidal Erythrocyte Death in Arteritis. Cell. Physiol. Biochem. 2016, 39, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Giannouli, S.; Voulgarelis, M.; Ziakas, P.D.; Tzioufas, A.G. Anaemia in systemic lupus erythematosus: From pathophysiology to clinical assessment. Ann. Rheum. Dis. 2006, 65, 144–148. [Google Scholar] [CrossRef]
- Jiang, P.; Bian, M.; Ma, W.; Liu, C.; Yang, P.; Zhu, B.; Xu, Y.; Zheng, M.; Qiao, J.; Shuai, Z.; et al. Eryptosis as an Underlying Mechanism in Systemic Lupus Erythematosus-Related Anemia. Cell Physiol. Biochem. 2016, 40, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Artz, A.S.; Thirman, M.J. Unexplained anemia predominates despite an intensive evaluation in a racially diverse cohort of older adults from a referral anemia clinic. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 925–932. [Google Scholar] [CrossRef]
- Lupescu, A.; Bissinger, R.; Goebel, T.; Salker, M.S.; Alzoubi, K.; Liu, G.; Chirigiu, L.; Mack, A.F.; Qadri, S.M.; Lang, F. Enhanced suicidal erythrocyte death contributing to anemia in the elderly. Cell Physiol. Biochem. 2015, 36, 773–783. [Google Scholar] [CrossRef]
- Kempe, D.S.; Ackermann, T.F.; Fischer, S.S.; Koka, S.; Boini, K.M.; Mahmud, H.; Foller, M.; Rosenblatt, K.P.; Kuro, O.M.; Lang, F. Accelerated suicidal erythrocyte death in Klotho-deficient mice. Pflugers Arch. 2009, 458, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Alfhili, M.A.; Alsughayyir, J.; Basudan, A.B. Epidemic dropsy toxin, sanguinarine chloride, stimulates sucrose-sensitive hemolysis and breakdown of membrane phospholipid asymmetry in human erythrocytes. Toxicon 2021, 199, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Cilla, A.; Alegria, A.; Attanzio, A.; Garcia-Llatas, G.; Tesoriere, L.; Livrea, M.A. Dietary phytochemicals in the protection against oxysterol-induced damage. Chem. Phys. Lipids 2017, 207, 192–205. [Google Scholar] [CrossRef]
- Yu, M.; Gouvinhas, I.; Rocha, J.; Barros, A. Phytochemical and antioxidant analysis of medicinal and food plants towards bioactive food and pharmaceutical resources. Sci. Rep. 2021, 11, 10041. [Google Scholar] [CrossRef]
- Perez-Jimenez, J.; Neveu, V.; Vos, F.; Scalbert, A. Identification of the 100 richest dietary sources of polyphenols: An application of the Phenol-Explorer database. Eur. J. Clin. Nutr. 2010, 64 (Suppl. 3), S112–S120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque, B.R.; Heleno, S.A.; Oliveira, M.; Barros, L.; Ferreira, I. Phenolic compounds: Current industrial applications, limitations and future challenges. Food Funct. 2021, 12, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, C. The protective effects of polyphenols on Alzheimer’s disease: A systematic review. Alzheimers Dement. 2019, 5, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Restivo, I.; Attanzio, A.; Tesoriere, L.; Allegra, M.; Garcia-Llatas, G.; Cilla, A. Anti-Eryptotic Activity of Food-Derived Phytochemicals and Natural Compounds. Int. J. Mol. Sci. 2022, 23, 3019. [Google Scholar] [CrossRef]
- Qiu, S.; Sun, H.; Zhang, A.H.; Xu, H.Y.; Yan, G.L.; Han, Y.; Wang, X.J. Natural alkaloids: Basic aspects, biological roles, and future perspectives. Chin. J. Nat. Med. 2014, 12, 401–406. [Google Scholar] [CrossRef]
- Dang, T.T.; Onoyovwi, A.; Farrow, S.C.; Facchini, P.J. Biochemical genomics for gene discovery in benzylisoquinoline alkaloid biosynthesis in opium poppy and related species. Methods Enzymol. 2012, 515, 231–266. [Google Scholar] [CrossRef]
- Shan, F.; Yang, R.; Ji, T.; Jiao, F. Vitamin C Inhibits Aggravated Eryptosis by Hydrogen Peroxide in Glucose-6-Phosphated Dehydrogenase Deficiency. Cell Physiol. Biochem. 2016, 39, 1453–1462. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghareeb, S.A.; Alfhili, M.A.; Fatima, S. Molecular Mechanisms and Pathophysiological Significance of Eryptosis. Int. J. Mol. Sci. 2023, 24, 5079. https://doi.org/10.3390/ijms24065079
Alghareeb SA, Alfhili MA, Fatima S. Molecular Mechanisms and Pathophysiological Significance of Eryptosis. International Journal of Molecular Sciences. 2023; 24(6):5079. https://doi.org/10.3390/ijms24065079
Chicago/Turabian StyleAlghareeb, Sumiah A., Mohammad A. Alfhili, and Sabiha Fatima. 2023. "Molecular Mechanisms and Pathophysiological Significance of Eryptosis" International Journal of Molecular Sciences 24, no. 6: 5079. https://doi.org/10.3390/ijms24065079