
The Potential Role of MiRs-139-5p and -454-3p in Endoglin-Knockdown-Induced Angiogenic Dysfunction in HUVECs

Abstract
:1. Introduction
2. Results
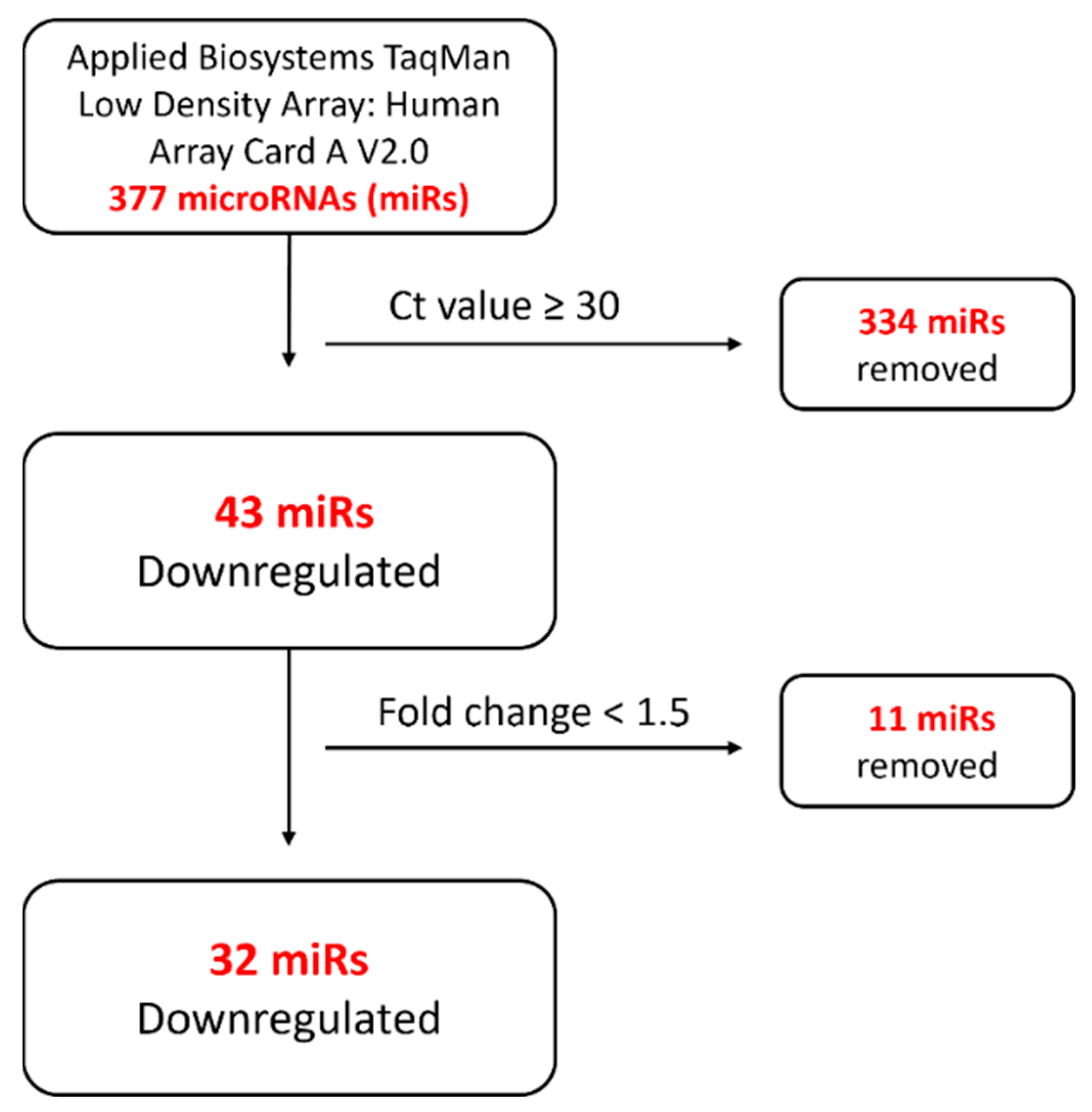
2.1. ENG-Knockdown HUVECs Demonstrated a Potentially Dysregulated MiRNA Profile
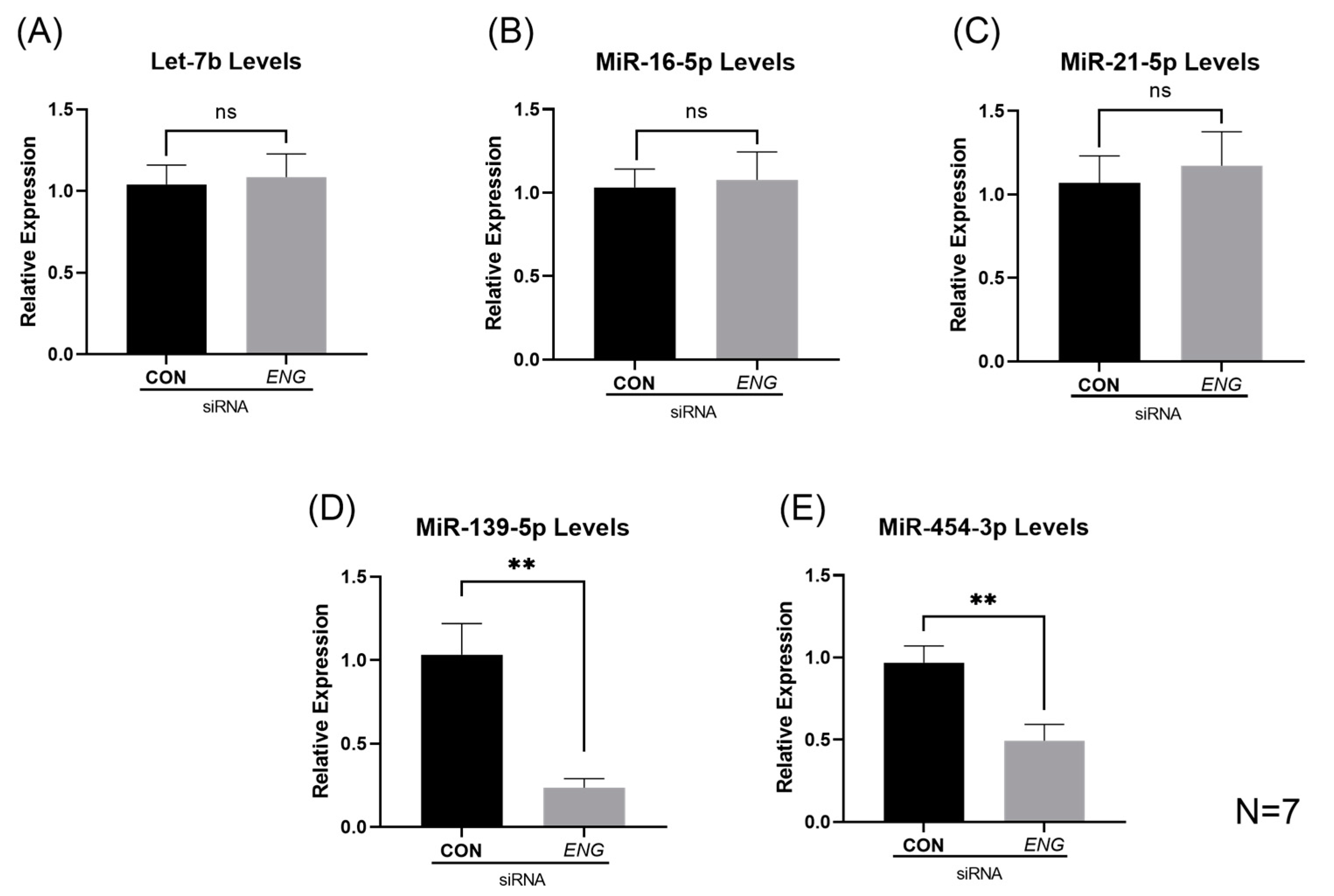
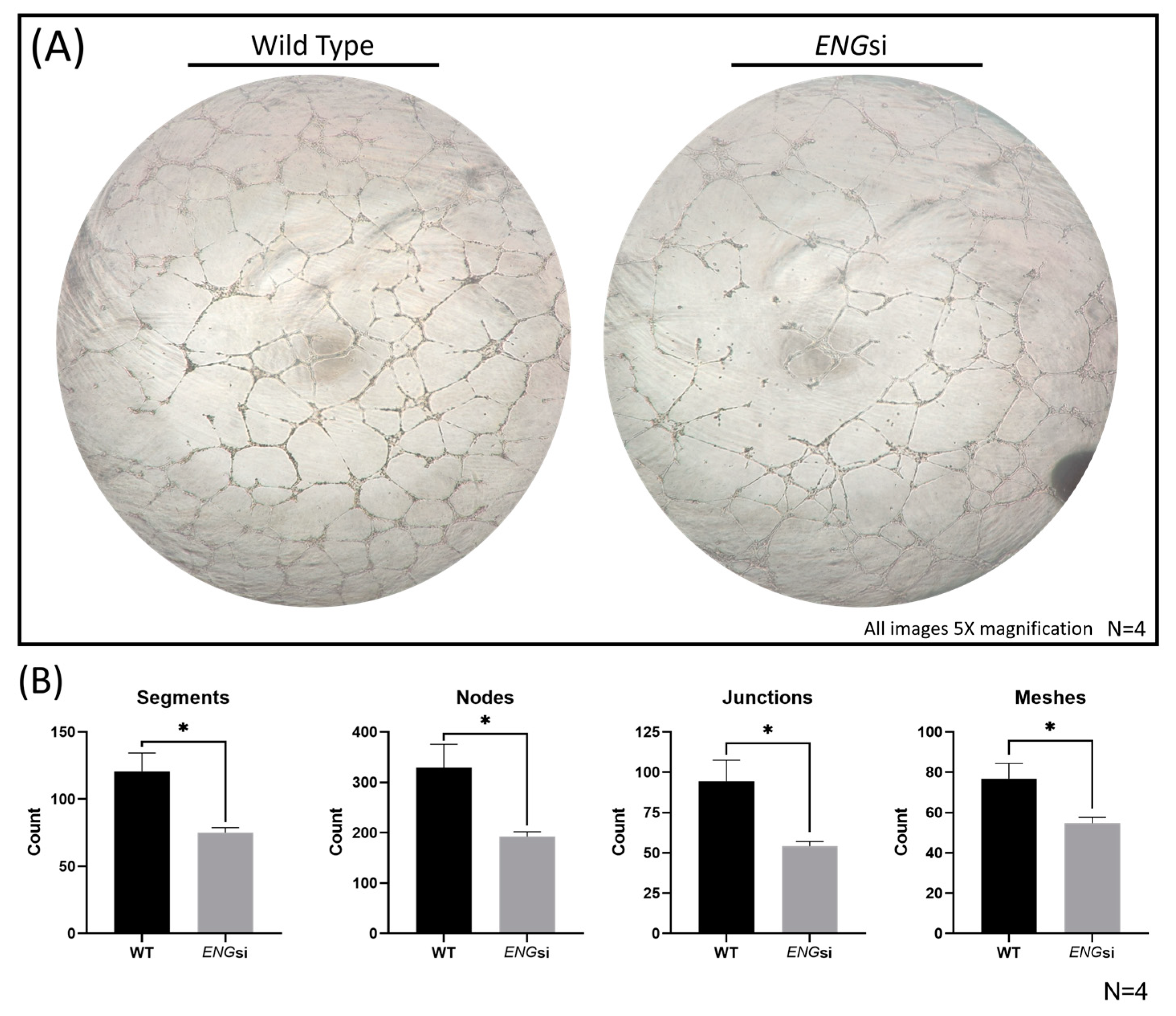
2.2. Significantly Reduced Levels of MiRs-139-5p and -454-3p in ENG-Knockdown HUVECs
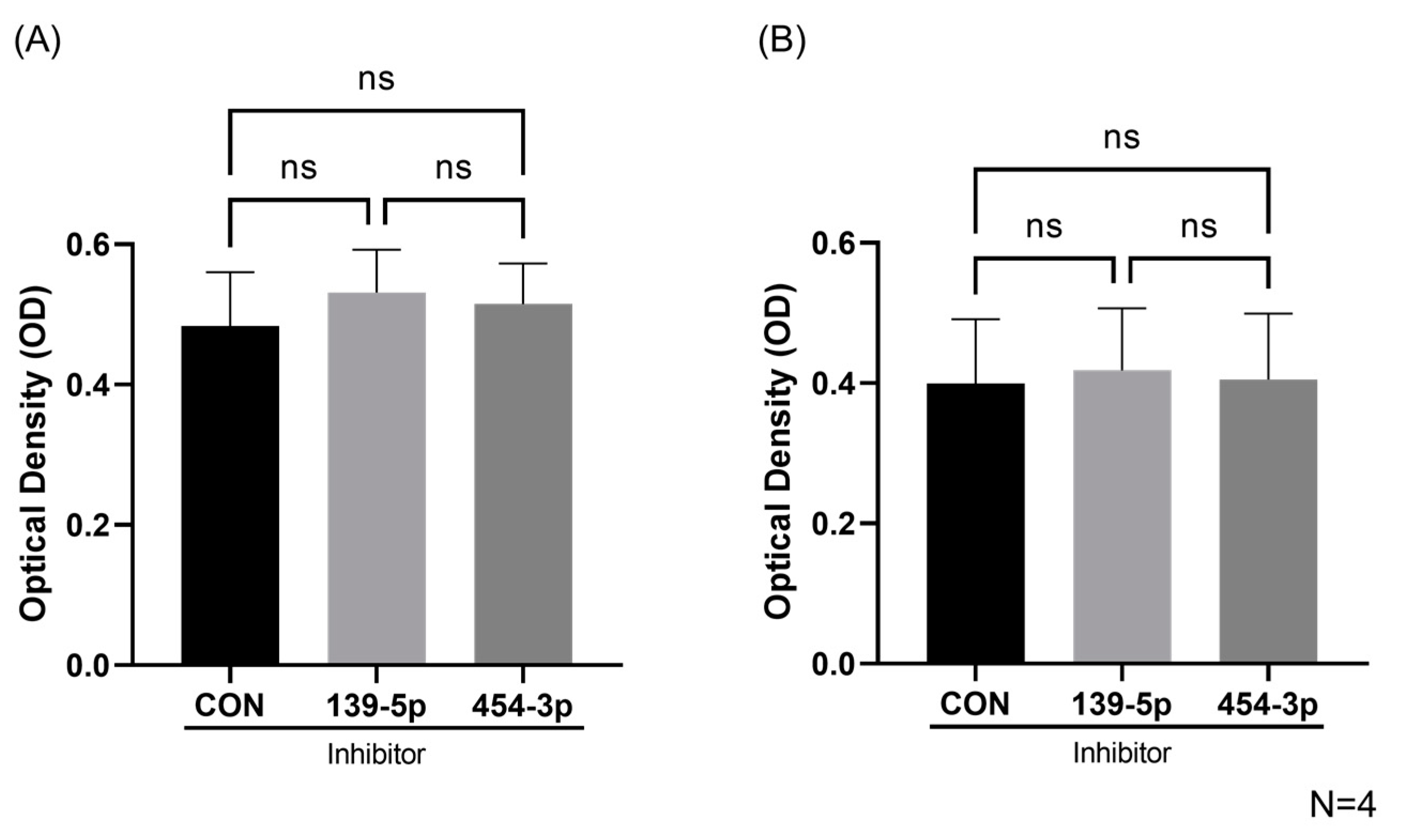
2.3. Inhibition of MiR-139-5p or MiR-454-3p Had No Effect on HUVEC Viability and Proliferation
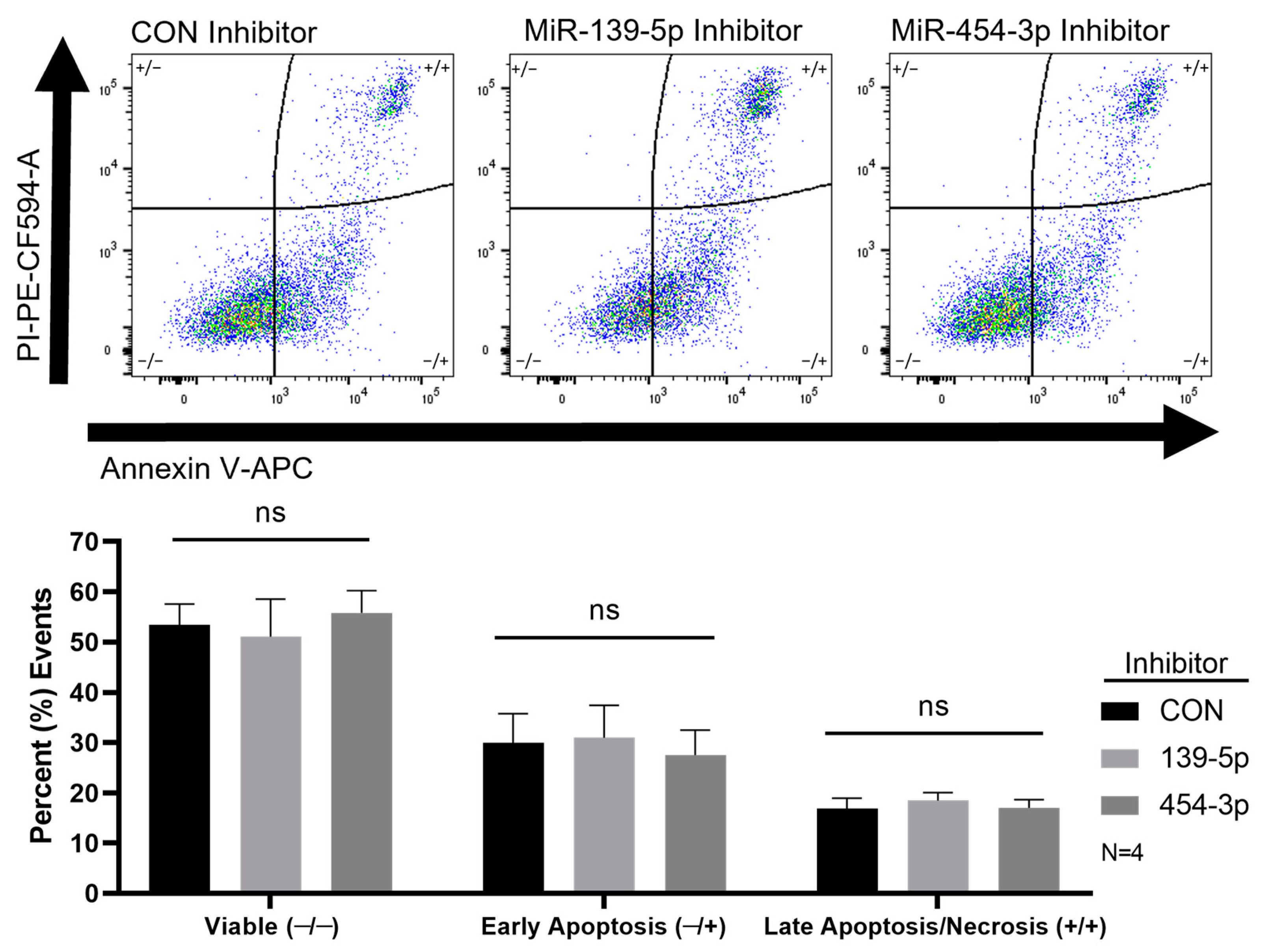
2.4. Inhibition of MiR-139-5p or MiR-454-3p Had No Effect on HUVEC Apoptosis
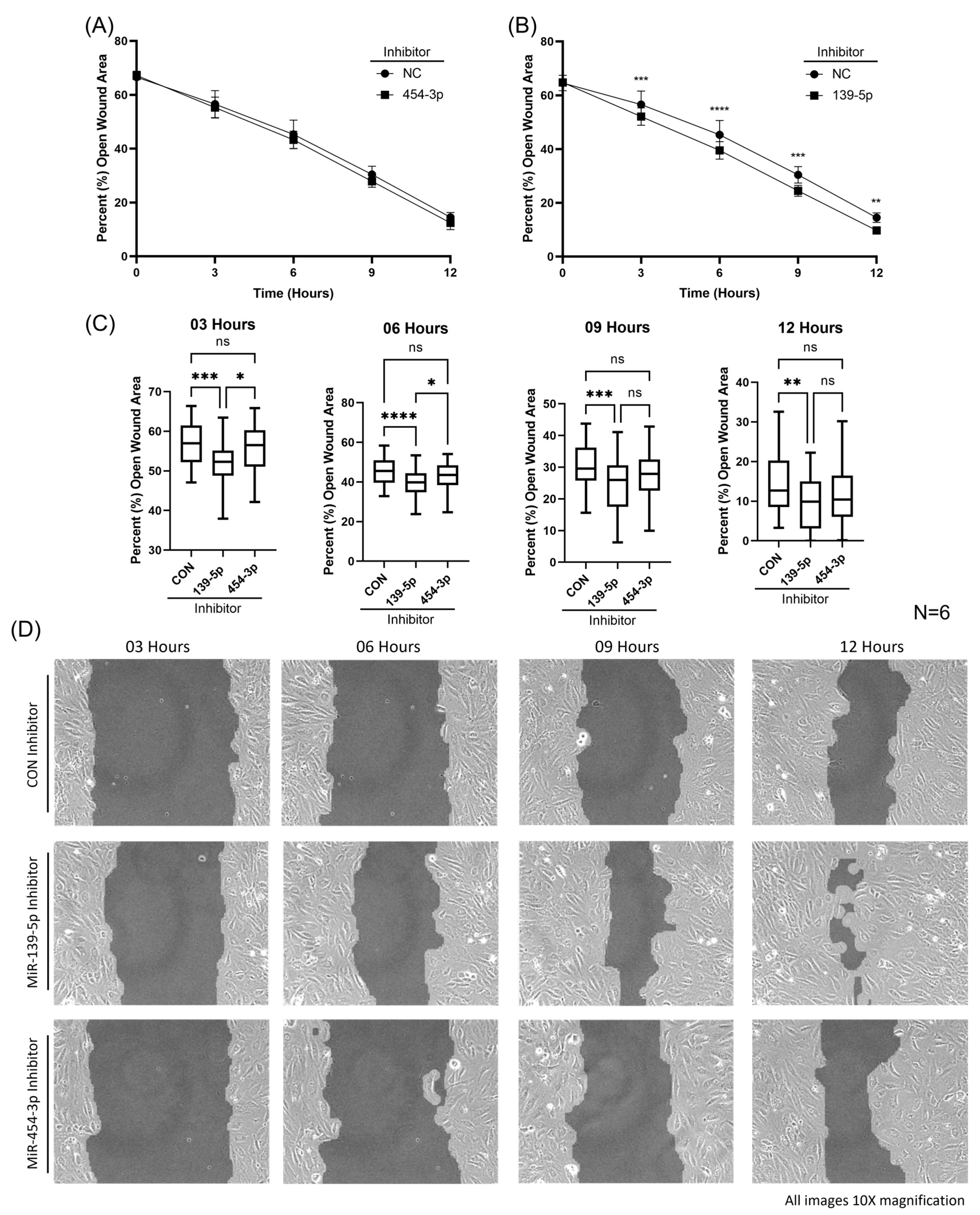
2.5. Inhibition of MiR-139-5p Augmented HUVEC Migration
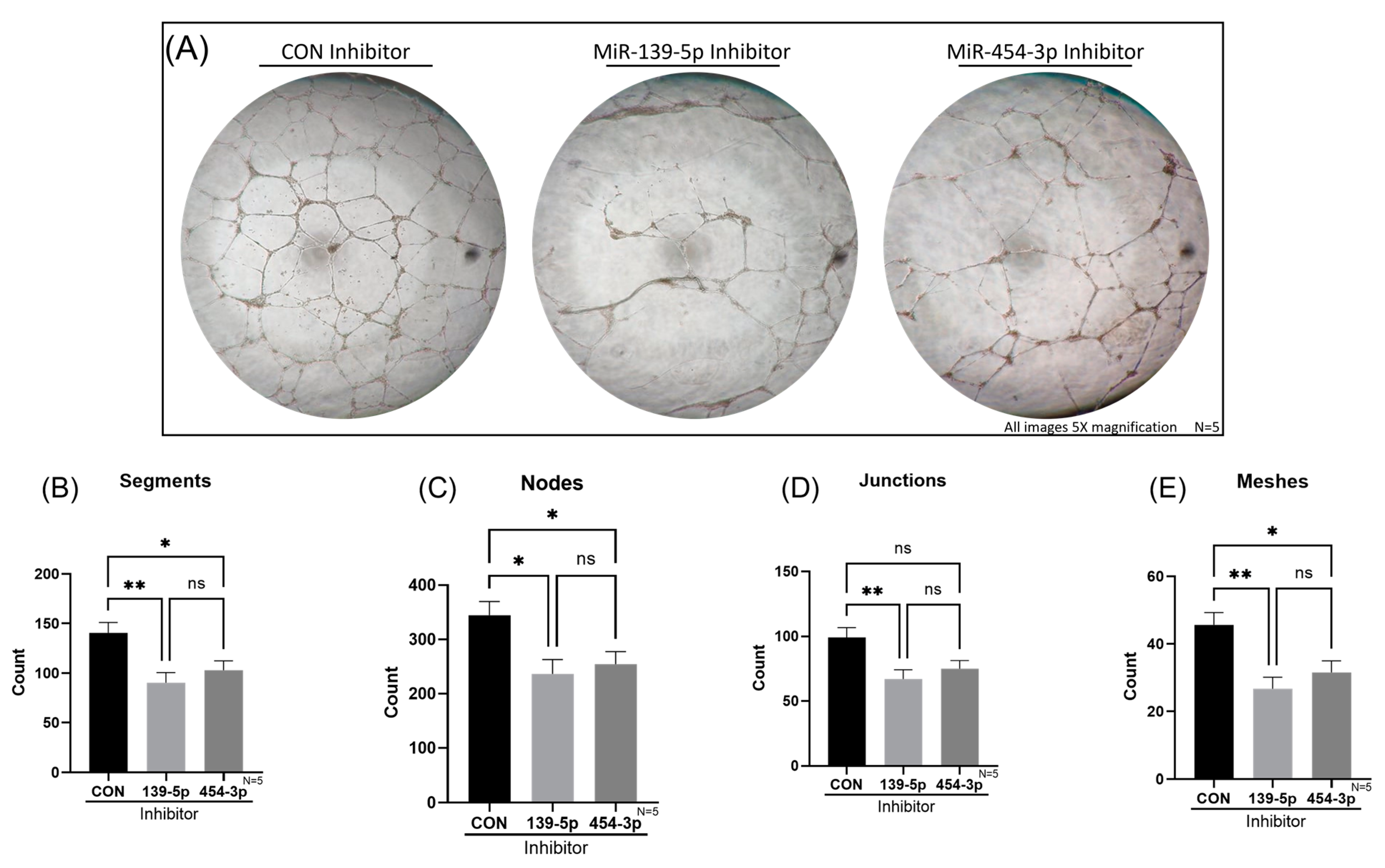
2.6. Reduction of MiR-139-5p or MiR-454-3p Impaired Tube Formation of HUVECs In Vitro
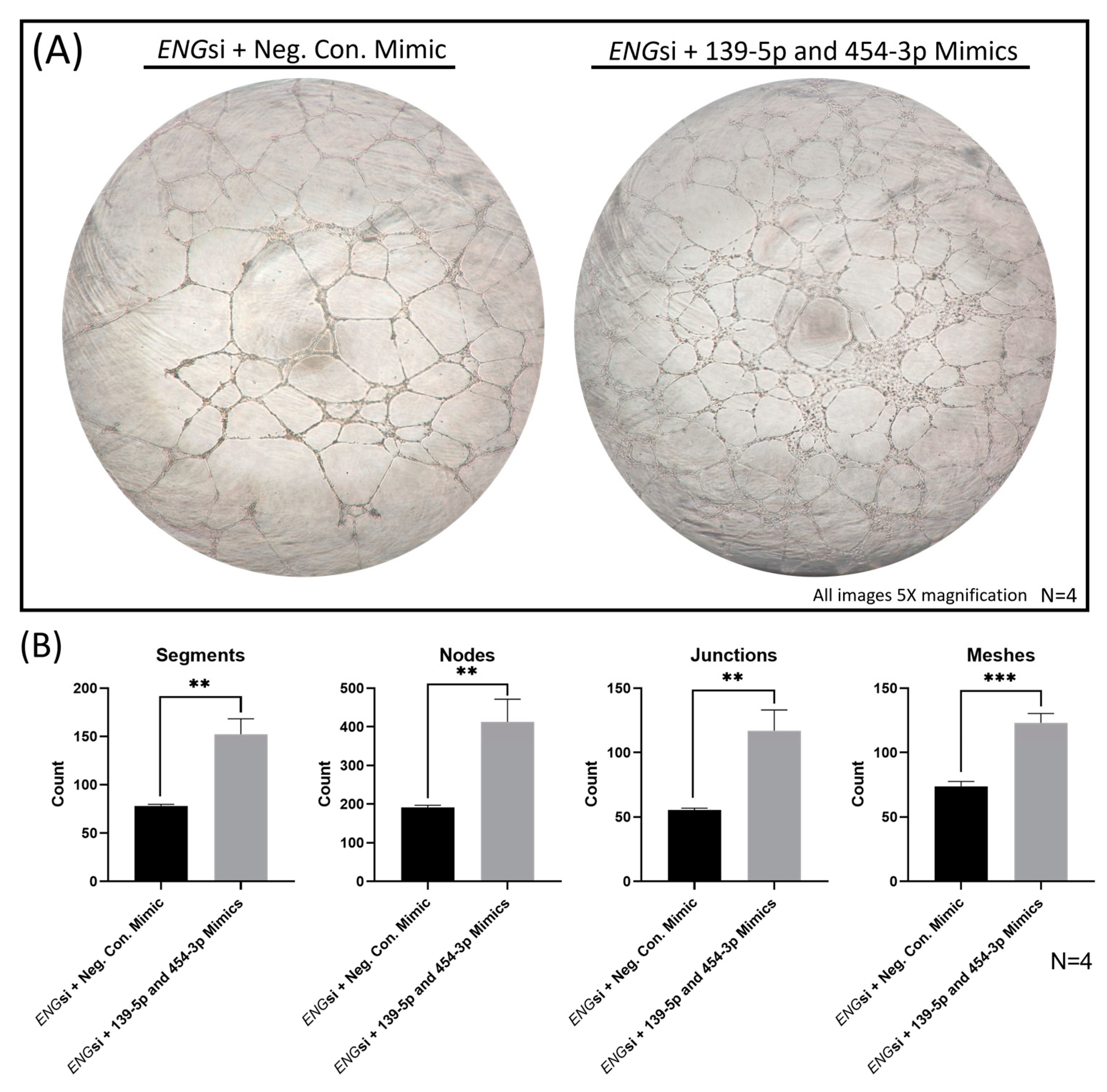
2.7. Overexpression of MiRs-139-5p and -454-3p Rescued ENG-knockdown-Induced HUVEC Dysfunction
3. Discussion
4. Materials and Methods
4.1. Culture of HUVECs
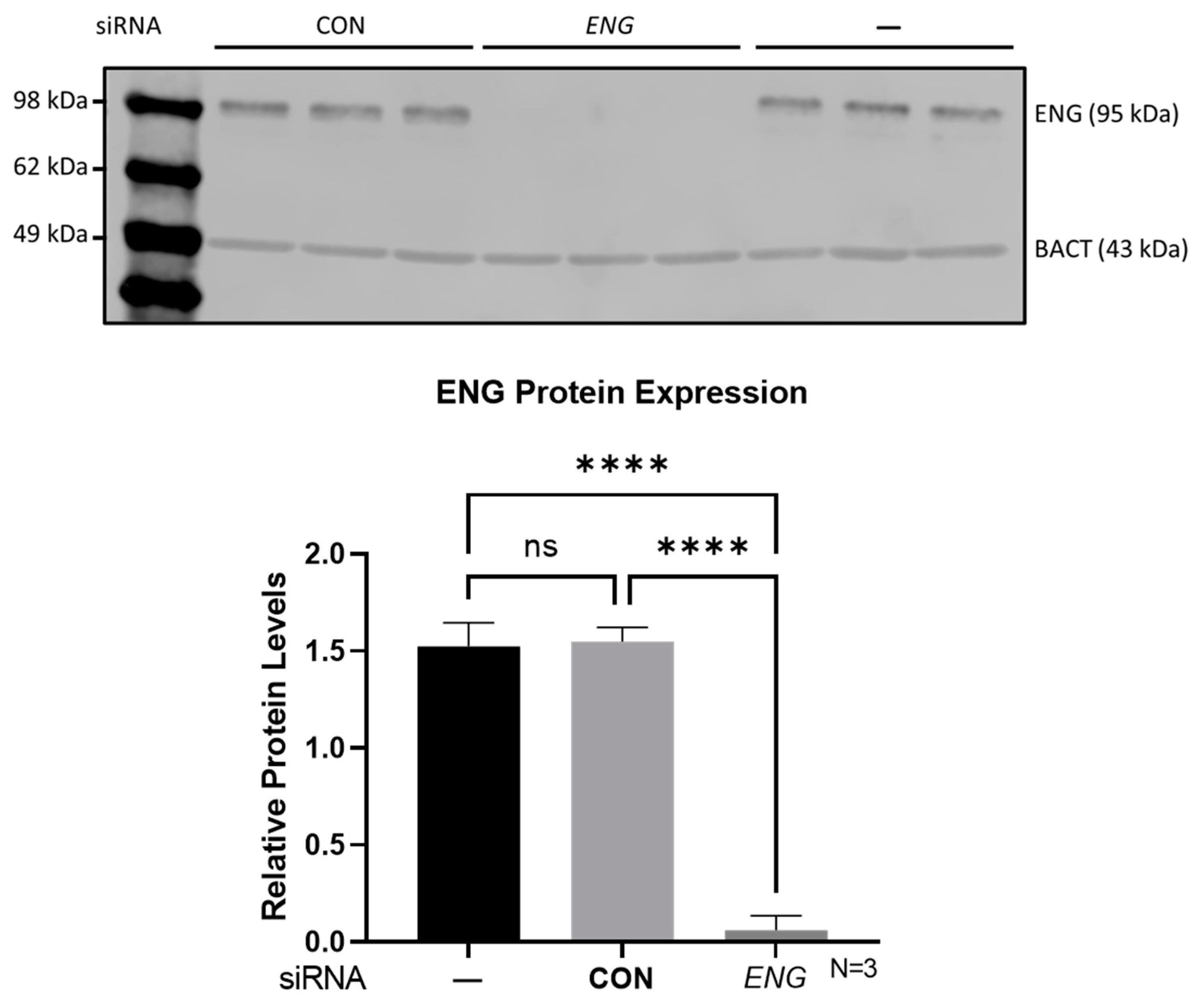
4.2. ENG Short Interfering RNA (siRNA) Transfection
4.3. Western Blotting
4.4. Total RNA Isolation from HUVECs
4.5. MiRNA Microarray of Total RNA from HUVECs
4.6. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR) for MiRNA Validation
4.7. MirVana miRNA Inhibitor or Mimic Transfection
4.8. Cell Viability and Proliferation Assay
4.9. Apoptosis Assay
4.10. Cellular Migration Assay
4.11. Tube Formation Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Govani, F.S.; Shovlin, C.L. Hereditary Haemorrhagic Telangiectasia: A Clinical and Scientific Review. Eur. J. Hum. 2009, 17, 860–871. [Google Scholar] [CrossRef] [Green Version]
- Guttmacher, A.E.; Marchuk, D.A.; White, R.I. Hereditary Hemorrhagic Telangiectasia. NEJM 1995, 333, 918–924. [Google Scholar] [CrossRef]
- Braverman, I.M.; Keh, A.; Jacobson, B.S. Ultrastructure and Three-Dimensional Organization of the Telangiectases of Hereditary Hemorrhagic Telangiectasia. J. Investig. Dermatol. 1990, 95, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Sharathkumar, A.A.; Shapiro, A. Hereditary Haemorrhagic Telangiectasia. Haemophilia 2008, 14, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L. Hereditary Haemorrhagic Telangiectasia: Pathophysiology, Diagnosis and Treatment. Blood Rev. 2010, 24, 203–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis-Girod, S.; Bailly, S.; Plauchu, H. Hereditary Hemorrhagic Telangiectasia: From Molecular Biology to Patient Care. J. Thromb. Haemost. 2010, 8, 1447–1456. [Google Scholar] [CrossRef]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J. Endoglin, a TGF-Beta Binding Protein of Endothelial Cells, Is the Gene for Hereditary Haemorrhagic Telangiectasia Type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the Activin Receptor-Like Kinase 1 Gene in Hereditary Haemorrhagic Telangiectasia Type 2. Nat. Gen. 1996, 13, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, É.; Westermann, C.J.J.; Marchuk, D.A. A Combined Syndrome of Juvenile Polyposis and Hereditary Haemorrhagic Telangiectasia Associated with Mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef]
- Hernandez, F.; Huether, R.; Carter, L.; Johnston, T.; Thompson, J.; Gossage, J.R.; Chao, E.; Elliott, A.M. Mutations in RASA1 and GDF2 Identified in Patients with Clinical Features of Hereditary Hemorrhagic Telangiectasia. Hum. Genome Var. 2015, 2, 15040. [Google Scholar] [CrossRef]
- Sánchez-Martínez, R.; Iriarte, A.; Mora-Luján, J.M.; Patier, J.L.; López-Wolf, D.; Ojeda, A.; Torralba, M.A.; Juyol, M.C.; Gil, R.; Anõn, S.; et al. Current HHT Genetic Overview in Spain and Its Phenotypic Correlation: Data from RiHHTa Registry. Orphanet J. Rare Dis. 2020, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Lesca, G.; Burnichon, N.; Raux, G.; Tosi, M.; Pinson, S.; Marion, M.J.; Babin, E.; Gilbert-Dussardier, B.; Rivière, S.; Goizet, C.; et al. Distribution of ENG and ACVRL1 (ALK1) Mutations in French HHT Patients. Hum. Mutat. 2006, 27, 598. [Google Scholar] [CrossRef] [PubMed]
- Schulte, C.; Geisthoff, U.; Lux, A.; Kupka, S.; Zenner, H.P.; Blin, N.; Pfister, M. High Frequency of ENG and ALK1/ACVRL1 Mutations in German HHT Patients. Hum. Mutat. 2005, 25, 595. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.; Aylsworth, A.S.; Beis, J.; Berk, T.; Bernhardt, B.; Clark, R.D.; Clericuzio, C.; Danesino, C.; Drautz, J.; Fahl, J.; et al. Overlapping Spectra of SMAD4 Mutations in Juvenile Polyposis (JP) and JP-HHT Syndrome. Am. J. Med. Genet. Part A 2010, 152, 333–339. [Google Scholar] [CrossRef]
- McDonald, J.; Wooderchak-Donahue, W.; VanSant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary Hemorrhagic Telangiectasia: Genetics and Molecular Diagnostics in a New Era. Front. Genet. 2015, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, Y.; Wang, Q.; Su, B.; Xu, H.; Sun, Y.; Sun, P.; Li, R.; Peng, X.; Cai, J. Role of RASA1 in Cancer: A Review and Update (Review). Oncol. Rep. 2020, 44, 2386–2396. [Google Scholar] [CrossRef]
- Tual-Chalot, S.; Oh, S.P.; Arthur, H.M. Mouse Models of Hereditary Hemorrhagic Telangiectasia: Recent Advances and Future Challenges. Front. Genet. 2015, 6, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Crist, A.M.; Lee, A.R.; Patel, N.R.; Westhoff, D.E.; Meadows, S.M. Vascular Deficiency of Smad4 Causes Arteriovenous Malformations: A Mouse Model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis 2018, 21, 363–380. [Google Scholar] [CrossRef] [Green Version]
- Çakmak, H.A.; Demir, M. Microrna and Cardiovascular Diseases. Balk. Med. J. 2020, 37, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional Regulation of the Heterochronic Gene Lin-14 by Lin-4 Mediates Temporal Pattern Formation in C. Elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An Overview of MicroRNAs: Biology, Functions, Therapeutics, and Analysis Methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, N.; Socci, N.D. Computational Identification of MicroRNA Targets. Dev. Biol. 2004, 267, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Fabbri, M.; Cimmino, A.; Calin, G.A.; Croce, C.M. MicroRNA Expression and Function in Cancer. Trends Mol. Med. 2006, 12, 580–587. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Cannavicci, A.; Zhang, Q.; Kutryk, M.J.B. Non-Coding RNAs and Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 3333. [Google Scholar] [CrossRef]
- Ha, T.Y. MicroRNAs in Human Diseases: From Cancer to Cardiovascular Disease. Immune Netw. 2011, 11, 135–154. [Google Scholar] [CrossRef] [Green Version]
- Sohel, M.M.H. Circulating MicroRNAs as Biomarkers in Cancer Diagnosis. Life Sci. 2020, 248, 117473. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for MicroRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [Green Version]
- Sugden, W.W.; Siekmann, A.F. Endothelial Cell Biology of Endoglin in Hereditary Hemorrhagic Telangiectasia. Curr. Opin. Hematol. 2018, 25, 237–244. [Google Scholar] [CrossRef]
- Gu, Y.; Ampofo, E.; Menger, M.D.; Laschke, M.W. MiR-191 Suppresses Angiogenesis by Activation of NF-κB Signaling. FASEB J. 2017, 31, 3321–3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Liang, P.; Zhang, P.; Zhang, M.; Huang, X. The Long Noncoding RNA PDK1-AS/miR-125b-5p/VEGFA Axis Modulates Human Dermal Microvascular Endothelial Cell And Human Umbilical Vein Endothelial Cell Angiogenesis After Thermal Injury. J. Cell. Physiol. 2021, 236, 3129–3142. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, K.S.; Choi, S.; Kim, J.; Lee, D.K.; Park, M.; Park, W.; Kim, T.H.; Hwang, J.Y.; Won, M.-H.; et al. NF-κB-responsive MiRNA-31-5p Elicits Endothelial Dysfunction Associated with Preeclampsia via Down-regulation of Endothelial Nitric-oxide Synthase. J. Biol. Chem. 2018, 293, 18989–19000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.R.; Srivastava, D. MiR-126 Regulates Angiogenic Signaling and Vascular Integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, C.; Cannavicci, A.; Faughnan, M.E.; Kutryk, M.J.B. Endoglin Deficiency Impairs VEGFR2 but Not FGFR1 or TIE2 Activation and Alters VEGF-Mediated Cellular Responses in Human Primary Endothelial Cells. Transl. Res. 2021, 235, 129–143. [Google Scholar] [CrossRef]
- Mahmoud, M.; Allinson, K.R.; Zhai, Z.; Oakenfull, R.; Ghandi, P.; Adams, R.H.; Fruttiger, M.; Arthur, H.M. Pathogenesis of Arteriovenous Malformations in the Absence of Endoglin. Circ. Res. 2010, 106, 1425–1433. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Lopez, A.; Garrido-Martin, E.M.; Sanz-Rodriguez, F.; Pericacho, M.; Rodriguez-Barbero, A.; Eleno, N.; Lopez-Novoa, J.M.; Düwell, A.; Vega, M.A.; Bernabeu, C.; et al. Gene Expression Fingerprinting for Human Hereditary Hemorrhagic Telangiectasia. Hum. Mol. Genet. 2007, 16, 1515–1533. [Google Scholar] [CrossRef] [Green Version]
- Bautch, V.L. Endoglin Moves and Shapes Endothelial Cells. Nat. Cell Biol. 2017, 19, 593–595. [Google Scholar] [CrossRef]
- Fernandez-L, A.; Sanz-Rodriguez, F.; Zarrabeitia, R.; Pérez-Molino, A.; Hebbel, R.P.; Nguyen, J.; Bernabéu, C.; Botella, L.M. Blood Outgrowth Endothelial Cells from Hereditary Haemorrhagic Telangiectasia Patients Reveal Abnormalities Compatible with Vascular Lesions. Cardiovasc. Res. 2005, 68, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Martin, E.M.; Nguyen, H.L.; Cunningham, T.A.; Choe, S.-W.; Jiang, Z.; Arthur, H.M.; Lee, Y.J.; Oh, S.P. Common and Distinctive Pathogenetic Features of Arteriovenous Malformations in Hereditary Hemorrhagic Telangiectasia 1 and Hereditary Hemorrhagic Telangiectasia 2 Animal Models. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2232–2236. [Google Scholar] [CrossRef] [Green Version]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.C.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin Promotes Endothelial Cell Proliferation and TGF-β/ALK1 Signal Transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin Prevents Vascular Malformation by Regulating Flow-Induced Cell Migration and Specification through VEGFR2 Signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez, Y.; Fernández-Hernando, C.; Yu, J.; Gerber, S.A.; Harrison, K.D.; Pober, J.S.; Iruela-Arispe, M.L.; Merkenschlager, M.; Sessa, W.C. Dicer-Dependent Endothelial MicroRNAs Are Necessary for Postnatal Angiogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 14082–14087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butz, H.; Rácz, K.; Hunyady, L.; Patócs, A. Crosstalk between TGF-B Signaling and the MicroRNA Machinery. Trends Pharmacol. Sci. 2012, 33, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I. MicroRNA Control of TGF-β Signaling. . Int. J. Mol. Sci. 2018, 19, 1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.H.; Zhang, Q.S.; Duan, Y.L.; Zhang, J.L.; Li, G.F.; Zheng, D.L. TGF-β Induced MiR-132 Enhances the Activation of TGF-β Signaling through Inhibiting SMAD7 Expression in Glioma Cells. Biochem. Biophys. Res. Commun. 2015, 463, 187–192. [Google Scholar] [CrossRef]
- Zhang, Q.; Kandic, I.; Faughnan, M.E.; Kutryk, M.J. Elevated Circulating MicroRNA-210 Levels in Patients with Hereditary Hemorrhagic Telangiectasia and Pulmonary Arteriovenous Malformations: A Potential New Biomarker. Biomarkers 2013, 18, 23–29. [Google Scholar] [CrossRef]
- Cannavicci, A.; Zhang, Q.; Dai, S.C.; Faughnan, M.E.; Kutryk, M.J.B. Decreased Levels of MiR-28-5p and MiR-361-3p and Increased Levels of Insulin-like Growth Factor 1 MRNA in Mononuclear Cells from Patients with Hereditary Hemorrhagic Telangiectasia. Can. J. Physiol. Pharmacol. 2019, 97, 562–569. [Google Scholar] [CrossRef]
- Cannavicci, A.; Zhang, Q.; Faughnan, M.E.; Kutryk, M.J.B. MicroRNA-132-3p, Downregulated in Myeloid Angiogenic Cells from Hereditary Hemorrhagic Telangiectasia Patients, Is Enriched in the TGFβ and PI3K/AKT Signalling Pathways. Genes 2022, 13, 665. [Google Scholar] [CrossRef]
- Tabruyn, S.P.; Hansen, S.; Ojeda-Fernández, M.L.; Bovy, N.; Zarrabeitia, R.; Recio-Poveda, L.; Bernabéu, C.; Martial, J.A.; Botella, L.M.; Struman, I. MiR-205 Is Downregulated in Hereditary Hemorrhagic Telangiectasia and Impairs TGF-Beta Signaling Pathways in Endothelial Cells. Angiogenesis 2013, 16, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Llorente, L.; Albiñana, V.; Botella, L.M.; Bernabeu, C. Differential Expression of Circulating Plasma MiRNA-370 and MiRNA-10a from Patients with Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 2855. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Klicka, K.; Włodarski, P.K. Regulators at Every Step—How MicroRNAs Drive Tumor Cell Invasiveness and Metastasis. Cancers 2020, 12, 3709. [Google Scholar] [CrossRef]
- Khalili, N.; Nouri-Vaskeh, M.; Hasanpour Segherlou, Z.; Baghbanzadeh, A.; Halimi, M.; Rezaee, H.; Baradaran, B. Diagnostic, Prognostic, and Therapeutic Significance of MiR-139-5p in Cancers. Life Sci. 2020, 256, 117865. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, J.; Zhi, L.; Cai, F. Linc00887 Suppresses Tumorigenesis of Cervical Cancer through Regulating the MiR-454-3p/FRMD6-Hippo Axis. Cancer Cell Int. 2021, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wang, W.C.; Shen, W.H.; Xu, K.; Hu, Y.Y.; Han, G.H.; Liu, Y.B. Thalidomide Suppresses Angiogenesis and Immune Evasion via LncRNA FGD5-AS1/MiR-454-3p/ZEB1 Axis-Mediated VEGFA Expression and PD-1/ PD-L1 Checkpoint in NSCLC. Chem. Biol. Interact. 2021, 349, 109652. [Google Scholar] [CrossRef] [PubMed]
- Aiting, Y.A.N.; Wang, C.; Zheng, L.; Zhou, J.; Zhang, Y. MicroRNA-454-3p Inhibits Cell Proliferation and Invasion in Esophageal Cancer by Targeting Insulin-like Growth Factor 2 MRNA-Binding Protein 1. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, C.; Wang, T.; Sun, L.; Qin, L.; Ju, J. MiR-139-5p Promotes Neovascularization in Diabetic Retinopathy by Regulating the Phosphatase and Tensin Homolog. Arch. Pharm. Res. 2021, 44, 205–218. [Google Scholar] [CrossRef]
- Luo, Y.F.; Wan, X.X.; Zhao, L.L.; Guo, Z.; Shen, R.T.; Zeng, P.Y.; Wang, L.H.; Yuan, J.J.; Yang, W.J.; Yue, C.; et al. MicroRNA-139-5p Upregulation Is Associated with Diabetic Endothelial Cell Dysfunction by Targeting c-Jun. Aging 2021, 13, 1186–1211. [Google Scholar] [CrossRef]
- Papangeli, I.; Kim, J.; Maier, I.; Park, S.; Lee, A.; Kang, Y.; Tanaka, K.; Khan, O.F.; Ju, H.; Kojima, Y.; et al. MicroRNA 139-5p Coordinates APLNR-CXCR4 Crosstalk during Vascular Maturation. Nat. Commun. 2016, 7, 11268. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, B.; Chen, D.; Liu, L.; Huang, C.; Lu, Z.; Lun, L.; Wan, X. MiR-139 and MiR-200c Regulate Pancreatic Cancer Endothelial Cell Migration and Angiogenesis. Oncol. Rep. 2015, 34, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Liao, L.; Yang, Q.; Li, H.; Meng, R.; Li, Y. MiR-454-3p Prevents Ox-LDL-induced Apoptosis in HAECs by Targeting TRPC3. Exp. Ther. Med. 2021, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Cheng, W.; Gao, Y.; Wang, H.; Liu, Z. Microarray Analysis of MicroRNA Expression Patterns in the Semen of Infertile Men with Semen Abnormalities. Mol. Med. 2012, 6, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolov, M.V.; Panyutin, I.V.; Neumann, R.D. Unraveling the Global MicroRNAome Responses to Ionizing Radiation in Human Embryonic Stem Cells. PLoS ONE 2012, 7, e31028. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNAs | Accession | Assay Target Sequence | Fold Decrease |
|---|---|---|---|
| hsa-miR-16-5p | MIMAT0000069 | UAGCAGCACGUAAAUAUUGGCG | 1.8 ± 0.43 |
| hsa-miR-17-5p | MIMAT0000070 | CAAAGUGCUUACAGUGCAGGUAG | 1.6 ± 0.11 |
| hsa-miR-19b-3p | MIMAT0000074 | UGUGCAAAUCCAUGCAAAACUGA | 1.7 ± 0.55 |
| hsa-miR-20a-5p | MIMAT0000075 | UAAAGUGCUUAUAGUGCAGGUAG | 1.6 ± 0.57 |
| hsa-miR-21-5p | MIMAT0000076 | UAGCUUAUCAGACUGAUGUUGA | 2.4 ± 0.88 |
| hsa-miR-24-3p | MIMAT0000080 | UGGCUCAGUUCAGCAGGAACAG | 2.0 ± 1.16 |
| hsa-miR-26a-5p | MIMAT0000082 | UUCAAGUAAUCCAGGAUAGGCU | 1.7 ± 0.45 |
| hsa-miR-28-3p | MIMAT0004502 | CACUAGAUUGUGAGCUCCUGGA | 1.7 ± 0.45 |
| hsa-miR-29a-3p | MIMAT0000086 | UAGCACCAUCUGAAAUCGGUUA | 2.0 ± 0.51 |
| hsa-miR-30b-5p | MIMAT0000420 | UGUAAACAUCCUACACUCAGCU | 1.7 ± 0.46 |
| hsa-miR-30c-5p | MIMAT0000244 | UGUAAACAUCCUACACUCUCAGC | 1.9 ± 0.68 |
| hsa-miR-31-5p | MIMAT0000089 | AGGCAAGAUGCUGGCAUAGCU | 1.9 ± 0.90 |
| hsa-miR-99a-5p | MIMAT0000097 | AACCCGUAGAUCCGAUCUUGUG | 2.0 ± 0.68 |
| hsa-miR-99b-5p | MIMAT0000689 | CACCCGUAGAACCGACCUUGCG | 4.6 ± 1.64 |
| hsa-miR-125b-5p | MIMAT0000423 | UCCCUGAGACCCUAACUUGUGA | 1.8 ± 0.64 |
| hsa-miR-126-3p | MIMAT0000445 | UCGUACCGUGAGUAAUAAUGCG | 2.0 ± 0.37 |
| hsa-miR-139-5p | MIMAT0000250 | UCUACAGUGCACGUGUCUCCAG | 2.1 ± 0.40 |
| hsa-miR-146a-5p | MIMAT0000449 | UGAGAACUGAAUUCCAUGGGUU | 1.9 ± 0.49 |
| hsa-miR-186-5p | MIMAT0000456 | CAAAGAAUUCUCCUUUUGGGCU | 2.5 ± 0.70 |
| hsa-miR-191-5p | MIMAT0000440 | CAACGGAAUCCCAAAAGCAGCUG | 1.7 ± 0.33 |
| hsa-miR-193b-3p | MIMAT0002819 | AACUGGCCCUCAAAGUCCCGCU | 2.0 ± 0.56 |
| hsa-miR-218-5p | MIMAT0000275 | UUGUGCUUGAUCUAACCAUGU | 1.7 ± 0.46 |
| hsa-miR-222-3p | MIMAT0000279 | AGCUACAUCUGGCUACUGGGU | 1.7 ± 0.39 |
| hsa-miR-320a | MI0000542 | AAAAGCUGGGUUGAGAGGGCGA | 1.6 ± 0.26 |
| hsa-let-7b-5p | MIMAT0000063 | UGAGGUAGUAGGUUGUGUGGUU | 3.3 ± 0.60 |
| hsa-miR-324-3p | MIMAT0000762 | ACUGCCCCAGGUGCUGCUGG | 2.1 ± 0.41 |
| hsa-miR-345-5p | MIMAT0000772 | GCUGACUCCUAGUCCAGGGCUC | 2.0 ± 0.73 |
| hsa-miR-454-3p | MIMAT0003885 | UAGUGCAAUAUUGCUUAUAGGGU | 1.5 ± 0.09 |
| hsa-miR-484 | MIMAT0002174 | UCAGGCUCAGUCCCCUCCCGAU | 1.5 ± 0.16 |
| hsa-miR-574-3p | MIMAT0003239 | CACGCUCAUGCACACACCCACA | 1.7 ± 0.22 |
| hsa-miR-590-5p | MIMAT0003258 | GAGCUUAUUCAUAAAAGUGCAG | 1.6 ± 0.12 |
| hsa-miR-376c-3p | MIMAT0000720 | AACAUAGAGGAAAUUCCACGU | 2.0 ± 0.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannavicci, A.; Zhang, Q.; Kutryk, M.J.B. The Potential Role of MiRs-139-5p and -454-3p in Endoglin-Knockdown-Induced Angiogenic Dysfunction in HUVECs. Int. J. Mol. Sci. 2023, 24, 4916. https://doi.org/10.3390/ijms24054916
Cannavicci A, Zhang Q, Kutryk MJB. The Potential Role of MiRs-139-5p and -454-3p in Endoglin-Knockdown-Induced Angiogenic Dysfunction in HUVECs. International Journal of Molecular Sciences. 2023; 24(5):4916. https://doi.org/10.3390/ijms24054916
Chicago/Turabian StyleCannavicci, Anthony, Qiuwang Zhang, and Michael J. B. Kutryk. 2023. "The Potential Role of MiRs-139-5p and -454-3p in Endoglin-Knockdown-Induced Angiogenic Dysfunction in HUVECs" International Journal of Molecular Sciences 24, no. 5: 4916. https://doi.org/10.3390/ijms24054916