
De Novo Assembly and Characterization of the Transcriptome of an Omnivorous Camel Cricket (Tachycines meditationis)


Abstract
:1. Introduction
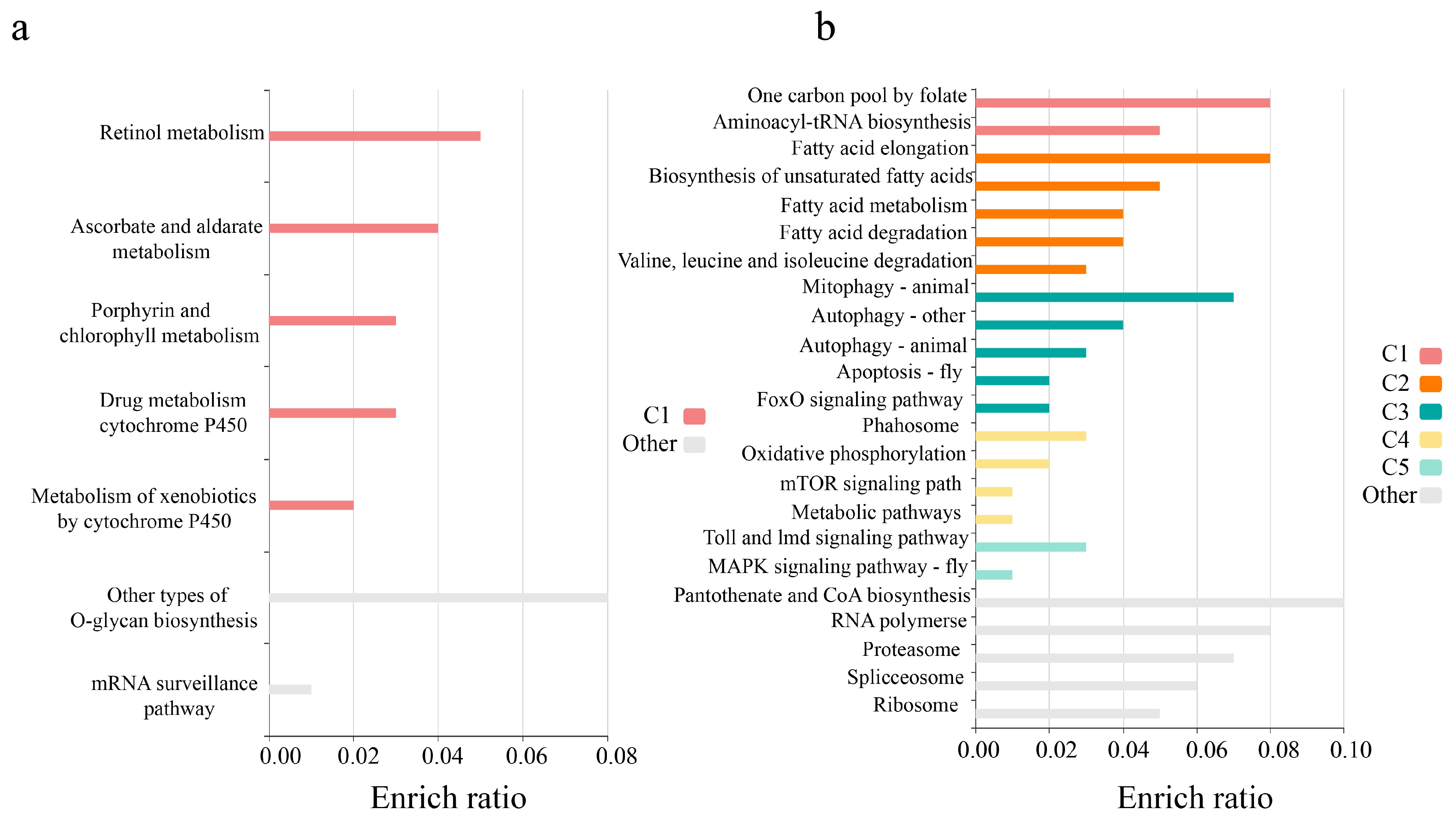
2. Results
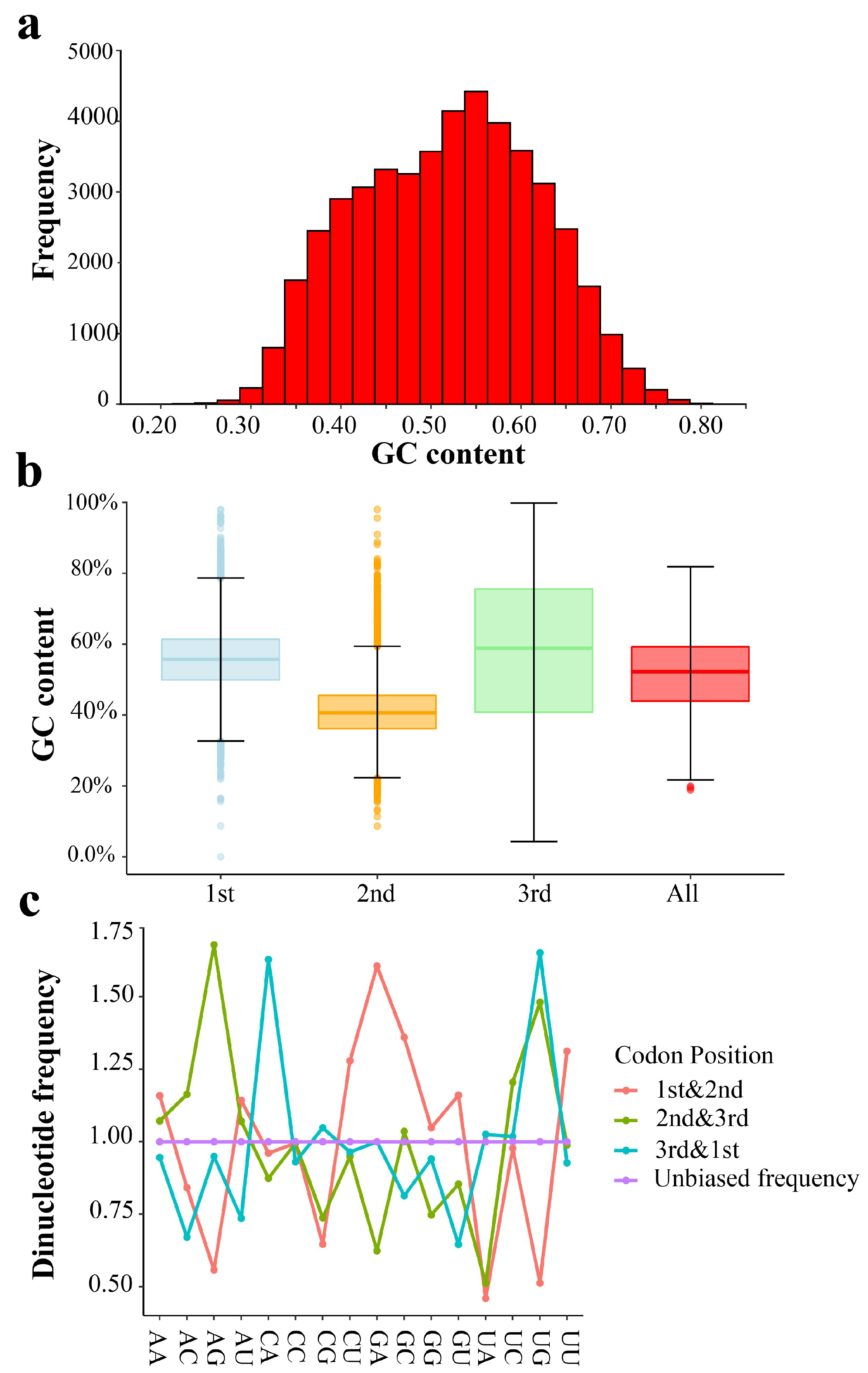
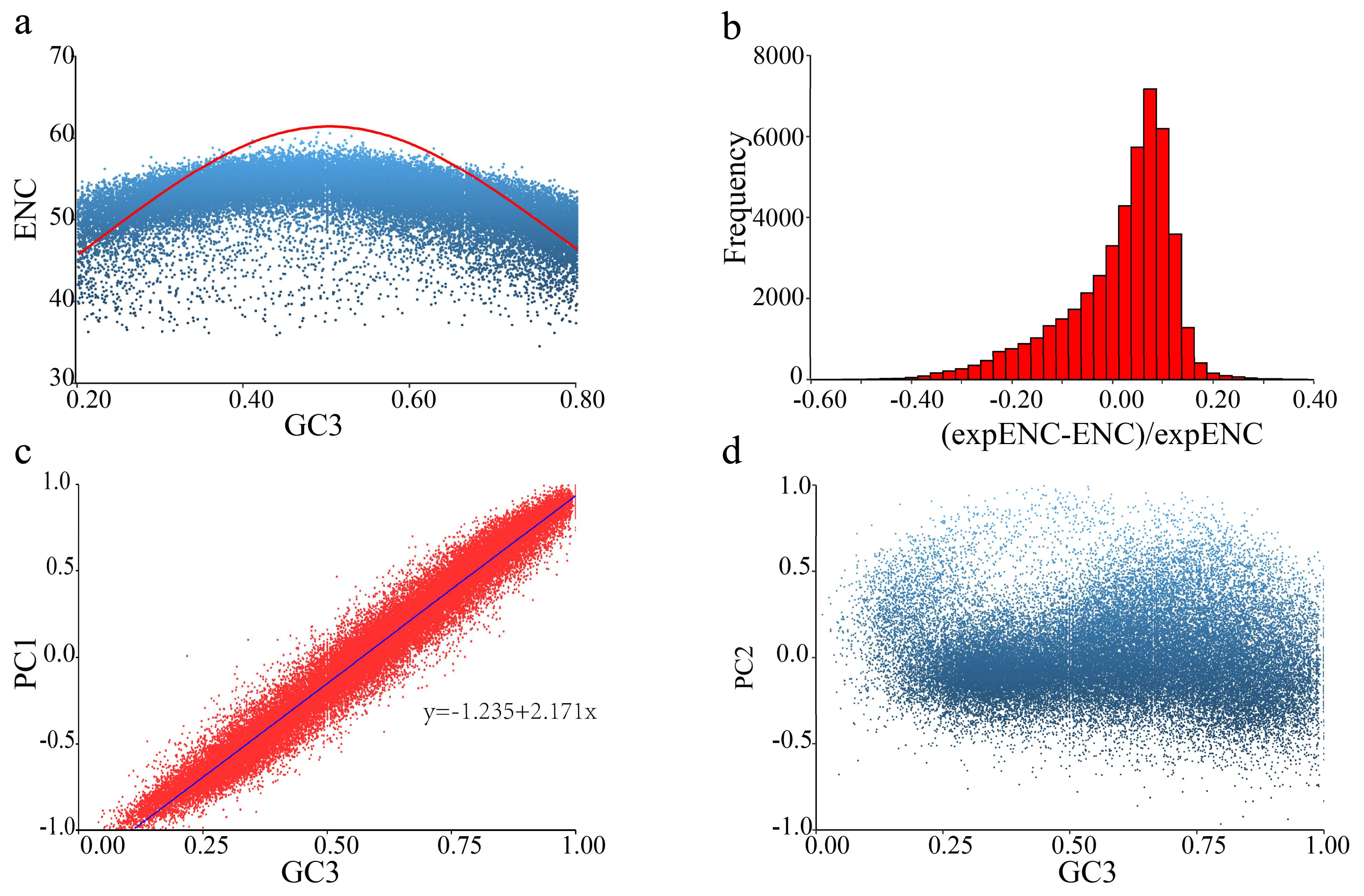
2.1. Codon Usage Bias in T. meditationis
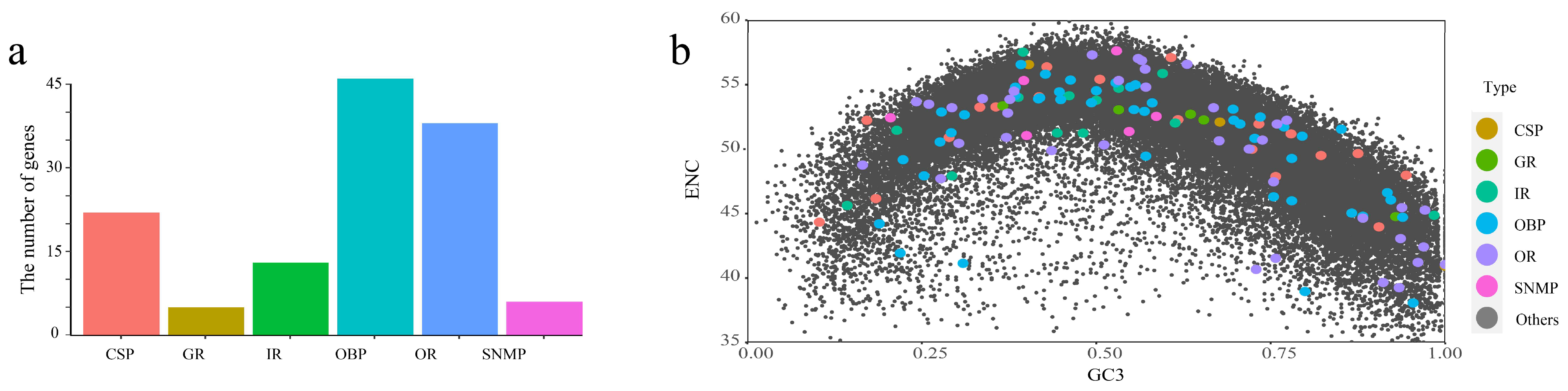
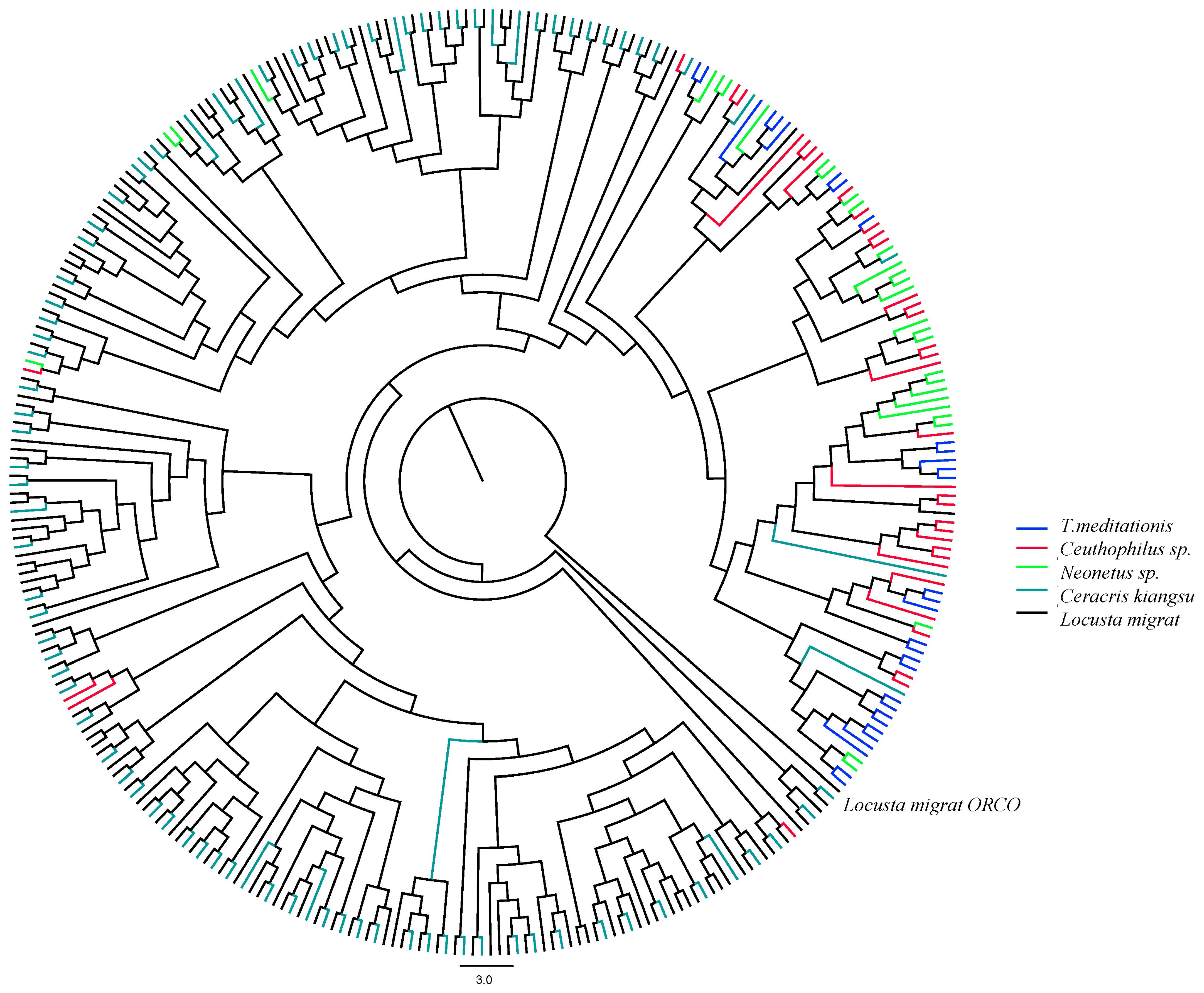
2.2. Evolution of Chemosensory Genes in T. meditationis
2.3. Genes under Positive Selection in T. meditationis
3. Discussion
3.1. Weak Codon Usage in T. meditationis
3.2. Chemosensory Gene Evolution in Camel Crickets
4. Materials and Methods
4.1. T. meditationis Transcriptome Sequencing and Assembly
4.2. CDS Identification and Annotation
4.3. Codon Usage Analysis
4.4. Genome-Wide Codon Usage Bias
4.5. Variation of Codon Bias among Genes
4.6. Chemosensory Gene Analysis
4.7. dN/dS Analysis to Identify Genes under Positive Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AROMO | Aromaticity |
| CDS | Coding sequences |
| COG/egg-NOG | Clusters of orthologous groups of proteins |
| ENC | Effective number of codons |
| FPKM | Fragments per kilobase million |
| GC1 | GC content at the first codon positions |
| GC2 | GC content at the second codon positions |
| GC3 | GC content at the third codon positions |
| GO | Gene Ontology |
| GRAVY | General average of hydrophobicity |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| Nr | NCBI non-redundant protein sequences |
| PCA | Principal component analysis |
| Pfam | Protein family |
| RSCU | Relative synonymous codon usage |
| OBP | Odorant-binding protein |
| CSP | Chemosensory protein |
| OR | Odorant receptor |
| IR | Ionotropic receptor |
| SNMP | Sensory neuron membrane protein |
| GR | Gustatory receptor |
References
- Qin, Y.; Wang, H.; Liu, X.; Li, K. Divided the genus Tachycines Adelung (Orthoptera, Rhaphidophoridae: Aemodogryllinae; Aemodogryllini) from China. Zootaxa 2018, 4374, 451–475. [Google Scholar] [CrossRef]
- Isaac, C.; Orue, P.O.; Iyamu, M.I.; Ehiaghe, J.I.; Isaac, O. Comparative Analysis of Pathogenic Organisms in Cockroaches from Different Community Settings in Edo State, Nigeria. Korean J. Parasitol. 2014, 52, 177–181. [Google Scholar] [CrossRef]
- Qin, Y.; Liu, X.; Li, K. Review of the subgenus Tachycines (Gymnaeta) Adelung, 1902 (Orthoptera, Rhaphidophoridae, Aemodogryllinae, Aemodogryllini). Zootaxa 2019, 4560, 273–310. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Y.; Chen, Q.; Sun, Y.; Lu, Z. WGDdetector: A pipeline for detecting whole genome duplication events using the genome or transcriptome annotations. BMC Bioinform. 2019, 20, 1–6. [Google Scholar] [CrossRef]
- Majewska, M.; Lipka, A.; Paukszto, L.; Jastrzebski, J.P.; Myszczynski, K.; Gowkielewicz, M.; Jozwik, M.; Majewski, M.K. Transcriptome profile of the human placenta. Funct. Integr. Genom. 2017, 17, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Morey, J.S.; Huntington, K.A.B.; Campbell, M.; Clauss, T.M.; Goertz, C.E.; Hobbs, R.C.; Lunardi, D.; Moors, A.J.; Neely, M.G.; Schwacke, L.H.; et al. De novo transcriptome assembly and RNA-Seq expression analysis in blood from beluga whales of Bristol Bay, AK. Mar. Genom. 2017, 35, 77–92. [Google Scholar] [CrossRef]
- Xue, P.; Zhao, X.; Qin, M.; Shi, Z.; Zhang, M.; Gu, W. Transcriptome Analysis of Male Drosophila melanogaster Exposed to Ethylparaben Using Digital Gene Expression Profiling. J. Insect Sci. 2017, 17, 87. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.-L.; Ma, L.-B.; Khan, M.S.; Zhang, X.-X.; Xu, S.-Q.; Xie, J.-Y. Analysis of codon usage patterns in Hirudinaria manillensis reveals a preference for GC-ending codons caused by dominant selection constraints. BMC Genom. 2018, 19, 542. [Google Scholar] [CrossRef]
- Bahiri-Elitzur, S.; Tuller, T. Codon-based indices for modeling gene expression and transcript evolution. Comput. Struct. Biotechnol. J. 2021, 19, 2646–2663. [Google Scholar] [CrossRef]
- Rocha, E.P. Codon usage bias from tRNA’s point of view: Redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004, 14, 2279–2286. [Google Scholar] [CrossRef] [Green Version]
- Eyre-Walker, A. An analysis of codon usage in mammals: Selection or mutation bias? J. Mol. Evol. 1991, 33, 442–449. [Google Scholar] [CrossRef]
- Komar, A.A. The Yin and Yang of codon usage. Hum. Mol. Genet. 2016, 25, R77–R85. [Google Scholar] [CrossRef]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [CrossRef]
- Lloyd, A.T.; Sharp, P. CODONS: A Microcomputer Program for Codon Usage Analysis. J. Hered. 1992, 83, 239–240. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Vang, S.; Yu, J.; Wong, G.K.-S.; Wang, J. Correlation Between Ka/Ks and Ks is Related to Substitution Model and Evolutionary Lineage. J. Mol. Evol. 2009, 68, 414–423. [Google Scholar] [CrossRef]
- Drummond, D.A.; Wilke, C.O. Mistranslation-Induced Protein Misfolding as a Dominant Constraint on Coding-Sequence Evolution. Cell 2008, 134, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Cole, N.C.; Wang, A.A.; Donovan, S.M.; Lee, S.-Y.; Teran-Garcia, M. Strong Kids the STRONG Kids Team Variants in Chemosensory Genes Are Associated with Picky Eating Behavior in Preschool-Age Children. Lifestyle Genom. 2017, 10, 84–92. [Google Scholar] [CrossRef]
- Sun, H.; Bu, L.-A.; Su, S.-C.; Guo, D.; Gao, C.-F.; Wu, S.-F. Knockout of the odorant receptor co-receptor, orco, impairs feeding, mating and egg-laying behavior in the fall armyworm Spodoptera frugiperda. Insect Biochem. Mol. Biol. 2023, 152, 103889. [Google Scholar] [CrossRef]
- Llopis-Giménez, A.; Carrasco-Oltra, T.; Jacquin-Joly, E.; Herrero, S.; Crava, C.M. Coupling Transcriptomics and Behaviour to Unveil the Olfactory System of Spodoptera exigua Larvae. J. Chem. Ecol. 2020, 46, 1017–1031. [Google Scholar] [CrossRef]
- Wang, X.; Kang, L. Molecular Mechanisms of Phase Change in Locusts. Annu. Rev. Èntomol. 2014, 59, 225–244. [Google Scholar] [CrossRef] [Green Version]
- Benton, R. Multigene Family Evolution: Perspectives from Insect Chemoreceptors. Trends Ecol. Evol. 2015, 30, 590–600. [Google Scholar] [CrossRef]
- Arguello, J.R.; Cardoso-Moreira, M.; Grenier, J.K.; Gottipati, S.; Clark, A.G.; Benton, R. Extensive local adaptation within the chemosensory system following Drosophila melanogaster’s global expansion. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Nei, M.; Niimura, Y.; Nozawa, M. The evolution of animal chemosensory receptor gene repertoires: Roles of chance and necessity. Nat. Rev. Genet. 2008, 9, 951–963. [Google Scholar] [CrossRef]
- Mombaerts, P. Seven-Transmembrane Proteins as Odorant and Chemosensory Receptors. Science 1999, 286, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Monahan, K.; Lomvardas, S. Monoallelic Expression of Olfactory Receptors. Annu. Rev. Cell Dev. Biol. 2015, 31, 721–740. [Google Scholar] [CrossRef] [Green Version]
- Wada-Katsumata, A.; Robertson, H.M.; Silverman, J.; Schal, C. Changes in the Peripheral Chemosensory System Drive Adaptive Shifts in Food Preferences in Insects. Front. Cell. Neurosci. 2018, 12, 281. [Google Scholar] [CrossRef] [Green Version]
- Robertson, H.M.; Baits, R.L.; Walden, K.K.; Wada-Katsumata, A.; Schal, C. Enormous expansion of the chemosensory gene repertoire in the omnivorous German cockroach Blattella germanica. J. Exp. Zool. Part B Mol. Dev. Evol. 2018, 330, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Fang, X.; Yang, P.; Jiang, X.; Jiang, F.; Zhao, D.; Li, B.; Cui, F.; Wei, J.; Ma, C.; et al. The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 2014, 5, 2957. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.-T.; Li, L.; Zhou, X.-R.; Tan, Y.; Pang, B.-P. Identification and expression profiling of candidate chemosensory membrane proteins in the band-winged grasshopper, Oedaleus asiaticus. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 30, 33–44. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Fages-Lartaud, M.; Hundvin, K.; Hohmann-Marriott, M.F. “Mechanisms governing codon usage bias and the implications for protein expression in the chloroplast of Chlamydomonas reinhardtii ”. Plant J. 2022, 112, 919–945. [Google Scholar] [CrossRef] [PubMed]
- Dedon, P.C.; Begley, T.J. Dysfunctional tRNA reprogramming and codon-biased translation in cancer. Trends Mol. Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Yang, Q.; Li, L.; Dang, Y.; Zhou, Z.; Chen, S.; Liu, Y. Adaptation of codon usage to tRNA I34 modification controls translation kinetics and proteome landscape. PLOS Genet. 2020, 16, e1008836. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.-F.; Xu, N.; Kleinhofs, A.; Zhou, J. Quantitative relationship between synonymous codon usage bias and GC composition across unicellular genomes. BMC Evol. Biol. 2004, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wint, R.; Salamov, A.; Grigoriev, I.V. Kingdom-Wide Analysis of Fungal Protein-Coding and tRNA Genes Reveals Conserved Patterns of Adaptive Evolution. Mol. Biol. Evol. 2022, 39, msab372. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, A.; Vitale, L.; Pelleri, M.C.; Strippoli, P. Universal tight correlation of codon bias and pool of RNA codons (codonome): The genome is optimized to allow any distribution of gene expression values in the transcriptome from bacteria to humans. Genomics 2013, 101, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, Q.; Zhao, F. Synonymous but Not Silent: The Codon Usage Code for Gene Expression and Protein Folding. Annu. Rev. Biochem. 2021, 90, 375–401. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Subramanian, S. Nearly Neutrality and the Evolution of Codon Usage Bias in Eukaryotic Genomes. Genetics 2008, 178, 2429–2432. [Google Scholar] [CrossRef] [Green Version]
- Galtier, N.; Roux, C.; Rousselle, M.; Romiguier, J.; Figuet, E.; Glémin, S.; Bierne, N.; Duret, L. Codon Usage Bias in Animals: Disentangling the Effects of Natural Selection, Effective Population Size, and GC-Biased Gene Conversion. Mol. Biol. Evol. 2018, 35, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Chen, H.; Shi, F. Remarks on the genus Tachycines Adelung, 1902 (Orthoptera: Rhaphidophoridae: Aemodogryllinae) with description of eight new species from caves in southern China. Zootaxa 2020, 4809, 71–94. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhou, Z.; Zheng, X.; Wang, T.; Ma, L.; Shi, F. Phylogeny and phylogeography of Diestramima cave crickets (Orthoptera: Rhaphidophoridae): Speciation driven by multiple dispersal and vicariance events. Syst. Èntomol. 2022, 47, 179–201. [Google Scholar] [CrossRef]
- Zhou, Y.-T.; Li, L.; Zhou, X.-R.; Tan, Y.; Pang, B.-P. Three Chemosensory Proteins Involved in Chemoreception of Oedaleus asiaticus (Orthopera: Acridoidea). J. Chem. Ecol. 2019, 46, 138–149. [Google Scholar] [CrossRef]
- Argüello, D.; Chavez, E.; Lauryssen, F.; Vanderschueren, R.; Smolders, E.; Montalvo, D. Soil properties and agronomic factors affecting cadmium concentrations in cacao beans: A nationwide survey in Ecuador. Sci. Total. Environ. 2019, 649, 120–127. [Google Scholar] [CrossRef]
- He, M.; Ma, Y.-F.; Guo, H.; Liu, X.-Z.; Long, G.-J.; Wang, Q.; Dewer, Y.; Zhang, F.; He, P. Genome-wide identification and expression pattern analysis of novel chemosensory genes in the German cockroach Blattella germanica. Genomics 2022, 114, 110310. [Google Scholar] [CrossRef]
- Brand, P.; Ramírez, S.R.; Leese, F.; Quezada-Euan, J.J.G.; Tollrian, R.; Eltz, T. Rapid evolution of chemosensory receptor genes in a pair of sibling species of orchid bees (Apidae: Euglossini). BMC Evol. Biol. 2015, 15, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.N.; Keeling, C.I.; Mitchell, R.F. Genomic content of chemosensory genes correlates with host range in wood-boring beetles (Dendroctonus ponderosae, Agrilus planipennis, and Anoplophora glabripennis). BMC Genom. 2019, 20, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Arensburger, P.; Megy, K.; Waterhouse, R.M.; Abrudan, J.; Amedeo, P.; Antelo, B.; Bartholomay, L.; Bidwell, S.; Caler, E.; Camara, F.; et al. Sequencing of Culex quinquefasciatus Establishes a Platform for Mosquito Comparative Genomics. Science 2010, 330, 86–88. [Google Scholar] [CrossRef] [Green Version]
- McBride, C.S. Rapid evolution of smell and taste receptor genes during host specialization in Drosophila sechellia. Proc. Natl. Acad. Sci. USA 2007, 104, 4996–5001. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, Y.-T.; Jiang, G.-F. The transcriptome analysis of the bamboo grasshopper provides insights into hypothermic stress acclimation. Int. J. Biol. Macromol. 2019, 134, 237–246. [Google Scholar] [CrossRef]
- Sanno, R.; Kataoka, K.; Hayakawa, S.; Ide, K.; Nguyen, C.N.; Nguyen, T.P.; Le, B.T.N.; Kim, O.T.P.; Mineta, K.; Takeyama, H.; et al. Comparative Analysis of Mitochondrial Genomes in Gryllidea (Insecta: Orthoptera): Implications for Adaptive Evolution in Ant-Loving Crickets. Genome Biol. Evol. 2021, 13, evab222. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Minei, R.; Ide, K.; Ogura, A.; Takeyama, H.; Takeda, M.; Suzuki, T.; Yura, K.; Asahi, T. The Draft Genome Dataset of the Asian Cricket Teleogryllus occipitalis for Molecular Research Toward Entomophagy. Front. Genet. 2020, 11, 470. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Rombel, I.T.; Sykes, K.F.; Rayner, S.; Johnston, S.A. ORF-FINDER: A vector for high-throughput gene identification. Gene 2002, 282, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Réau, M.; Lagarde, N.; Zagury, J.-F.; Montes, M. Nuclear Receptors Database Including Negative Data (NR-DBIND): A Database Dedicated to Nuclear Receptors Binding Data Including Negative Data and Pharmacological Profile. J. Med. Chem. 2018, 62, 2894–2904. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. DAMBE5: A Comprehensive Software Package for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2021, 49, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Bhattacharyya, T.K.; Ghosh, T.C. Synonymous Codon Usage in Lactococcus lactis: Mutational Bias Versus Translational Selection. J. Biomol. Struct. Dyn. 2004, 21, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Khelifi, A.; Meunier, J.; Duret, L.; Mouchiroud, D. GC Content Evolution of the Human and Mouse Genomes: Insights from the Study of Processed Pseudogenes in Regions of Different Recombination Rates. J. Mol. Evol. 2006, 62, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Nag, D.; Mazumder, T.H.; Uddin, A. Codon usage pattern and prediction of gene expression level in Bungarus species. Gene 2017, 604, 48–60. [Google Scholar] [CrossRef]
- Duret, L. Evolution of synonymous codon usage in metazoans. Curr. Opin. Genet. Dev. 2002, 12, 640–649. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z. DAMBE: Software Package for Data Analysis in Molecular Biology and Evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Scharf, M.E.; Bennett, G.W. Behavioral and Physiological Resistance of the German Cockroach to Gel Baits (Blattodea: Blattellidae). J. Econ. Èntomol. 2004, 97, 2067–2072. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | Codon | RSCU | AA | Codon | RSCU | AA | Codon | RSCU | AA | Codon | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ala | GCU | 1.122 | His | CAC | 1.003 | Pro | CCA | 1.076 | Ser | UCG | 0.768 |
| GCG | 0.687 | CAU | 0.997 | CCC | 1.014 | UCU | 1.187 | ||||
| GCC | 1.301 | Ile | AUU | 1.190 | CCU | 1.182 | Thr | ACC | 1.223 | ||
| GCA | 0.891 | AUA | 0.465 | CCG | 0.727 | ACA | 1.021 | ||||
| Cys | UGU | 1.010 | AUC | 1.345 | Gln | CAA | 0.849 | ACG | 0.674 | ||
| UGC | 0.990 | Lys | AAA | 0.784 | CAG | 1.151 | ACU | 1.082 | |||
| Asp | GAU | 1.023 | AAG | 1.216 | Arg | AGA | 1.187 | GUU | 1.048 | ||
| GAC | 0.977 | Leu | CUA | 0.392 | AGG | 0.813 | GUG | 1.282 | |||
| Glu | GAG | 1.067 | CUC | 1.148 | CGA | 0.772 | GUC | 1.126 | |||
| GAA | 0.933 | CUG | 1.478 | CGC | 1.460 | GUA | 0.545 | ||||
| Phe | UUU | 0.847 | CUU | 0.982 | CGG | 0.668 | Trp | UGG | 1.000 | ||
| UUC | 1.153 | UUA | 0.793 | CGU | 1.100 | Tyr | UAC | 1.083 | |||
| Gly | GGU | 1.153 | UUG | 1.207 | Ser | AGC | 1.060 | UAU | 0.917 | ||
| GGG | 0.519 | Met | AUG | 1.000 | AGU | 0.940 | |||||
| GGC | 1.388 | Asn | AAC | 1.062 | UCA | 1.048 | |||||
| GGA | 0.941 | AAU | 0.938 | UCC | 0.998 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.-H.; Guan, D.-L.; Xu, S.-Q.; Huang, H. De Novo Assembly and Characterization of the Transcriptome of an Omnivorous Camel Cricket (Tachycines meditationis). Int. J. Mol. Sci. 2023, 24, 4005. https://doi.org/10.3390/ijms24044005
Lu J-H, Guan D-L, Xu S-Q, Huang H. De Novo Assembly and Characterization of the Transcriptome of an Omnivorous Camel Cricket (Tachycines meditationis). International Journal of Molecular Sciences. 2023; 24(4):4005. https://doi.org/10.3390/ijms24044005
Chicago/Turabian StyleLu, Jun-Hui, De-Long Guan, Sheng-Quan Xu, and Huateng Huang. 2023. "De Novo Assembly and Characterization of the Transcriptome of an Omnivorous Camel Cricket (Tachycines meditationis)" International Journal of Molecular Sciences 24, no. 4: 4005. https://doi.org/10.3390/ijms24044005