
Identification and Characterization of p300-Mediated Lysine Residues in Cardiac SERCA2a
 ,
,
Abstract
:1. Introduction
2. Results
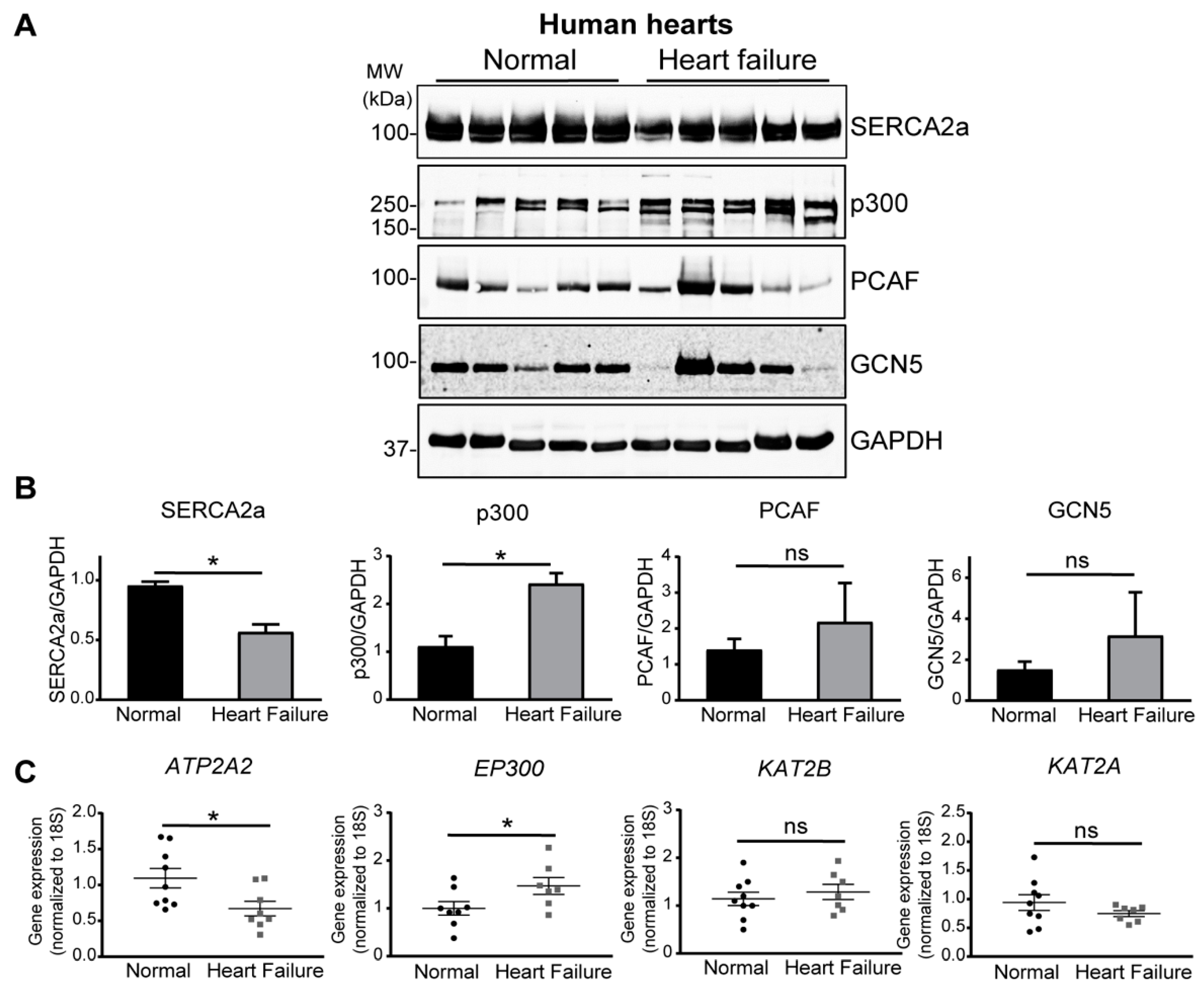
2.1. Increase of p300 Is More Prominent in Human Heart Failure
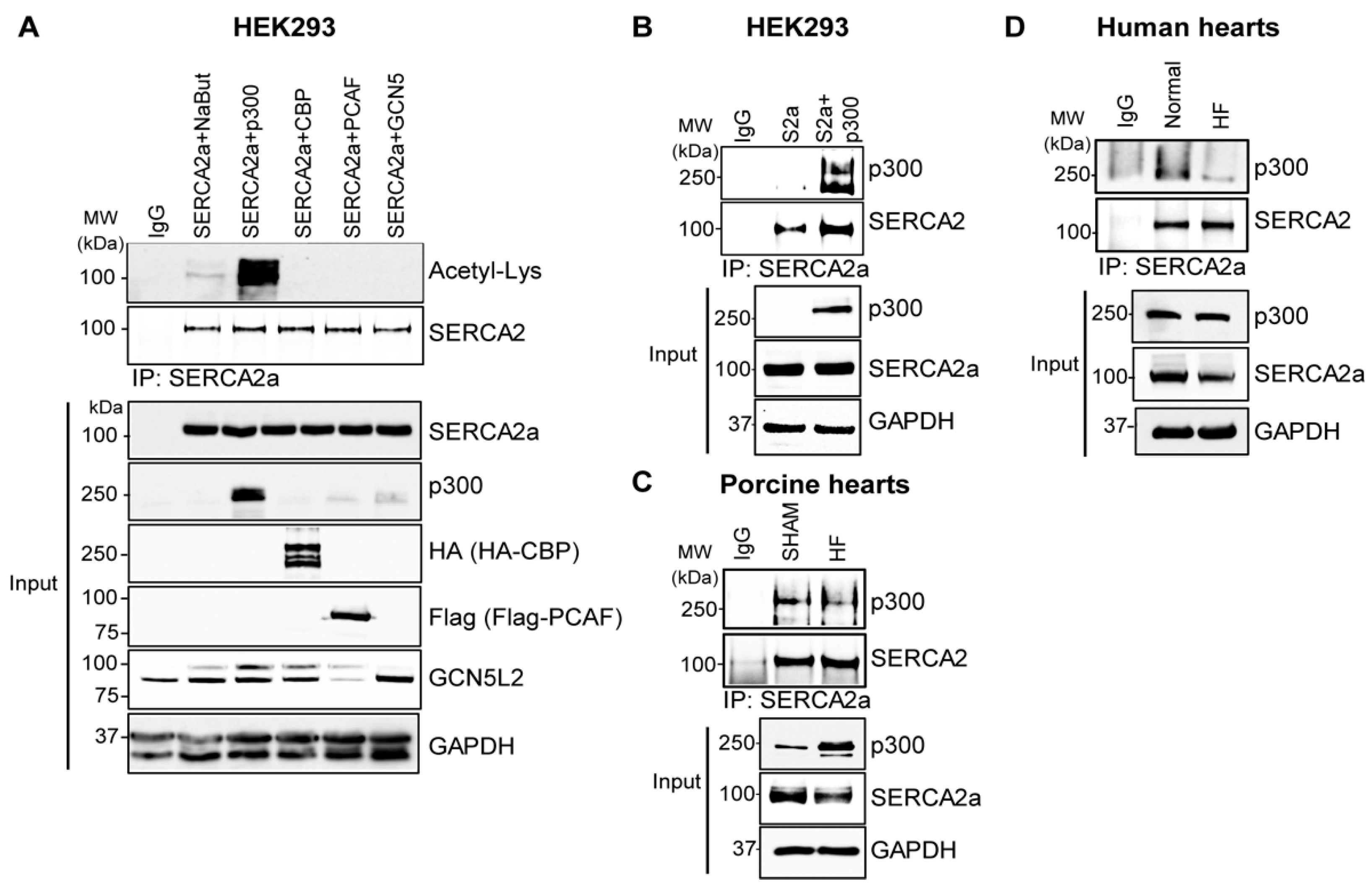
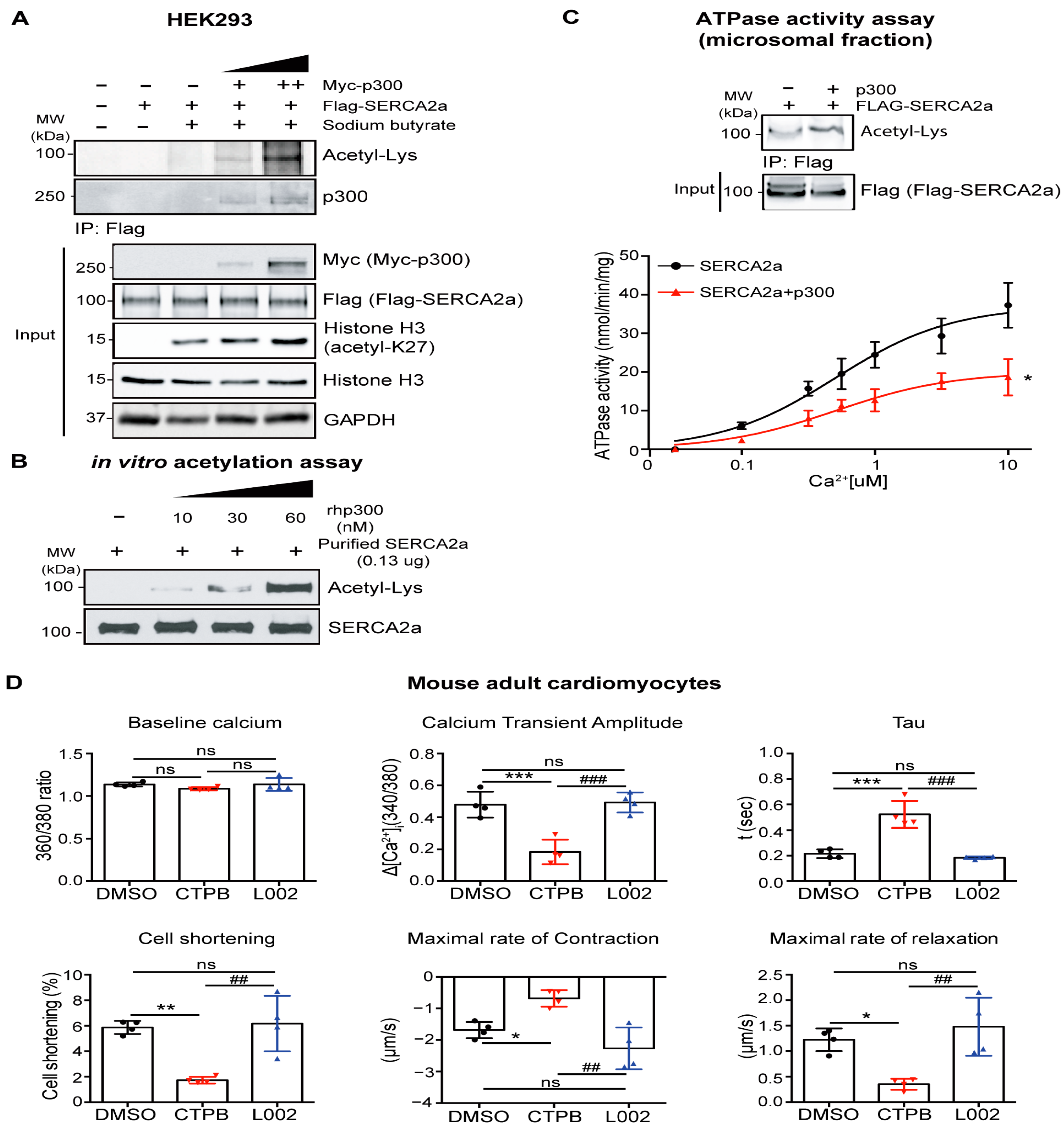
2.2. p300 Directly Interacts with and Acetylates SERCA2a Resulting in Diminished Calcium Transport Activity
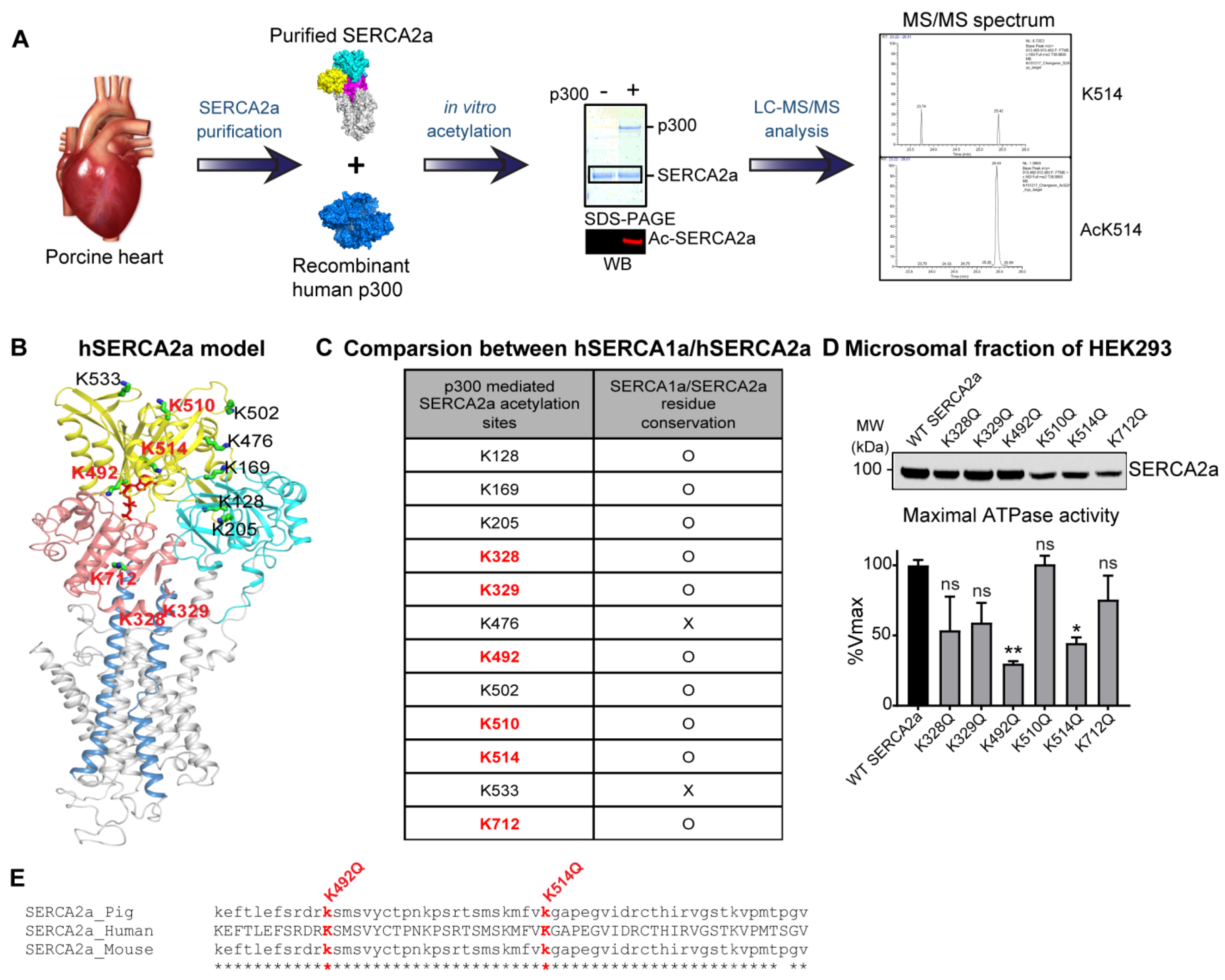
2.3. p300 Acetylates SERCA2a at Multiple Lysine Residues
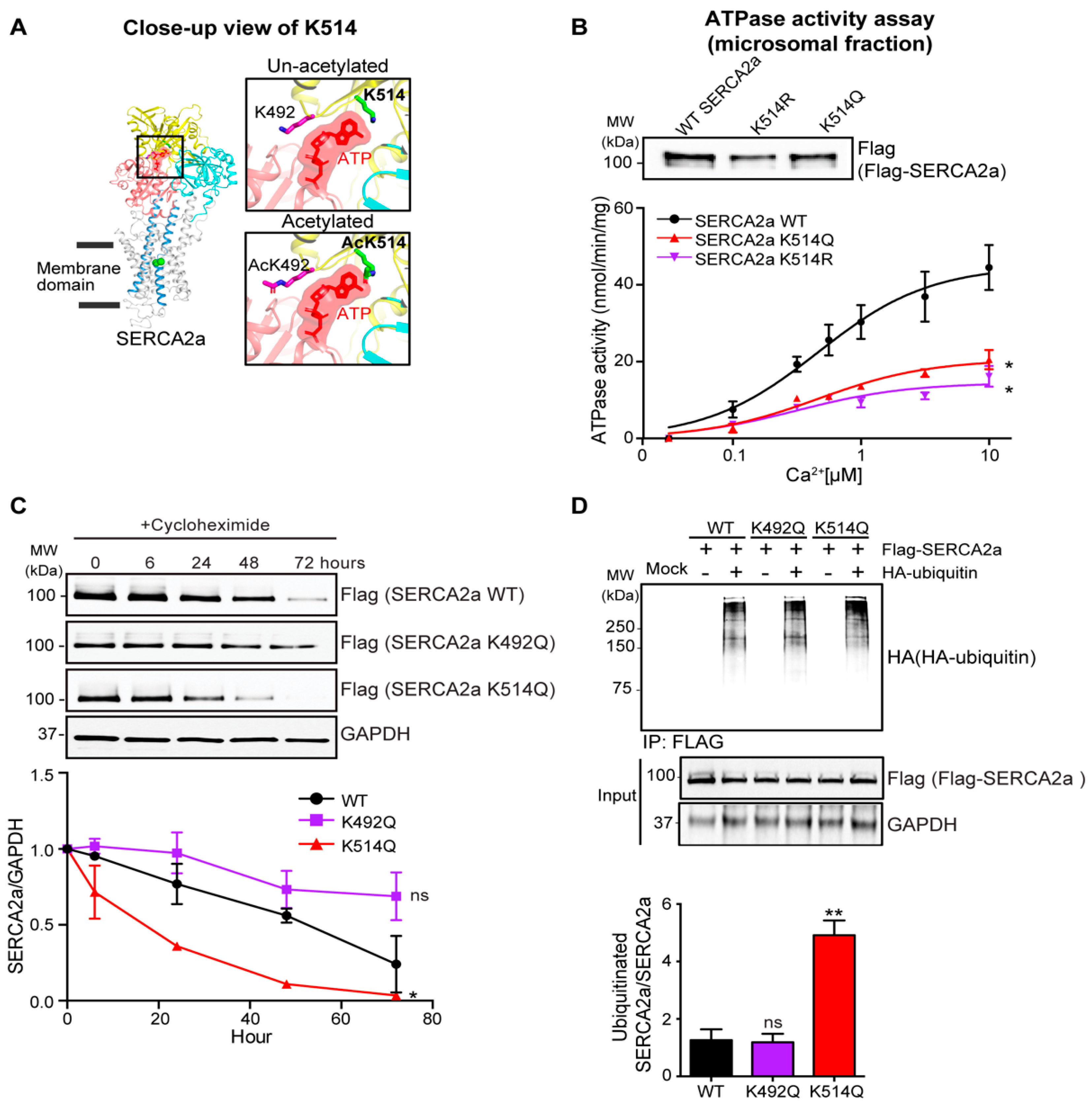
2.4. Acetylation on Lys514 Has a Profound Effect on SERCA2a Function and Stability
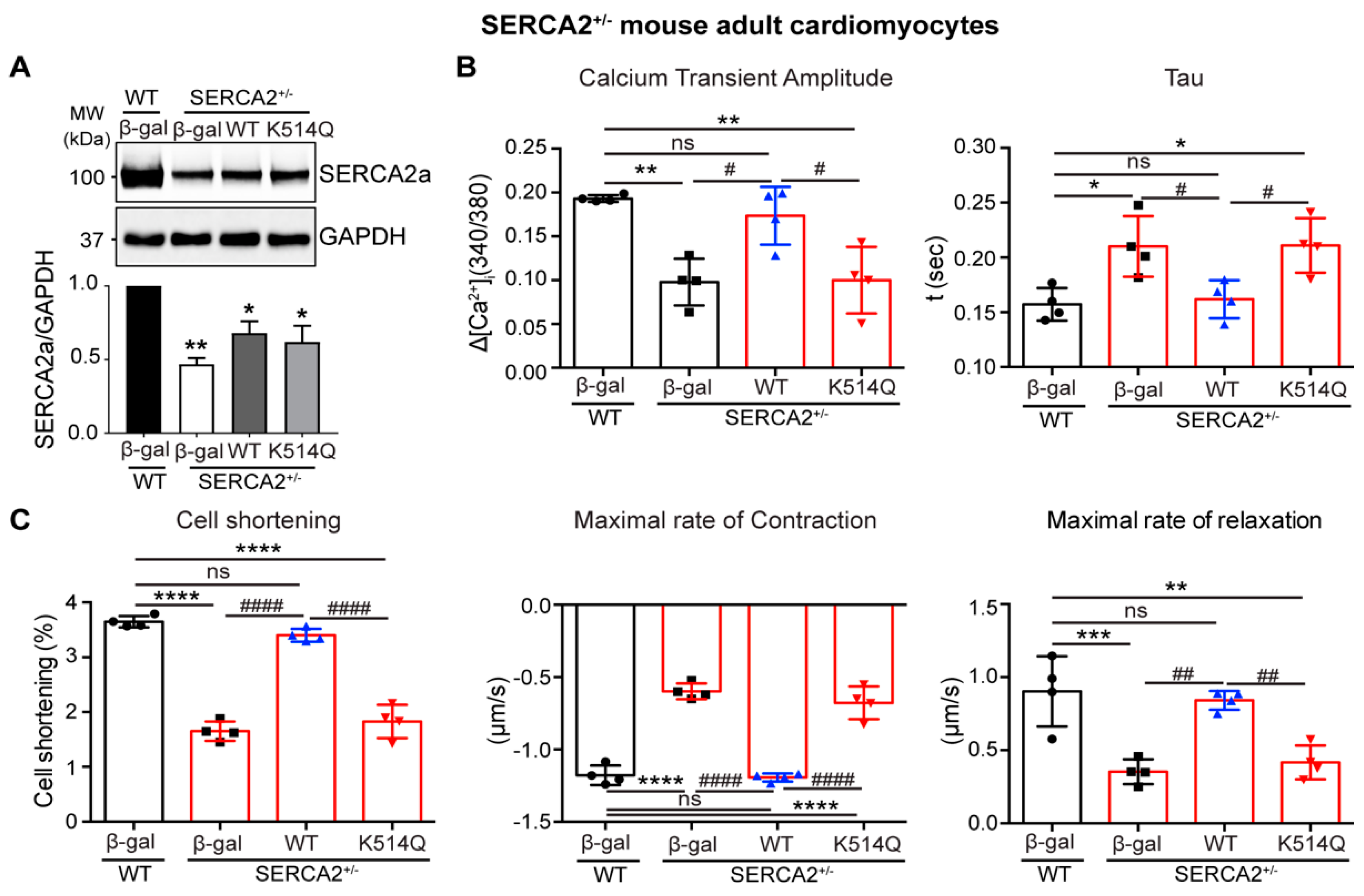
2.5. Acetylation on Lys514 Impairs Cardiomyocyte Function
3. Discussion
4. Materials and Methos
4.1. Adenovirus Generation
4.2. Calcium-Dependent ATPase Activity Assay
4.3. Cardiomyocyte Isolation and Physiology
4.4. Human Heart Samples
4.5. Immunoblotting
4.6. Immunoprecipitation
4.7. In Vitro Acetylation Assay
4.8. Mass Spectrometry Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bragazzi, N.L.; Zhong, W.; Shu, J.; Abu Much, A.; Lotan, D.; Grupper, A.; Younis, A.; Dai, H. Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur. J. Prev. Cardiol. 2021, 28, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Eur. J. Heart Fail 2020, 22, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Hasenfuss, G.; Reinecke, H.; Studer, R.; Meyer, M.; Pieske, B.; Holtz, J.; Holubarsch, C.; Posival, H.; Just, H.; Drexler, H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994, 75, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, F.; Williams, E.; Lebeche, D.; Schmidt, U.; Rosenzweig, A.; Gwathmey, J.K.; Lewandowski, E.D.; Hajjar, R.J. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca2+-ATPase in a rat model of heart failure. Circulation 2001, 104, 1424–1429. [Google Scholar] [CrossRef]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef]
- Lyon, A.R.; Bannister, M.L.; Collins, T.; Pearce, E.; Sepehripour, A.H.; Dubb, S.S.; Garcia, E.; O’Gara, P.; Liang, L.; Kohlbrenner, E.; et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythmia Electrophysiol. 2011, 4, 362–372. [Google Scholar] [CrossRef]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef]
- Park, J.H.; Kho, C. MicroRNAs and Calcium Signaling in Heart Disease. Int. J. Mol. Sci. 2021, 22, 10582. [Google Scholar] [CrossRef]
- Dan, G.A. Thyroid hormones and the heart. Heart Fail Rev. 2016, 21, 357–359. [Google Scholar] [CrossRef]
- Lee, A.; Oh, J.G.; Gorski, P.A.; Hajjar, R.J.; Kho, C. Post-translational Modifications in Heart Failure: Small Changes, Big Impact. Heart Lung Circ. 2016, 25, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Beltrao, P.; Bork, P.; Krogan, N.J.; van Noort, V. Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 2013, 22, 714. [Google Scholar] [CrossRef]
- Huang, K.Y.; Lee, T.Y.; Kao, H.J.; Ma, C.T.; Lee, C.C.; Lin, T.H.; Chang, W.C.; Huang, H.D. dbPTM in 2019: Exploring disease association and cross-talk of post-translational modifications. Nucleic Acids Res. 2019, 47, D298–D308. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, U.D.; Besch, H.R., Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar] [CrossRef]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef]
- Viner, R.I.; Ferrington, D.A.; Williams, T.D.; Bigelow, D.J.; Schöneich, C. Protein modification during biological aging: Selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem. J. 1999, 340, 657–669. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef]
- Lee, A.; Jeong, D.; Mitsuyama, S.; Oh, J.G.; Liang, L.; Ikeda, Y.; Sadoshima, J.; Hajjar, R.J.; Kho, C. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid Redox Signal. 2014, 21, 1986–2001. [Google Scholar] [CrossRef]
- Tilemann, L.; Lee, A.; Ishikawa, K.; Aguero, J.; Rapti, K.; Santos-Gallego, C.; Kohlbrenner, E.; Fish, K.M.; Kho, C.; Hajjar, R.J. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci. Transl. Med. 2013, 5, 211ra159. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; DeVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Yanazume, T.; Hasegawa, K.; Morimoto, T.; Kawamura, T.; Wada, H.; Matsumori, A.; Kawase, Y.; Hirai, M.; Kita, T. Cardiac p300 is involved in myocyte growth with decompensated heart failure. Mol. Cell. Biol. 2003, 23, 3593–3606. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.Q.; Shehadeh, L.A.; Mitrani, J.M.; Pessanha, M.; Slepak, T.I.; Webster, K.A.; Bishopric, N.H. Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation 2008, 118, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1602–H1609. [Google Scholar] [CrossRef] [PubMed]
- Gorski, P.A.; Jang, S.P.; Jeong, D.; Lee, A.; Lee, P.; Oh, J.G.; Chepurko, V.; Yang, D.K.; Kwak, T.H.; Eom, S.H.; et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca2+-ATPase in Heart Failure. Circ. Res. 2019, 124, e63–e80. [Google Scholar] [CrossRef]
- Andersson, K.B.; Birkeland, J.A.; Finsen, A.V.; Louch, W.E.; Sjaastad, I.; Wang, Y.; Chen, J.; Molkentin, J.D.; Chien, K.R.; Sejersted, O.M.; et al. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J. Mol. Cell. Cardiol. 2009, 47, 180–187. [Google Scholar] [CrossRef]
- Li, P.; Ge, J.; Li, H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 96–115. [Google Scholar] [CrossRef]
- Sabari, B.R.; Tang, Z.; Huang, H.; Yong-Gonzalez, V.; Molina, H.; Kong, H.E.; Dai, L.; Shimada, M.; Cross, J.R.; Zhao, Y.; et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 2015, 58, 203–215. [Google Scholar] [CrossRef]
- Wang, Y.; Tu, K.; Liu, D.; Guo, L.; Chen, Y.; Li, Q.; Maiers, J.L.; Liu, Z.; Shah, V.H.; Dou, C.; et al. p300 Acetyltransferase Is a Cytoplasm-to-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor β-Stimulated Hepatic Stellate Cells. Hepatology 2019, 70, 1409–1423. [Google Scholar] [CrossRef]
- Dai, Y.S.; Markham, B.E. p300 Functions as a coactivator of transcription factor GATA-4. J. Biol. Chem. 2001, 276, 37178–37185. [Google Scholar] [CrossRef] [Green Version]
- Slepak, T.I.; Webster, K.A.; Zang, J.; Prentice, H.; O’Dowd, A.; Hicks, M.N.; Bishopric, N.H. Control of cardiac-specific transcription by p300 through myocyte enhancer factor-2D. J. Biol. Chem. 2001, 276, 7575–7585. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Yuan, W.; Mori, Y.; Varga, J. Smad-dependent stimulation of type I collagen gene expression in human skin fibroblasts by TGF-beta involves functional cooperation with p300/CBP transcriptional coactivators. Oncogene 2000, 19, 3546–3555. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kawamura, T.; Morimoto, T.; Ono, K.; Wada, H.; Kawase, Y.; Matsumori, A.; Nishio, R.; Kita, T.; Hasegawa, K. Histone acetyltransferase activity of p300 is required for the promotion of left ventricular remodeling after myocardial infarction in adult mice in vivo. Circulation 2006, 113, 679–690. [Google Scholar] [CrossRef]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, A.K. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell. Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef]
- Morimoto, T.; Sunagawa, Y.; Kawamura, T.; Takaya, T.; Wada, H.; Nagasawa, A.; Komeda, M.; Fujita, M.; Shimatsu, A.; Kita, T.; et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J. Clin. Investig. 2008, 118, 868–878. [Google Scholar] [CrossRef]
- Rai, R.; Verma, S.K.; Kim, D.; Ramirez, V.; Lux, E.; Li, C.; Sahoo, S.; Wilsbacher, L.D.; Vaughan, D.E.; Quaggin, S.E.; et al. A novel acetyltransferase p300 inhibitor ameliorates hypertension-associated cardio-renal fibrosis. Epigenetics 2017, 12, 1004–1013. [Google Scholar] [CrossRef]
- Foster, D.B.; Liu, T.; Rucker, J.; O’Meally, R.N.; Devine, L.R.; Cole, R.N.; O’Rourke, B. The cardiac acetyl-lysine proteome. PLoS ONE 2013, 8, e67513. [Google Scholar] [CrossRef]
- Sitsel, A.; De Raeymaecker, J.; Drachmann, N.D.; Derua, R.; Smaardijk, S.; Andersen, J.L.; Vandecaetsbeek, I.; Chen, J.; De Maeyer, M.; Waelkens, E.; et al. Structures of the heart specific SERCA2a Ca(2+)-ATPase. EMBO J. 2019, 38, e100020. [Google Scholar] [CrossRef]
- Zhang, Y.; Inaba, K. Structural basis of the conformational and functional regulation of human SERCA2b, the ubiquitous endoplasmic reticulum calcium pump. Bioessays 2022, 44, e2200052. [Google Scholar] [CrossRef]
- Morita, T.; Hussain, D.; Asahi, M.; Tsuda, T.; Kurzydlowski, K.; Toyoshima, C.; Maclennan, D.H. Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca(2+)-ATPase. Biochem. Biophys. Res. Commun. 2008, 369, 188–194. [Google Scholar] [CrossRef]
- Akin, B.L.; Jones, L.R. Characterizing phospholamban to sarco(endo)plasmic reticulum Ca2+-ATPase 2a (SERCA2a) protein binding interactions in human cardiac sarcoplasmic reticulum vesicles using chemical cross-linking. J. Biol. Chem. 2012, 287, 7582–7593. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, C.; Zhang, Y.; Xu, Y.; Zhang, S.; Liu, Z.; Xue, Y. GPS-PAIL: Prediction of lysine acetyltransferase-specific modification sites from protein sequences. Sci. Rep. 2016, 6, 39787. [Google Scholar] [CrossRef] [PubMed]
- Makarewich, C.A.; Munir, A.Z.; Schiattarella, G.G.; Bezprozvannaya, S.; Raguimova, O.N.; Cho, E.E.; Vidal, A.H.; Robia, S.L.; Bassel-Duby, R.; Olson, E.N. The dworf micropeptide enhances contractility and prevents heart failure in a mouse model of dilated cardiomyopathy. eLife 2018, 7, e38319. [Google Scholar] [CrossRef] [PubMed]
- Vangheluwe, P.; Raeymaekers, L.; Dode, L.; Wuytack, F. Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: Cell biological implications. Cell Calcium 2005, 38, 291–302. [Google Scholar] [CrossRef]
- Gorski, P.A.; Kho, C.; Oh, J.G. Measuring Cardiomyocyte Contractility and Calcium Handling In Vitro. Methods Mol. Biol. 2018, 1816, 93–104. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acetylated Lysine | Peptide Sequence | Detected Monoisotopic Mass (m/z) | Actual Mass (m/z) | Delta Da | n |
|---|---|---|---|---|---|
| K128 | EYEPEMGK*VYR | 729.83 | 1457.65 | 0.0024 | 3 |
| K169 | LTSIK*STTLR | 581.35 | 1160.68 | 0.0018 | 3 |
| K205 | K*NMLFSGTNIAAGK | 755.39 | 1508.77 | 0.0027 | 3 |
| K328 | MAK*KNAIVR | 565.82 | 1129.63 | 0.00082 | 2 |
| K329 | MAKK*NAIVR | 565.82 | 1129.63 | 0.00082 | 2 |
| K476 | ANACNSVIK*QLMK | 767.89 | 1533.77 | 0.0031 | 2 |
| K492 | K*SMSVYCTPNKPSR | 856.91 | 1711.80 | 0.0027 | 3 |
| K502 | KSMSVYCTPNK*PSR | 856.91 | 1711.80 | 0.0027 | 2 |
| K510 | TSMSK*MFVK | 566.77 | 1131.53 | 0.0019 | 4 |
| K514 | MFVK*GAPEGVIDR | 738.88 | 1475.75 | 0.0042 | 4 |
| K533 | VGSTK*VPMTPGVK | 679.87 | 1357.73 | −0.00061 | 3 |
| K541 | VPMTPGVK*QK | 571.82 | 1141.62 | 0.0014 | 2 |
| K712 | K*SEIGIAMGSGTAVAK | 789.42 | 1576.82 | 0.0029 | 4 |
| SERCA2a Form | Vmax (%) | KCa (µM) | nH | N | |||
|---|---|---|---|---|---|---|---|
| AVR | SEM | AVR | SEM | AVR | SEM | ||
| WT | 100.0 | 3.7 | 0.45 | 0.0 | 1.3 | 0.2 | 5 |
| K328Q | 53.7 | 24 | 0.47 | 0.0 | 1.5 | 0.1 | 3 |
| K329Q | 59.2 | 14 | 0.44 | 0.1 | 1.8 | 0.3 | 3 |
| K492Q | 30.0 | 1.7 | 0.25 | 0.0 | 1.8 | 0.4 | 3 |
| K510Q | 100.7 | 6.0 | 0.43 | 0.1 | 1.3 | 0.3 | 3 |
| K514Q | 44.6 | 4.0 | 0.48 | 0.1 | 1.4 | 0.4 | 3 |
| K712Q | 75.6 | 17 | 0.40 | 0.0 | 1.3 | 0.0 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorski, P.A.; Lee, A.; Lee, P.; Oh, J.G.; Vangheluwe, P.; Ishikawa, K.; Hajjar, R.; Kho, C. Identification and Characterization of p300-Mediated Lysine Residues in Cardiac SERCA2a. Int. J. Mol. Sci. 2023, 24, 3502. https://doi.org/10.3390/ijms24043502
Gorski PA, Lee A, Lee P, Oh JG, Vangheluwe P, Ishikawa K, Hajjar R, Kho C. Identification and Characterization of p300-Mediated Lysine Residues in Cardiac SERCA2a. International Journal of Molecular Sciences. 2023; 24(4):3502. https://doi.org/10.3390/ijms24043502
Chicago/Turabian StyleGorski, Przemek A., Ahyoung Lee, Philyoung Lee, Jae Gyun Oh, Peter Vangheluwe, Kiyotake Ishikawa, Roger Hajjar, and Changwon Kho. 2023. "Identification and Characterization of p300-Mediated Lysine Residues in Cardiac SERCA2a" International Journal of Molecular Sciences 24, no. 4: 3502. https://doi.org/10.3390/ijms24043502