
Zinc: From Biological Functions to Therapeutic Potential

Abstract
:1. Introduction
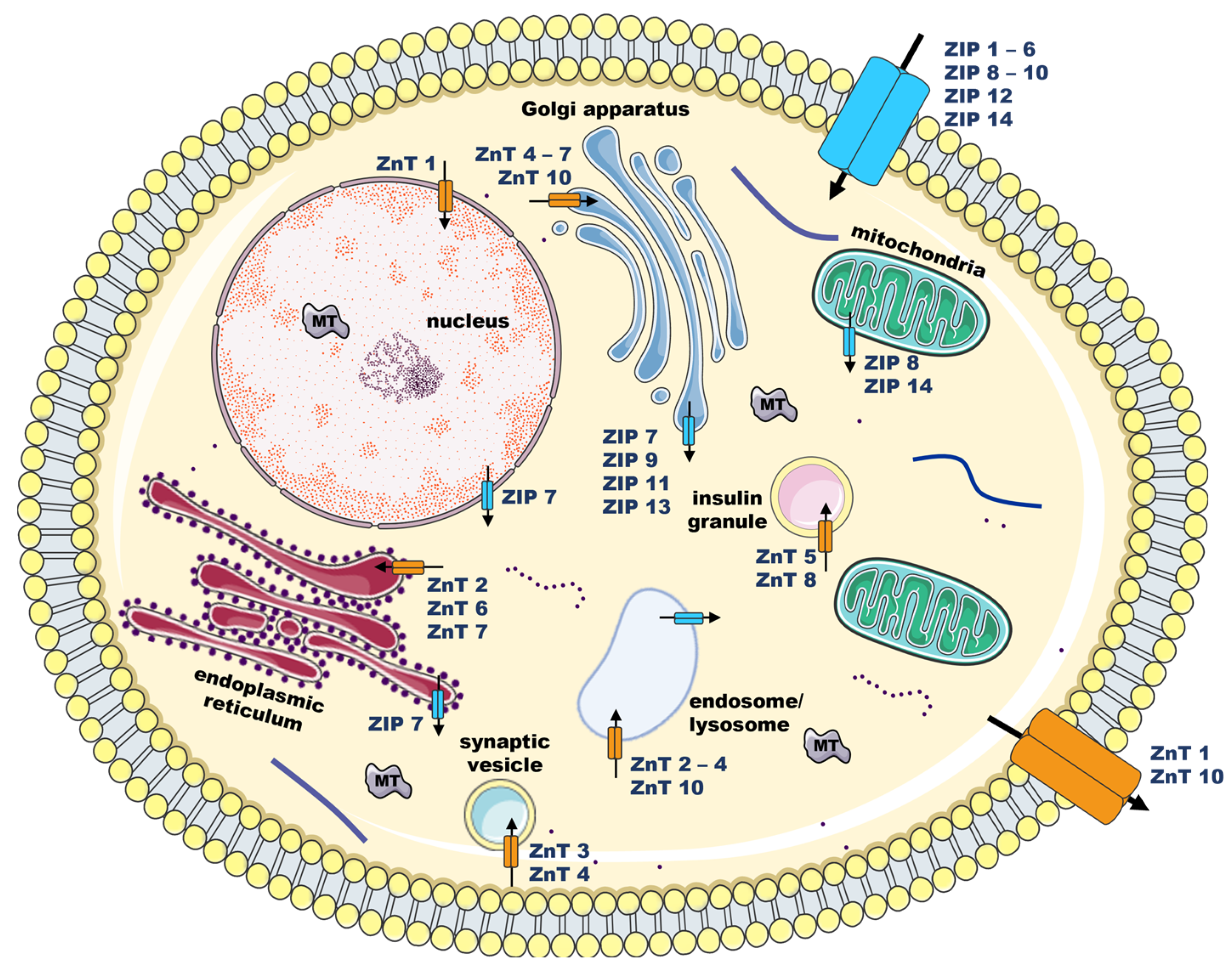
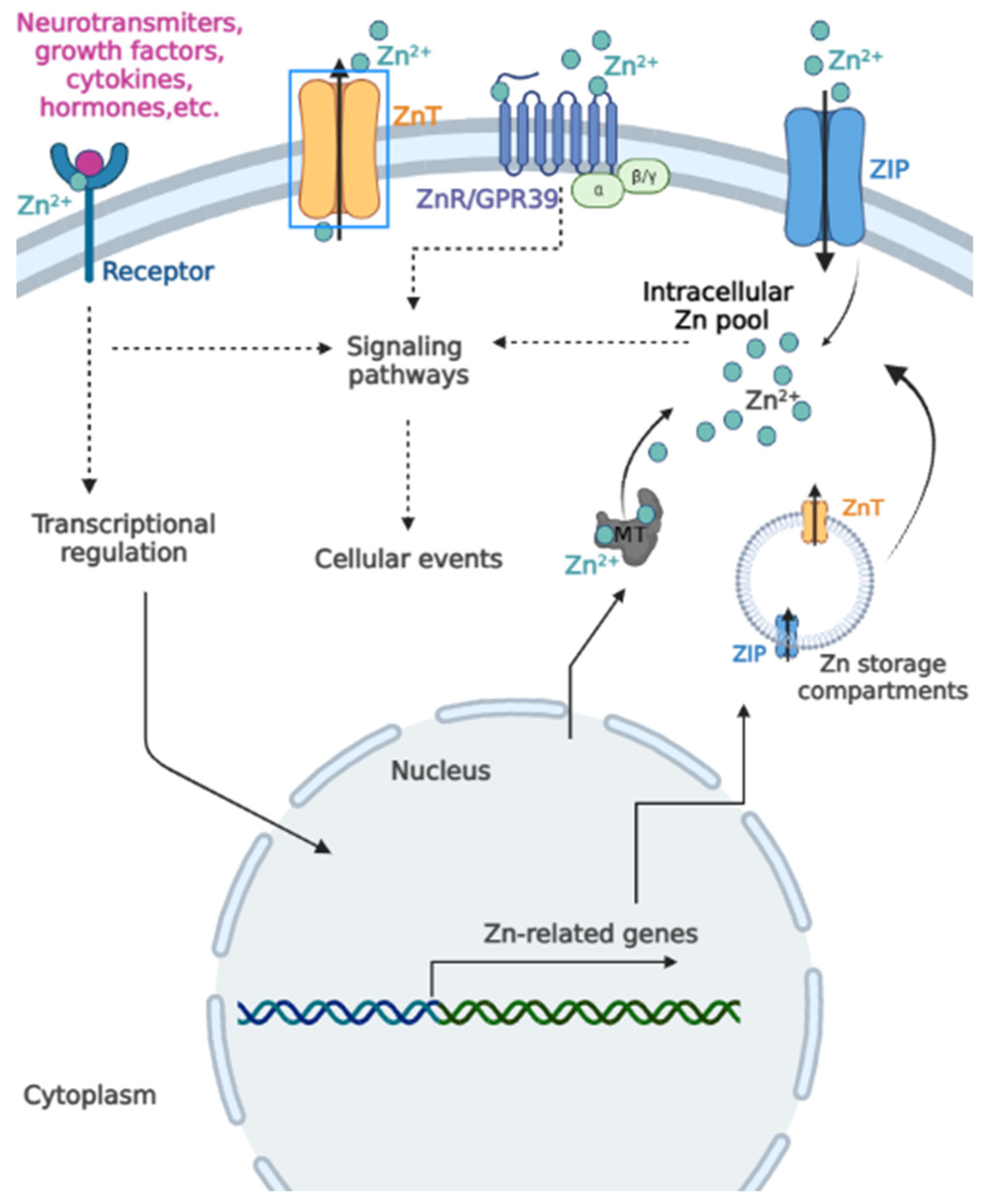
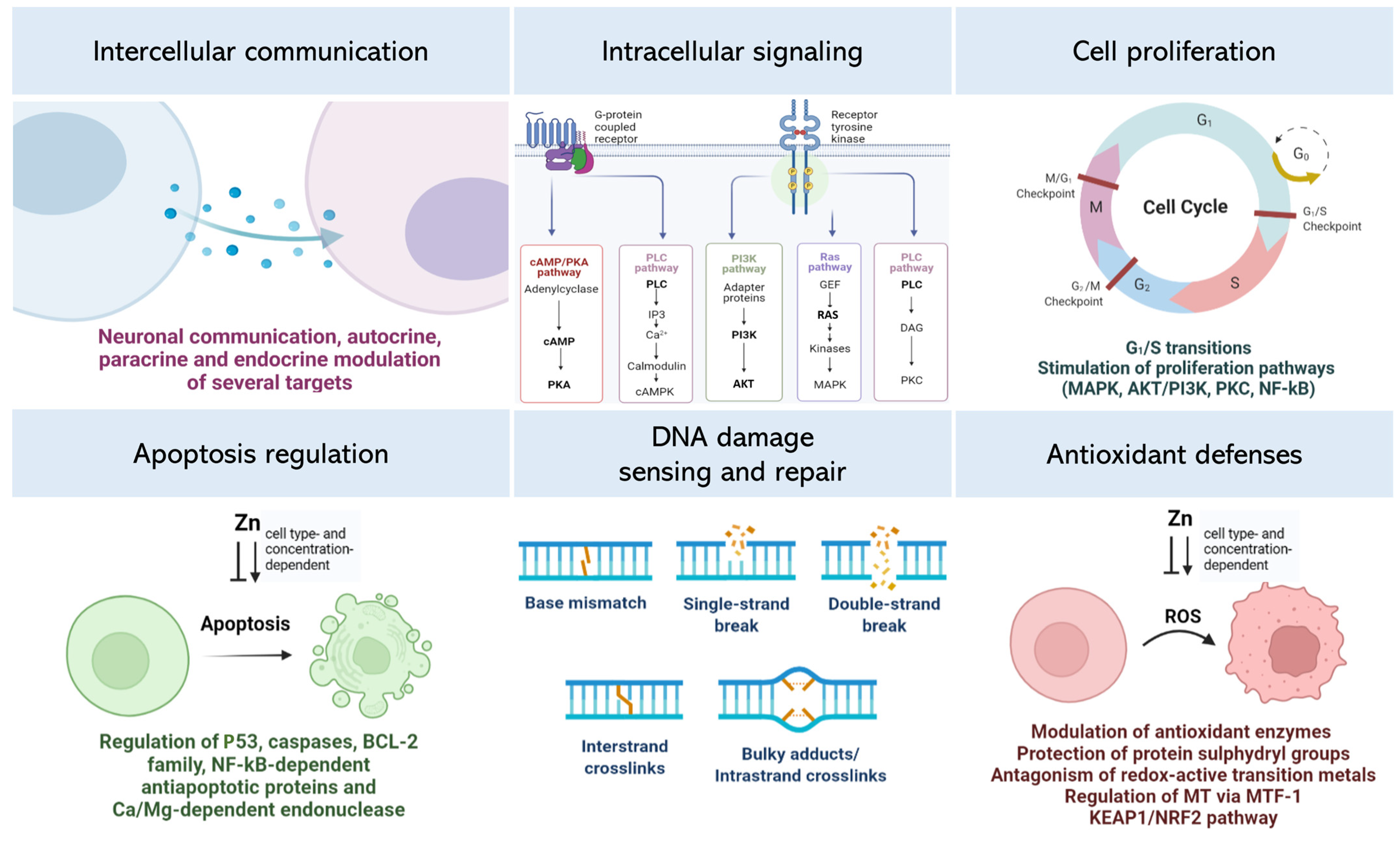
2. Zinc (Zn) and Cell Signaling
2.1. Extracellular Zn Targets
2.2. Intracellular Zn Signaling
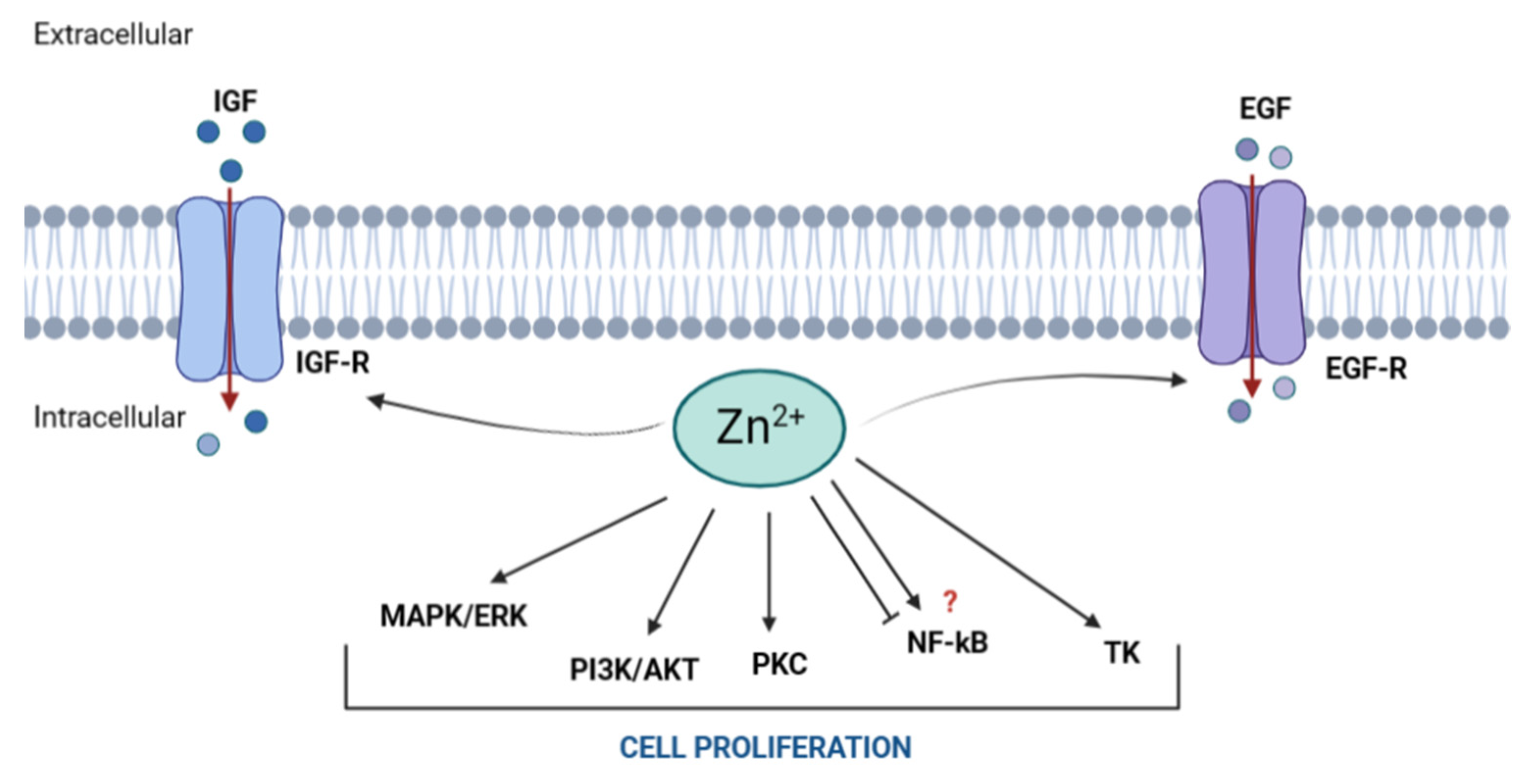
3. Zn and Cell Proliferation
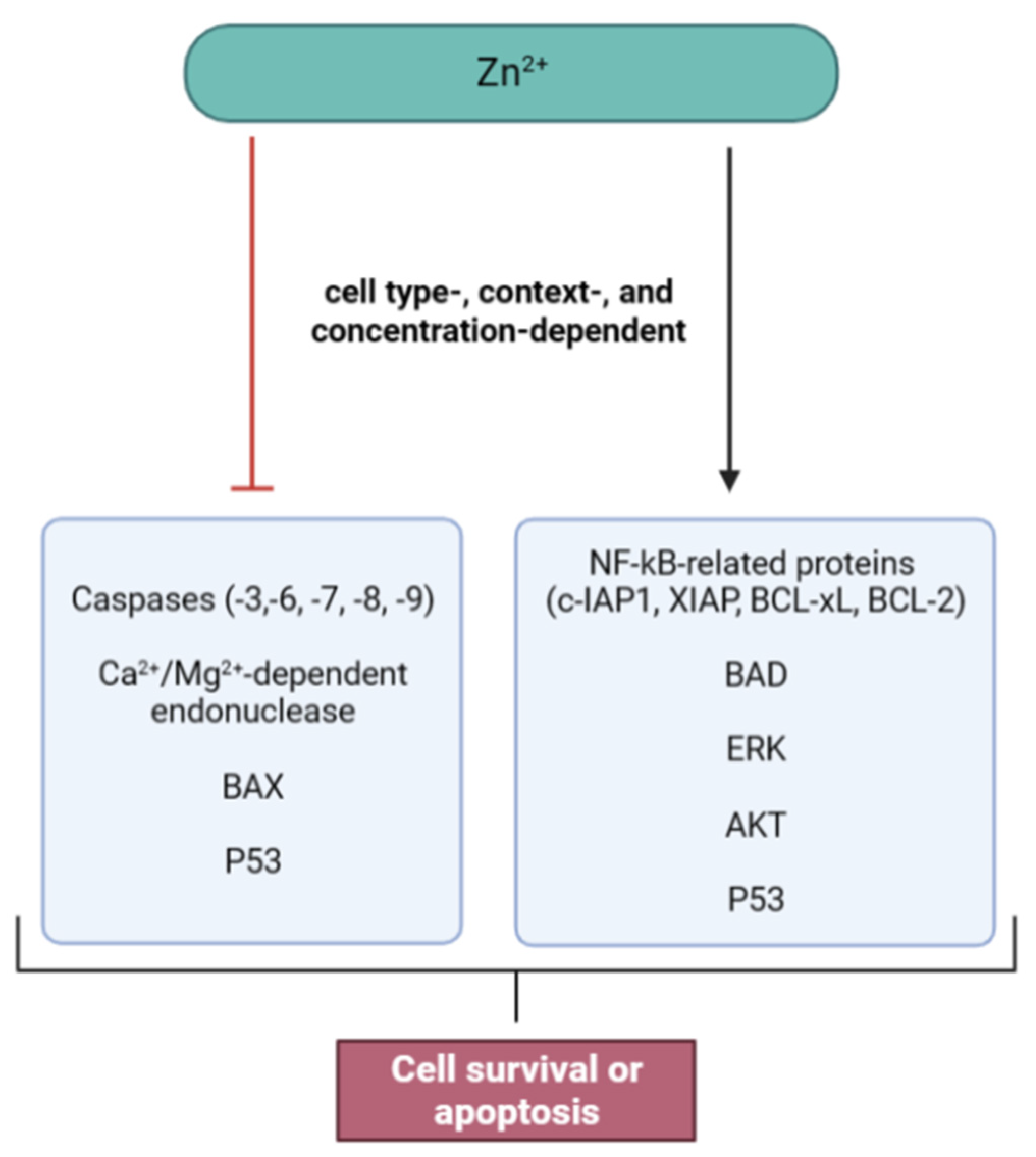
4. Zn and Cell Death
5. Zn and DNA Repair
6. The Therapeutic Potential of Zn
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Maret, W. Zinc biochemistry: From a single zinc enzyme to a key element of life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Maret, W. Zinc in Cellular Regulation: The Nature and Significance of “Zinc Signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef] [Green Version]
- Beyersmann, D.; Haase, H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 2001, 14, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Taylor, K.M.; Fu, D. Zinc transporters and their functional integration in mammalian cells. J. Biol. Chem. 2021, 296, 100320. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Matsunaga, M.; Takeda, T.-A. Understanding the Contribution of Zinc Transporters in the Function of the Early Secretory Pathway. Int. J. Mol. Sci. 2017, 18, 2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübner, C.; Haase, H. Interactions of zinc- and redox-signaling pathways. Redox. Biol. 2021, 41, 101916. [Google Scholar] [CrossRef] [PubMed]
- Grzywacz, A.; Gdula-Argasińska, J.; Muszyńska, B.; Tyszka-Czochara, M.; Librowski, T.; Opoka, W. Metal responsive transcription factor 1 (MTF-1) regulates zinc dependent cellular processes at the molecular level. Acta Biochim. Pol. 2015, 62, 491–498. [Google Scholar] [CrossRef]
- Krall, R.F.; Tzounopoulos, T.; Aizenman, E. The function and regulation of zinc in the brain. Neuroscience 2021, 457, 235–258. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of Zinc in the Central Nervous System: The Zinc-Containing Neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Dischler, A.; Glover, K.; Qin, Y. Neuronal signalling of zinc: From detection and modulation to function. Open Biol. 2022, 12, 220188. [Google Scholar] [CrossRef]
- Fukada, T.; Yamasaki, S.; Nishida, K.; Murakami, M.; Hirano, T. Zinc homeostasis and signaling in health and diseases: Zinc signaling. J. Biol. Inorg. Chem. 2011, 16, 1123–1134. [Google Scholar] [CrossRef] [Green Version]
- Sunuwar, L.; Gilad, D.; Hershfinkel, M. The zinc sensing receptor, ZnR/GPR39, in health and disease. Front. Biosci. Landmark Ed. 2017, 22, 1469–1492. [Google Scholar] [CrossRef] [Green Version]
- Hoang, B.X.; Han, B.; Shaw, D.G.; Nimni, M. Zinc as a possible preventive and therapeutic agent in pancreatic, prostate, and breast cancer. Eur. J. Cancer Prev. 2016, 25, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Laitakari, A.; Liu, L.; Frimurer, T.M.; Holst, B. The Zinc-Sensing Receptor GPR39 in Physiology and as a Pharmacological Target. Int. J. Mol. Sci. 2021, 22, 3872. [Google Scholar] [CrossRef] [PubMed]
- Popovics, P.; Stewart, A.J. GPR39: A Zn2+-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell. Mol. Life Sci. 2011, 68, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Asraf, H.; Salomon, S.; Nevo, A.; Sekler, I.; Mayer, D.; Hershfinkel, M. The ZnR/GPR39 Interacts with the CaSR to Enhance Signaling in Prostate and Salivary Epithelia. J. Cell. Physiol. 2014, 229. [Google Scholar] [CrossRef]
- Azriel-Tamir, H.; Sharir, H.; Schwartz, B.; Hershfinkel, M. Extracellular Zinc Triggers ERK-dependent Activation of Na+/H+ Exchange in Colonocytes Mediated by the Zinc-sensing Receptor. J. Biol. Chem. 2004, 279, 51804–51816. [Google Scholar] [CrossRef] [Green Version]
- Mero, M.; Asraf, H.; Sekler, I.; Taylor, K.M.; Hershfinkel, M. ZnR/GPR39 upregulation of K+/Cl−-cotransporter 3 in tamoxifen resistant breast cancer cells. Cell Calcium 2019, 81, 12–20. [Google Scholar] [CrossRef]
- Murakami, M.; Hirano, T. Intracellular zinc homeostasis and zinc signaling. Cancer Sci. 2008, 99, 1515–1522. [Google Scholar] [CrossRef]
- Haase, H.; Maret, W. Protein tyrosine phosphatases as targets of the combined insulinomimetic effects of zinc and oxidants. Biometals 2005, 18, 333–338. [Google Scholar] [CrossRef]
- Kim, B.; Lee, W.-W. Regulatory Role of Zinc in Immune Cell Signaling. Mol. Cells 2021, 44, 335–341. [Google Scholar] [CrossRef]
- Kabu, K.; Yamasaki, S.; Kamimura, D.; Ito, Y.; Hasegawa, A.; Sato, E.; Kitamura, H.; Nishida, K.; Hirano, T. Zinc Is Required for FcεRI-Mediated Mast Cell Activation1. J. Immunol. 2006, 177, 1296–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, T.J. Intracellular free zinc and zinc buffering in human red blood cells. J. Membr. Biol. 1991, 123, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Alker, W.; Haase, H. Comparison of Free Zinc Levels Determined by Fluorescent Probes in THP1 Cells Using Microplate Reader and Flow Cytometer. Biol. Trace Elem. Res. 2021, 199, 2414–2419. [Google Scholar] [CrossRef]
- Liu, R.; Kowada, T.; Du, Y.; Amagai, Y.; Matsui, T.; Inaba, K.; Mizukami, S. Organelle-Level Labile Zn2+ Mapping Based on Targetable Fluorescent Sensors. ACS Sens. 2022, 7, 748–757. [Google Scholar] [CrossRef]
- Maret, W. Metallothionein redox biology in the cytoprotective and cytotoxic functions of zinc. Exp. Gerontol. 2008, 43, 363–369. [Google Scholar] [CrossRef]
- Pitt, S.J.; Stewart, A.J. Examining a new role for zinc in regulating calcium release in cardiac muscle. Biochem. Soc. Trans. 2015, 43, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Tuncay, E.; Bilginoglu, A.; Sozmen, N.N.; Zeydanli, E.N.; Ugur, M.; Vassort, G.; Turan, B. Intracellular free zinc during cardiac excitation–contraction cycle: Calcium and redox dependencies. Cardiovasc. Res. 2011, 89, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, I.; Fieuw-Makaroff, S.; Hall, A.L.; Sim, A.T.; Rostas, J.A.; Dunkley, P.R. Modulation of the phosphorylation and activity of calcium/calmodulin-dependent protein kinase II by zinc. J. Neurochem. 2000, 75, 594–605. [Google Scholar] [CrossRef]
- Skrajnowska, D.; Bobrowska-Korczak, B. Role of Zinc in Immune System and Anti-Cancer Defense Mechanisms. Nutrients 2019, 11, 2273. [Google Scholar] [CrossRef] [Green Version]
- von Bülow, V.; Rink, L.; Haase, H. Zinc-mediated inhibition of cyclic nucleotide phosphodiesterase activity and expression suppresses TNF-alpha and IL-1 beta production in monocytes by elevation of guanosine 3′,5′-cyclic monophosphate. J. Immunol. 2005, 175, 4697–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korichneva, I.; Hoyos, B.; Chua, R.; Levi, E.; Hammerling, U. Zinc Release from Protein Kinase C as the Common Event during Activation by Lipid Second Messenger or Reactive Oxygen*. J. Biol. Chem. 2002, 277, 44327–44331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, K.-M.; Kim, Y.H.; Koh, J.-Y. Mediation by Membrane Protein Kinase C of Zinc-Induced Oxidative Neuronal Injury in Mouse Cortical Cultures. J. Neurochem. 1999, 72, 1609–1616. [Google Scholar] [CrossRef]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef] [PubMed]
- Chasapis, C.T.; Loutsidou, A.C.; Spiliopoulou, C.A.; Stefanidou, M.E. Zinc and human health: An update. Arch. Toxicol. 2012, 86, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp. Cell Res. 2003, 291, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.M.; Zago, M.P.; Mackenzie, G.G.; Aimo, L.; Keen, C.L.; Keenan, A.; Oteiza, P.I. The role of zinc in the modulation of neuronal proliferation and apoptosis. Neurotox Res. 2010, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, R.S. The role of zinc in growth and cell proliferation. J. Nutr. 2000, 130, 1500S–1508S. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, K.; Nagata, Y.; Wada, E.; Zammit, P.S.; Shiozuka, M.; Matsuda, R. Zinc promotes proliferation and activation of myogenic cells via the PI3K/Akt and ERK signaling cascade. Exp. Cell Res. 2015, 333, 228–237. [Google Scholar] [CrossRef]
- Cossack, Z.T. Decline in somatomedin-C (insulin-like growth factor-1) with experimentally induced zinc deficiency in human subjects. Clin. Nutr. 1991, 10, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Dørup, I.; Flyvbjerg, A.; Everts, M.E.; Clausen, T. Role of insulin-like growth factor-1 and growth hormone in growth inhibition induced by magnesium and zinc deficiencies. Br. J. Nutr. 1991, 66, 505–521. [Google Scholar] [CrossRef] [PubMed]
- McCusker, R.H.; Kaleko, M.; Sackett, R.L. Multivalent cations and ligand affinity of the type 1 insulin-like growth factor receptor on P2A2-LISN muscle cells. J. Cell Physiol. 1998, 176, 392–401. [Google Scholar] [CrossRef]
- Lefebvre, D.; Boney, C.M.; Ketelslegers, J.M.; Thissen, J.P. Inhibition of insulin-like growth factor-I mitogenic action by zinc chelation is associated with a decreased mitogen-activated protein kinase activation in RAT-1 fibroblasts. FEBS Lett. 1999, 449, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Graves, L.M.; Jaspers, I.; Devlin, R.B.; Reed, W.; Samet, J.M. Activation of the EGF receptor signaling pathway in human airway epithelial cells exposed to metals. Am. J. Physiol. 1999, 277, L924–L931. [Google Scholar] [CrossRef]
- McNall, A.D.; Etherton, T.D.; Fosmire, G.J. The impaired growth induced by zinc deficiency in rats is associated with decreased expression of the hepatic insulin-like growth factor I and growth hormone receptor genes. J. Nutr. 1995, 125, 874–879. [Google Scholar] [CrossRef]
- Tang, X.; Shay, N.F. Zinc Has an Insulin-Like Effect on Glucose Transport Mediated by Phosphoinositol-3-Kinase and Akt in 3T3-L1 Fibroblasts and Adipocytes. J. Nutr. 2001, 131, 1414–1420. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-S.; Mukherjee, J.J.; Chung, T.; Crilly, K.S.; Kiss, Z. Extracellular calcium stimulates DNA synthesis in synergism with zinc, insulin and insulin-like growth factor I in fibroblasts. Eur. J. Biochem. 1999, 266, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Chesters, J.K.; Petrie, L.; Travis, A.J. A requirement for Zn2+ for the induction of thymidine kinase but not ornithine decarboxylase in 3T3 cells stimulated from quiescence. Biochem. J. 1990, 272, 525–527. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.B.; Chesters, J.K. The effects of early zinc deficiency on DNA and protein synthesis in the rat. Br. J. Nutr. 1970, 24, 1053–1059. [Google Scholar] [CrossRef] [Green Version]
- Herbein, G.; Varin, A.; Fulop, T. NF-kappaB, AP-1, Zinc-deficiency and aging. Biogerontology 2006, 7, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Młyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 2017, 25, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, B.; Prasad, A.S.; Beck, F.W.J.; Sarkar, F.H. Zinc up-regulates NF-κB activation via phosphorylation of IκB in HUT-78 (Th0) cells. FEBS Lett. 2007, 581, 4507–4511. [Google Scholar] [CrossRef] [Green Version]
- Ho, E.; Quan, N.; Tsai, Y.-H.; Lai, W.; Bray, T.M. Dietary Zinc Supplementation Inhibits NFκB Activation and Protects Against Chemically Induced Diabetes in CD1 Mice. Exp. Biol. Med. Maywood 2001, 226, 103–111. [Google Scholar] [CrossRef]
- Andrews, G.K.; Gallant, K.R.; Cherian, M.G. Regulation of the ontogeny of rat liver metallothionein mRNA by zinc. Eur. J. Biochem. 1987, 166, 527–531. [Google Scholar] [CrossRef]
- Cai, L.; Wang, G.J.; Xu, Z.L.; Deng, D.X.; Chakrabarti, S.; Cherian, M.G. Metallothionein and apoptosis in primary human hepatocellular carcinoma (HCC) from northern China. Anticancer Res. 1998, 18, 4667–4672. [Google Scholar]
- Tsujikawa, K.; Suzuki, N.; Sagawa, K.; Itoh, M.; Sugiyama, T.; Kohama, Y.; Otaki, N.; Kimura, M.; Mimura, T. Induction and subcellular localization of metallothionein in regenerating rat liver. Eur. J. Cell Biol. 1994, 63, 240–246. [Google Scholar]
- Nagel, W.W.; Vallee, B.L. Cell cycle regulation of metallothionein in human colonic cancer cells. Proc. Natl. Acad. Sci. USA 1995, 92, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Tsujikawa, K.; Imai, T.; Kakutani, M.; Kayamori, Y.; Mimura, T.; Otaki, N.; Kimura, M.; Fukuyama, R.; Shimizu, N. Localization of metallothionein in nuclei of growing primary cultured adult rat hepatocytes. FEBS Lett. 1991, 283, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Nartey, N.; Cherian, M.G.; Banerjee, D. Immunohistochemical localization of metallothionein in human thyroid tumors. Am. J. Pathol. 1987, 129, 177–182. [Google Scholar] [PubMed]
- Kuo, S.M.; Kondo, Y.; DeFilippo, J.M.; Ernstoff, M.S.; Bahnson, R.R.; Lazo, J.S. Subcellular localization of metallothionein IIA in human bladder tumor cells using a novel epitope-specific antiserum. Toxicol. Appl. Pharm. 1994, 125, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Takagishi, T.; Hara, T.; Fukada, T. Recent Advances in the Role of SLC39A/ZIP Zinc Transporters In Vivo. Int. J. Mol. Sci. 2017, 18, 2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clegg, M.S.; Hanna, L.A.; Niles, B.J.; Momma, T.Y.; Keen, C.L. Zinc deficiency-induced cell death. IUBMB Life 2005, 57, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Chai, F.; Truong-Tran, A.Q.; Ho, L.H.; Zalewski, P.D. Regulation of caspase activation and apoptosis by cellular zinc fluxes and zinc deprivation: A review. Immunol. Cell Biol. 1999, 77, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Wang, Y.; Li, L.; Liu, Y. Neuronal death/apoptosis induced by intracellular zinc deficiency associated with changes in amino-acid neurotransmitters and glutamate receptor subtypes. J. Inorg. Biochem. 2018, 179, 54–59. [Google Scholar] [CrossRef]
- Chimienti, F.; Seve, M.; Richard, S.; Mathieu, J.; Favier, A. Role of cellular zinc in programmed cell death: Temporal relationship between zinc depletion, activation of caspases, and cleavage of Sp family transcription factors. Biochem. Pharmacol. 2001, 62, 51–62. [Google Scholar] [CrossRef]
- Lee, J.K.; Ha, J.-H.; Kim, D.-K.; Kwon, J.; Cho, Y.-E.; Kwun, I.-S. Depletion of Zinc Causes Osteoblast Apoptosis with Elevation of Leptin Secretion and Phosphorylation of JAK2/STAT3. Nutrients 2022, 15, 77. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, J.; Wang, Y.; Yang, M.; Guo, M. Zinc Deficiency Promotes Testicular Cell Apoptosis in Mice. Biol. Trace Elem. Res. 2020, 195, 142–149. [Google Scholar] [CrossRef]
- Hao, Y.; Ren, J.; Liu, C.; Li, H.; Liu, J.; Yang, Z.; Li, R.; Su, Y. Zinc Protects Human Kidney Cells from Depleted Uranium-induced Apoptosis. Basic Clin. Pharmacol. Toxicol. 2014, 114, 271–280. [Google Scholar] [CrossRef]
- Kontargiris, E.; Vadalouka, A.; Ragos, V.; Kalfakakou, V. Zinc Inhibits Apoptosis and Maintains NEP Downregulation, Induced by Ropivacaine, in HaCaT Cells. Biol. Trace Elem. Res. 2012, 150, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Li, J.; He, S. Zinc Protects against Heat Stress–Induced Apoptosis via the Inhibition of Endoplasmic Reticulum Stress in TM3 Leydig Cells. Biol. Trace Elem. Res. 2022, 200, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Benedetti, G.; Albarède, F.; Miossec, P. Zinc and its role in immunity and inflammation. Autoimmun. Rev. 2015, 14, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Gumulec, J.; Masarik, M.; Krizkova, S.; Adam, V.; Hubalek, J.; Hrabeta, J.; Eckschlager, T.; Stiborova, M.; Kizek, R. Insight to physiology and pathology of zinc(II) ions and their actions in breast and prostate carcinoma. Curr. Med. Chem. 2011, 18, 5041–5051. [Google Scholar] [CrossRef] [Green Version]
- Formigari, A.; Irato, P.; Santon, A. Zinc, antioxidant systems and metallothionein in metal mediated-apoptosis: Biochemical and cytochemical aspects. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2007, 146, 443–459. [Google Scholar] [CrossRef]
- Franklin, R.B.; Costello, L.C. The Important Role of the Apoptotic Effects of Zinc in the Development of Cancers. J. Cell Biochem. 2009, 106, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.N.; Lee, Y.S.; Kim, M.Y.; Kim, J.D.; Park, L.O. Antiproliferative and apoptotic effects of zinc-citrate compound (CIZAR(R)) on human epithelial ovarian cancer cell line, OVCAR-3. Gynecol. Oncol. 2006, 103, 127–136. [Google Scholar] [CrossRef]
- Donadelli, M.; Dalla Pozza, E.; Scupoli, M.T.; Costanzo, C.; Scarpa, A.; Palmieri, M. Intracellular zinc increase inhibits p53(-/-) pancreatic adenocarcinoma cell growth by ROS/AIF-mediated apoptosis. Biochim. Biophys. Acta 2009, 1793, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.Y.; Liu, Y.Y.; Zou, J.; Franklin, R.B.; Costello, L.C.; Feng, P. Inhibitory effect of zinc on human prostatic carcinoma cell growth. Prostate 1999, 40, 200–207. [Google Scholar] [CrossRef]
- Seth, R.; Corniola, R.S.; Gower-Winter, S.D.; Morgan, T.J.; Bishop, B.; Levenson, C.W. Zinc deficiency induces apoptosis via mitochondrial p53- and caspase-dependent pathways in human neuronal precursor cells. J. Trace Elem. Med. Biol. 2015, 30, 59–65. [Google Scholar] [CrossRef]
- Fukamachi, Y.; Karasaki, Y.; Sugiura, T.; Itoh, H.; Abe, T.; Yamamura, K.; Higashi, K. Zinc Suppresses Apoptosis of U937 Cells Induced by Hydrogen Peroxide through an Increase of the Bcl-2/Bax Ratio. Biochem. Biophys. Res. Commun. 1998, 246, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, D.K.; Chadha, V.D. Zinc: A promising agent in dietary chemoprevention of cancer. Indian J. Med. Res. 2010, 132, 676–682. [Google Scholar] [PubMed]
- Hainaut, P.; Mann, K. Zinc binding and redox control of p53 structure and function. Antioxid. Redox. Signal. 2001, 3, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Loh, S.N. The missing zinc: p53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Ames, B.N. Low intracellular zinc induces oxidative DNA damage, disrupts p53, NFκB, and AP1 DNA binding, and affects DNA repair in a rat glioma cell line. Proc. Natl. Acad. Sci. USA 2002, 99, 16770–16775. [Google Scholar] [CrossRef] [Green Version]
- Truong-Tran, A.Q.; Ho, L.H.; Chai, F.; Zalewski, P.D. Cellular zinc fluxes and the regulation of apoptosis/gene-directed cell death. J. Nutr. 2000, 130, 1459S–1466S. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.M.; Sun, X.M.; Snowden, R.T.; Dinsdale, D.; Skilleter, D.N. Key morphological features of apoptosis may occur in the absence of internucleosomal DNA fragmentation. Biochem. J. 1992, 286, 331–334. [Google Scholar] [CrossRef]
- Chai, F.; Truong-Tran, A.Q.; Evdokiou, A.; Young, G.P.; Zalewski, P.D. Intracellular Zinc Depletion Induces Caspase Activation and p21Waf1/Cip1 Cleavage in Human Epithelial Cell Lines. J. Infect. Dis. 2000, 182, S85–S92. [Google Scholar] [CrossRef] [Green Version]
- Eron, S.J.; MacPherson, D.J.; Dagbay, K.B.; Hardy, J.A. Multiple Mechanisms of Zinc-Mediated Inhibition for the Apoptotic Caspases-3, -6, -7, and -8. ACS Chem. Biol. 2018, 13, 1279–1290. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M.F. Dietary reference values of individual micronutrients and nutriomes for genome damage prevention: Current status and a road map to the future. Am. J. Clin. Nutr. 2010, 91, 1438S–1454S. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M.F. Nutriomes and personalised nutrition for DNA damage prevention, telomere integrity maintenance and cancer growth control. Cancer Treat. Res. 2014, 159, 427–441. [Google Scholar] [CrossRef]
- Arigony, A.L.V.; de Oliveira, I.M.; Machado, M.; Bordin, D.L.; Bergter, L.; Prá, D.; Henriques, J.A.P. The influence of micronutrients in cell culture: A reflection on viability and genomic stability. Biomed. Res. Int. 2013, 2013, 597282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.R.; Azqueta, A.; Langie, S.A.S. Effects of micronutrients on DNA repair. Eur. J. Nutr. 2012, 51, 261–279. [Google Scholar] [CrossRef]
- Reddy, V.S.; Palika, R.; Ismail, A.; Pullakhandam, R.; Reddy, G.B. Nutrigenomics: Opportunities & challenges for public health nutrition. Indian J. Med. Res. 2018, 148, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Ho, E. Zinc deficiency, DNA damage and cancer risk. J. Nutr. Biochem. 2004, 15, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Bao, B. Molecular Mechanisms of Zinc as a Pro-Antioxidant Mediator: Clinical Therapeutic Implications. Antioxidants 2019, 8, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Song, Y.; Wong, C.P.; Hardin, K.; Ho, E. Zinc deficiency alters DNA damage response genes in normal human prostate epithelial cells. J. Nutr. 2008, 138, 667–673. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tan, Y.; Dai, J.; Li, B.; Guo, L.; Cui, J.; Wang, G.; Shi, X.; Zhang, X.; Mellen, N.; et al. Exacerbation of diabetes-induced testicular apoptosis by zinc deficiency is most likely associated with oxidative stress, p38 MAPK activation, and p53 activation in mice. Toxicol. Lett. 2011, 200, 100–106. [Google Scholar] [CrossRef]
- Cortese, M.M.; Suschek, C.V.; Wetzel, W.; Kröncke, K.-D.; Kolb-Bachofen, V. Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free Radic. Biol. Med. 2008, 44, 2002–2012. [Google Scholar] [CrossRef]
- Samavarchi Tehrani, S.; Mahmoodzadeh Hosseini, H.; Yousefi, T.; Abolghasemi, M.; Qujeq, D.; Maniati, M.; Amani, J. The crosstalk between trace elements with DNA damage response, repair, and oxidative stress in cancer. J. Cell Biochem. 2018, 120. [Google Scholar] [CrossRef]
- Bruno, R.S.; Song, Y.; Leonard, S.W.; Mustacich, D.J.; Taylor, A.W.; Traber, M.G.; Ho, E. Dietary zinc restriction in rats alters antioxidant status and increases plasma F2 isoprostanes. J. Nutr. Biochem. 2007, 18, 509–518. [Google Scholar] [CrossRef]
- Song, Y.; Leonard, S.W.; Traber, M.G.; Ho, E. Zinc Deficiency Affects DNA Damage, Oxidative Stress, Antioxidant Defenses, and DNA Repair in Rats. J. Nutr. 2009, 139, 1626–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zyba, S.J.; Shenvi, S.V.; Killilea, D.W.; Holland, T.C.; Kim, E.; Moy, A.; Sutherland, B.; Gildengorin, V.; Shigenaga, M.K.; King, J.C. A moderate increase in dietary zinc reduces DNA strand breaks in leukocytes and alters plasma proteins without changing plasma zinc concentrations. Am. J. Clin. Nutr. 2017, 105, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Joray, M.L.; Yu, T.-W.; Ho, E.; Clarke, S.L.; Stanga, Z.; Gebreegziabher, T.; Hambidge, K.M.; Stoecker, B.J. Zinc supplementation reduced DNA breaks in Ethiopian women. Nutr. Res. 2015, 35, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.K.; van Attikum, H. DNA double-strand break repair: Putting zinc fingers on the sore spot. Semin. Cell Dev. Biol. 2021, 113, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhou, X.; Du, L.; Liu, W.; Liu, Y.; Hudson, L.G.; Liu, K.J. Arsenite binding-induced zinc loss from PARP-1 is equivalent to zinc deficiency in reducing PARP-1 activity, leading to inhibition of DNA repair. Toxicol. Appl. Pharm. 2014, 274, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-L.; Ra, H.; Kim, K.-R.; Lee, J.-M.; Im, H.; Kim, Y.-H. Poly(ADP-ribosyl)ation of p53 Contributes to TPEN-Induced Neuronal Apoptosis. Mol. Cells 2015, 38, 312–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, E.; Courtemanche, C.; Ames, B.N. Zinc deficiency induces oxidative DNA damage and increases p53 expression in human lung fibroblasts. J. Nutr. 2003, 133, 2543–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreosti, I.E. Zinc and the gene. Mutat. Res. 2001, 475, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, A.; Kaya, Y.; Tanriverdi, O. Effect of the Interaction Between Selenium and Zinc on DNA Repair in Association With Cancer Prevention. J. Cancer Prev. 2019, 24, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, H.; Xu, Z.; Cheng, X. Zinc dysregulation in cancers and its potential as a therapeutic target. Cancer Biol. Med. 2020, 17, 612–625. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Drake, V.J.; Ho, E. Zinc. Adv. Nutr. 2015, 6, 224–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Stovall, D.B.; Wang, W.; Sui, G. Advances of Zinc Signaling Studies in Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memon, A.-R.; Kazi, T.G.; Afridi, H.I.; Jamali, M.K.; Arain, M.B.; Jalbani, N.; Syed, N. Evaluation of zinc status in whole blood and scalp hair of female cancer patients. Clin. Chim. Acta 2007, 379, 66–70. [Google Scholar] [CrossRef]
- Costello, L.C.; Levy, B.A.; Desouki, M.M.; Zou, J.; Bagasra, O.; Johnson, L.A.; Hanna, N.; Franklin, R.B. Decreased zinc and downregulation of ZIP3 zinc uptake transporter in the development of pancreatic adenocarcinoma. Cancer Biol. 2011, 12, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christudoss, P.; Selvakumar, R.; Fleming, J.J.; Gopalakrishnan, G. Zinc status of patients with benign prostatic hyperplasia and prostate carcinoma. Indian J. Urol. 2011, 27, 14–18. [Google Scholar] [CrossRef]
- Kaba, M.; Pirincci, N.; Yuksel, M.B.; Gecit, I.; Gunes, M.; Ozveren, H.; Eren, H.; Demir, H. Serum levels of trace elements in patients with prostate cancer. Asian Pac. J. Cancer Prev. 2014, 15, 2625–2629. [Google Scholar] [CrossRef] [Green Version]
- Desouki, M.M.; Geradts, J.; Milon, B.; Franklin, R.B.; Costello, L.C. hZip2 and hZip3 zinc transporters are down regulated in human prostate adenocarcinomatous glands. Mol. Cancer 2007, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.B.; Feng, P.; Milon, B.; Desouki, M.M.; Singh, K.K.; Kajdacsy-Balla, A.; Bagasra, O.; Costello, L.C. hZIP1 zinc uptake transporter down regulation and zinc depletion in prostate cancer. Mol. Cancer 2005, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Henshall, S.M.; Afar, D.E.H.; Rasiah, K.K.; Horvath, L.G.; Gish, K.; Caras, I.; Ramakrishnan, V.; Wong, M.; Jeffry, U.; Kench, J.G.; et al. Expression of the zinc transporter ZnT4 is decreased in the progression from early prostate disease to invasive prostate cancer. Oncogene 2003, 22, 6005–6012. [Google Scholar] [CrossRef] [Green Version]
- Kagara, N.; Tanaka, N.; Noguchi, S.; Hirano, T. Zinc and its transporter ZIP10 are involved in invasive behavior of breast cancer cells. Cancer Sci. 2007, 98, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.B.; Levy, B.A.; Zou, J.; Hanna, N.; Desouki, M.M.; Bagasra, O.; Johnson, L.A.; Costello, L.C. ZIP14 Zinc Transporter Downregulation and Zinc Depletion in the Development and Progression of Hepatocellular Cancer. J. Gastrointest Cancer 2012, 43, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, L.C.; Zou, J.; Desouki, M.M.; Franklin, R.B. Evidence for Changes in RREB-1, ZIP3, and Zinc in the Early Development of Pancreatic Adenocarcinoma. J. Gastrointest Cancer 2012, 43, 570–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocdor, H.; Ates, H.; Aydin, S.; Cehreli, R.; Soyarat, F.; Kemanli, P.; Harmanci, D.; Cengiz, H.; Kocdor, M.A. Zinc supplementation induces apoptosis and enhances antitumor efficacy of docetaxel in non-small-cell lung cancer. Drug Des. Dev. 2015, 9, 3899–3909. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Li, Y.; Tang, X.; Guo, R.; Li, J.; Chen, Y.Y.; Guo, H.; Su, J.; Sun, L.; Liu, Y. Zinc enhances chemosensitivity to paclitaxel in PC-3 prostate cancer cells. Oncol. Rep. 2018, 40, 2269–2277. [Google Scholar] [CrossRef]
- Costa, M.I.; Lapa, B.S.; Jorge, J.; Alves, R.; Carreira, I.M.; Sarmento-Ribeiro, A.B.; Gonçalves, A.C. Zinc Prevents DNA Damage in Normal Cells but Shows Genotoxic and Cytotoxic Effects in Acute Myeloid Leukemia Cells. Int. J. Mol. Sci. 2022, 23, 2567. [Google Scholar] [CrossRef]
- Wysokinski, D.; Blasiak, J.; Wozniak, K. Zinc differentially modulates DNA damage induced by anthracyclines in normal and cancer cells. Exp. Oncol. 2012, 34, 327–331. [Google Scholar]
- Sliwinski, T.; Czechowska, A.; Kolodziejczak, M.; Jajte, J.; Wisniewska-Jarosinska, M.; Blasiak, J. Zinc salts differentially modulate DNA damage in normal and cancer cells. Cell Biol. Int. 2009, 33, 542–547. [Google Scholar] [CrossRef]
- Yan, M.; Hardin, K.; Ho, E. Differential response to zinc-induced apoptosis in benign prostate hyperplasia and prostate cancer cells. J. Nutr. Biochem. 2010, 21, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.-F.; Abubakar, S. High intracellular Zn2+ ions modulate the VHR, ZAP-70 and ERK activities of LNCaP prostate cancer cells. Cell Mol. Biol. Lett. 2008, 13, 375–390. [Google Scholar] [CrossRef]
- To, P.K.; Do, M.-H.; Cho, Y.-S.; Kwon, S.-Y.; Kim, M.S.; Jung, C. Zinc Inhibits Expression of Androgen Receptor to Suppress Growth of Prostate Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacioglu, C.; Kacar, S.; Kar, F.; Kanbak, G.; Sahinturk, V. Concentration-Dependent Effects of Zinc Sulfate on DU-145 Human Prostate Cancer Cell Line: Oxidative, Apoptotic, Inflammatory, and Morphological Analyzes. Biol. Trace Elem. Res. 2020, 195, 436–444. [Google Scholar] [CrossRef]
- Feng, P.; Li, T.; Guan, Z.; Franklin, R.B.; Costello, L.C. The Involvement of Bax in Zinc-Induced Mitochondrial Apoptogenesis in Malignant Prostate Cells. Mol. Cancer 2008, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, J.H.; Seo, S.Y.; Kwak, C.; Kim, H.H. The role of survivin and Bcl-2 in zinc-induced apoptosis in prostate cancer cells. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Bixenshpaner, H.; Asraf, H.; Chakraborty, M.; Elkabets, M.; Sekler, I.; Taylor, K.M.; Hershfinkel, M. Enhanced ZnR/GPR39 Activity in Breast Cancer, an Alternative Trigger of Signaling Leading to Cell Growth. Sci. Rep. 2018, 8, 8119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, N.; He, H.; Chai, J.; Cheng, X.; Zhao, H.; Zhou, D.; Teng, T.; Kong, X.; Yang, Q.; et al. The zinc transporter ZIP7 (Slc39a7) controls myocardial reperfusion injury by regulating mitophagy. Basic Res. Cardiol. 2021, 116, 54. [Google Scholar] [CrossRef]
- Tamura, Y. The Role of Zinc Homeostasis in the Prevention of Diabetes Mellitus and Cardiovascular Diseases. J. Atheroscler. Thromb. 2021, 28, 1109–1122. [Google Scholar] [CrossRef]
- Hara, T.; Yoshigai, E.; Ohashi, T.; Fukada, T. Zinc transporters as potential therapeutic targets: An updated review. J. Pharmacol. Sci. 2022, 148, 221–228. [Google Scholar] [CrossRef]
- Zhao, L.; Oliver, E.; Maratou, K.; Atanur, S.S.; Dubois, O.D.; Cotroneo, E.; Chen, C.-N.; Wang, L.; Arce, C.; Chabosseau, P.L.; et al. The zinc transporter ZIP12 regulates the pulmonary vascular response to chronic hypoxia. Nature 2015, 524, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Doboszewska, U.; Młyniec, K.; Wlaź, A.; Poleszak, E.; Nowak, G.; Wlaź, P. Zinc signaling and epilepsy. Pharmacol. Ther. 2019, 193, 156–177. [Google Scholar] [CrossRef]
- Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjbar, E.; Kasaei, M.S.; Mohammad-Shirazi, M.; Nasrollahzadeh, J.; Rashidkhani, B.; Shams, J.; Mostafavi, S.-A.; Mohammadi, M.R. Effects of Zinc Supplementation in Patients with Major Depression: A Randomized Clinical Trial. Iran. J. Psychiatry 2013, 8, 73–79. [Google Scholar] [PubMed]
- Lai, J.; Moxey, A.; Nowak, G.; Vashum, K.; Bailey, K.; McEvoy, M. The efficacy of zinc supplementation in depression: Systematic review of randomised controlled trials. J. Affect. Disord. 2012, 136, e31–e39. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.C.; Reinhart, V.; Lanz, T.A.; Weber, M.L.; Pang, J.; Le, K.X.V.; Bell, R.D.; O’Donnell, P.; Buhl, D.L. Schizophrenia-associated SLC39A8 polymorphism is a loss-of-function allele altering glutamate receptor and innate immune signaling. Transl. Psychiatry 2021, 11, 136. [Google Scholar] [CrossRef]
- Xie, Z.; Wu, H.; Zhao, J. Multifunctional roles of zinc in Alzheimer’s disease. NeuroToxicology 2020, 80, 112–123. [Google Scholar] [CrossRef]
- Kim, J.W.; Byun, M.S.; Yi, D.; Lee, J.H.; Kim, M.J.; Jung, G.; Lee, J.-Y.; Kang, K.M.; Sohn, C.-H.; Lee, Y.-S.; et al. Serum zinc levels and in vivo beta-amyloid deposition in the human brain. Alzheimer’s Res. Ther. 2021, 13, 190. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Zinc ions promote Alzheimer Aβ aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. USA 2010, 107, 9490–9495. [Google Scholar] [CrossRef] [Green Version]
- Read, S.A.; Obeid, S.; Ahlenstiel, C.; Ahlenstiel, G. The Role of Zinc in Antiviral Immunity. Adv. Nutr. 2019, 10, 696–710. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Suzuki, T.; Watanabe, M.; Hatakeyama, S.; Kimura, S.; Nakazono, A.; Honma, A.; Nakamaru, Y.; Vreugde, S.; Homma, A. Role of intracellular zinc in molecular and cellular function in allergic inflammatory diseases. Allergol. Int. 2021, 70, 190–200. [Google Scholar] [CrossRef]
- Yazar, A.S.; Güven, Ş.; Dinleyici, E.Ç. Effects of zinc or synbiotic on the duration of diarrhea in children with acute infectious diarrhea. Turk. J. Gastroenterol. 2016, 27, 537–540. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kinoshita, M.; Shimada, S.; Kawamura, T. Zinc and Skin Disorders. Nutrients 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaka, A.; Fujitani, Y. Role of Zinc Homeostasis in the Pathogenesis of Diabetes and Obesity. Int. J. Mol. Sci. 2018, 19, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochmah, N.; Faizi, M.; Windarti, S.W. Zinc transporter 8 autoantibody in the diagnosis of type 1 diabetes in children. Clin. Exp. Pediatr. 2020, 63, 402–405. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Mechanisms | Zn Implication | Biological Effect |
|---|---|---|
| Antioxidant defenses | Cu/Zn SOD | Decreasing oxidative stress and oxidative DNA lesions |
| Fenton reactions | ||
| Binding to thiol and sulfhydryl groups | ||
| ROS buffering by MT | ||
| KEAP1-NRF2 | ||
| Replication | DNA and RNA polymerases | Cell cycle regulation |
| TK | ||
| RPA | ||
| Tumor suppression | P53 | Cell cycle and apoptosis regulation |
| DDR | PARP1 | BER and SSB repair pathways |
| OGG1 | ||
| APE2 | ||
| XPA | NER pathway |
| Zn Transporters | Disease Implications | References |
|---|---|---|
| ZIP1 | Alzheimer’s disease | [111,112,119,140,145] |
| Pancreatic cancer | ||
| Prostate cancer | ||
| Seizures | ||
| ZIP2 | Cardiovascular diseases | [111,112,118,137] |
| Pancreatic cancer | ||
| Prostate cancer | ||
| ZIP3 | Pancreatic cancer | [111,112,115,118,123,140] |
| Prostate cancer | ||
| Seizures | ||
| ZIP4 | Acrodermatitis enteropathica | [111,112,137,138,151] |
| Cancer cachexia | ||
| Hepatocellular carcinoma | ||
| Pancreatic cancer | ||
| ZIP6 | Breast cancer | [30,111,138] |
| Esophageal squamous cell carcinoma | ||
| ZIP7 | Breast cancer | [30,111,136,138] |
| Gastric cancer | ||
| ZIP8 | Cardiovascular diseases | [137,138,144] |
| Chron’s disease | ||
| Parkinson’s disease | ||
| Schizophrenia | ||
| ZIP9 | Breast cancer | [111] |
| ZIP10 | Atopic dermatitis | [30,111,121,138,151] |
| Breast cancer | ||
| Sepsis | ||
| ZIP12 | Cardiovascular diseases | [137,138,139] |
| Pulmonary hypertension | ||
| ZIP13 | Ehlers–Danlos syndrome (Spondylocheiro dysplastic form) | [137,138,151] |
| Type 2 diabetes | ||
| ZIP14 | Parkinson’s disease | [111,138] |
| Cancer cachexia | ||
| ZnT1 | Atrial fibrillation | [35,138,145,151] |
| Alzheimer’s disease | ||
| Epidermodysplasia verruciformis | ||
| ZnT3 | Alzheimer’s disease | [12,35,140,145] |
| Seizures | ||
| Behavioral and cognitive impairment | ||
| ZnT4 | Prostate cancer | [111,120] |
| ZnT6 | Alzheimer’s disease | [145] |
| ZnT7 | Alzheimer’s disease | [12,35] |
| Prostate cancer | ||
| ZnT8 | Autoimmune type 1 diabetes | [12,35,112,137,152,153] |
| Type 2 diabetes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, M.I.; Sarmento-Ribeiro, A.B.; Gonçalves, A.C. Zinc: From Biological Functions to Therapeutic Potential. Int. J. Mol. Sci. 2023, 24, 4822. https://doi.org/10.3390/ijms24054822
Costa MI, Sarmento-Ribeiro AB, Gonçalves AC. Zinc: From Biological Functions to Therapeutic Potential. International Journal of Molecular Sciences. 2023; 24(5):4822. https://doi.org/10.3390/ijms24054822
Chicago/Turabian StyleCosta, Maria Inês, Ana Bela Sarmento-Ribeiro, and Ana Cristina Gonçalves. 2023. "Zinc: From Biological Functions to Therapeutic Potential" International Journal of Molecular Sciences 24, no. 5: 4822. https://doi.org/10.3390/ijms24054822