
Investigation of Molecular Mechanisms Involved in Sensitivity to the Anti-Cancer Activity of Costunolide in Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
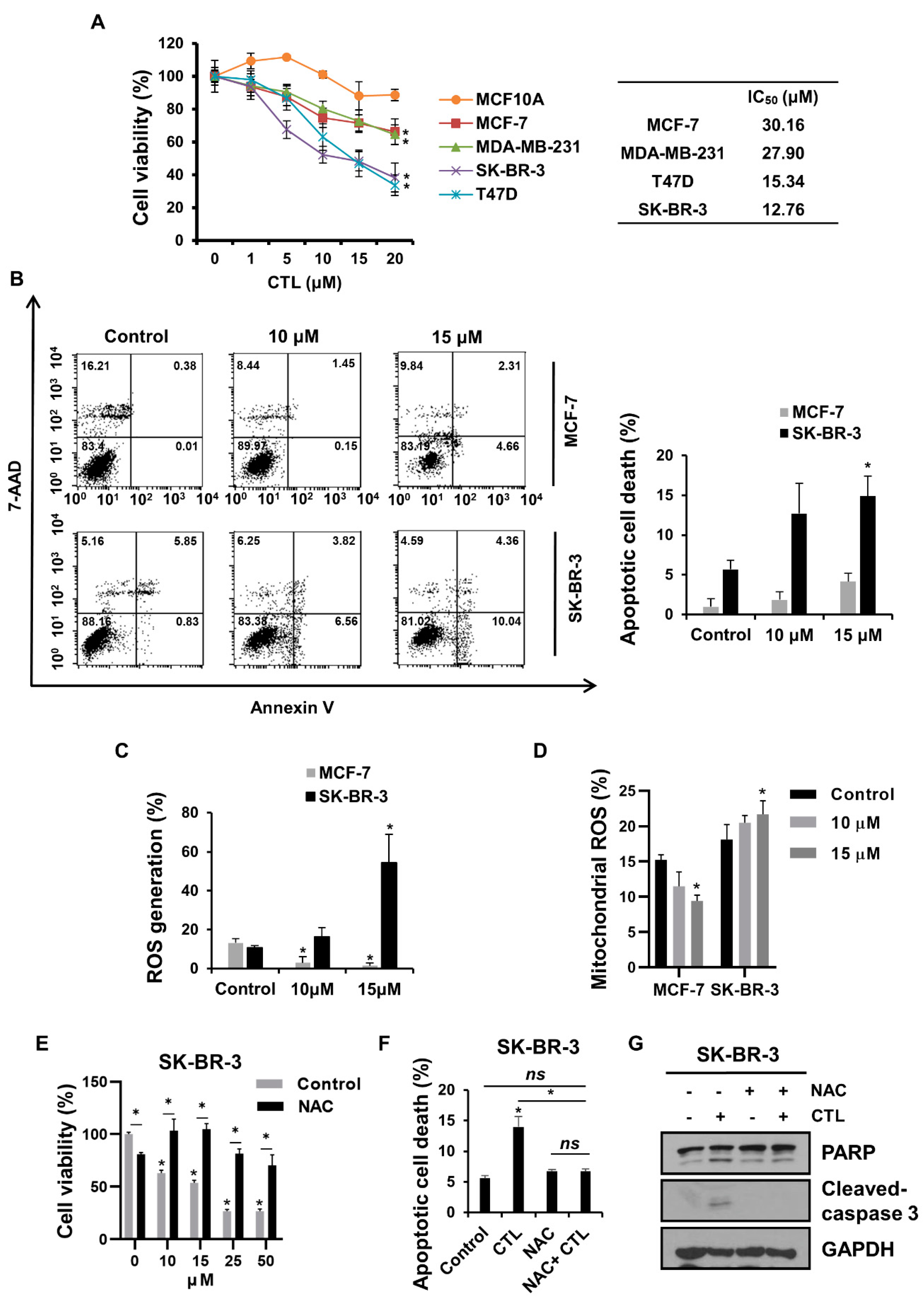
2.1. CTL Promotes Apoptotic Breast Cancer Cell Death in a ROS-Dependent Manner
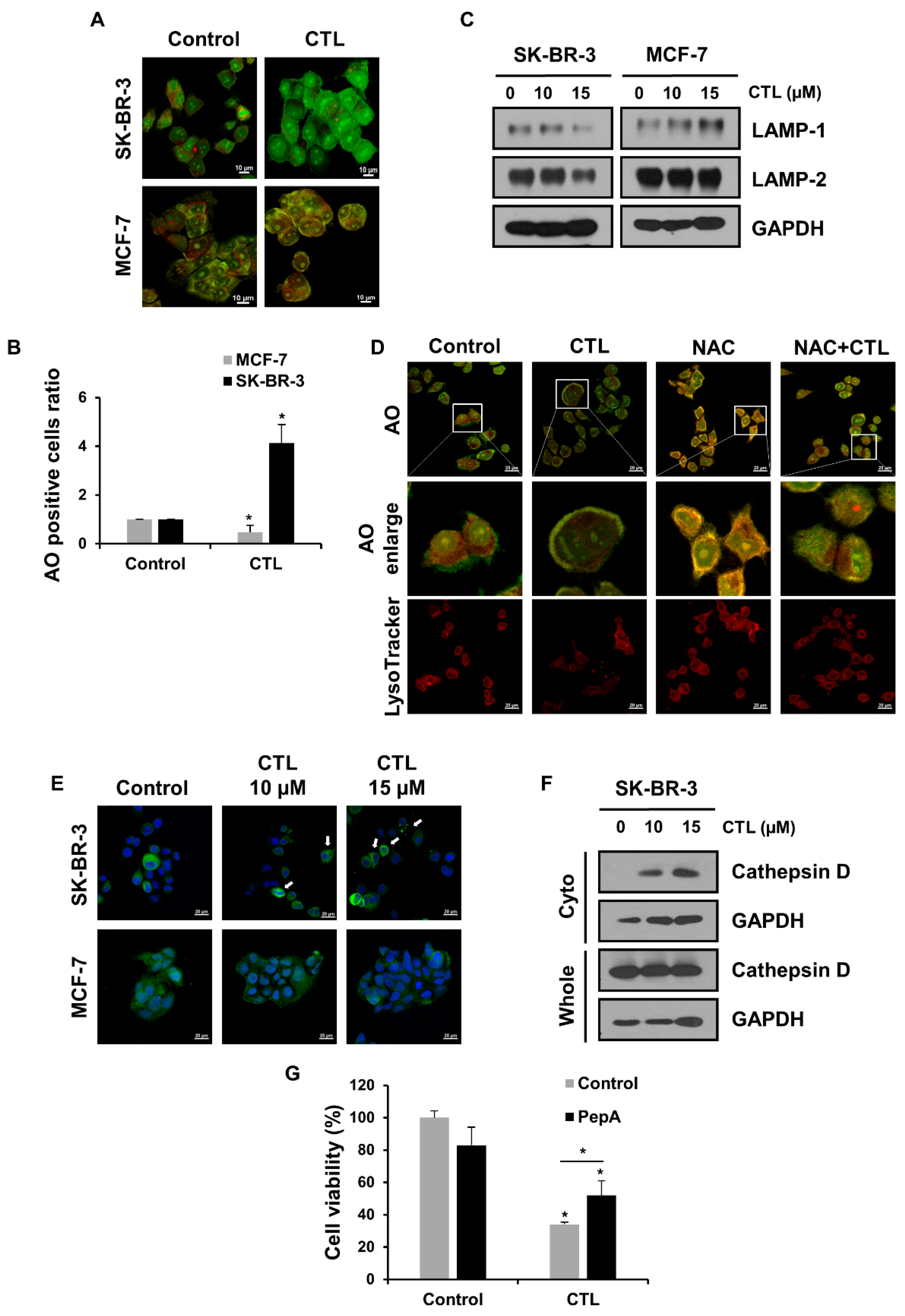
2.2. CTL-Mediated ROS Production Induces LMP and the Release of Cathepsin D in SK-BR-3 Cells
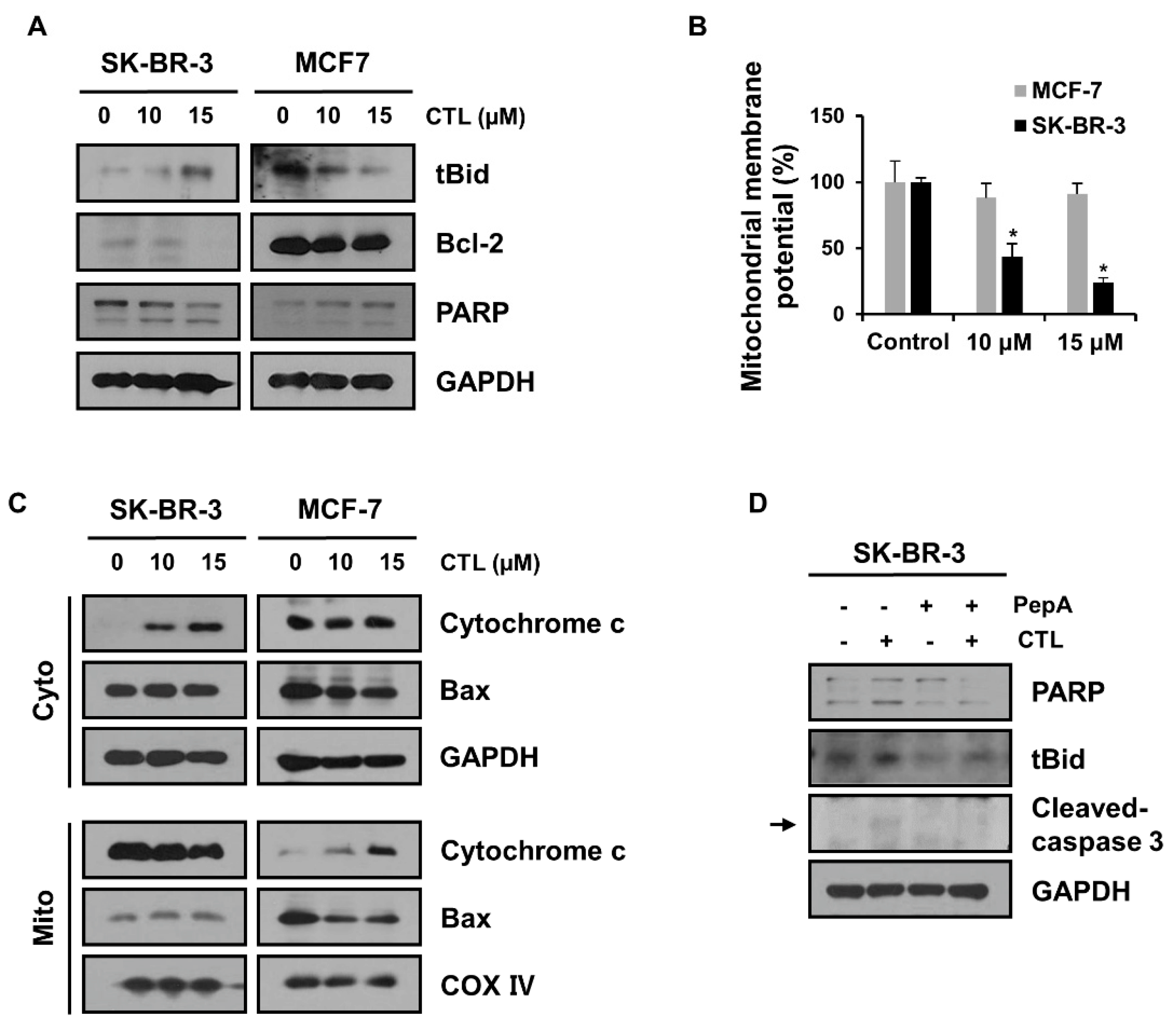
2.3. The Released Cathepsin D Leads to MOMP and Subsequent Mitochondrial Apoptosis in SK-BR-3 Cells
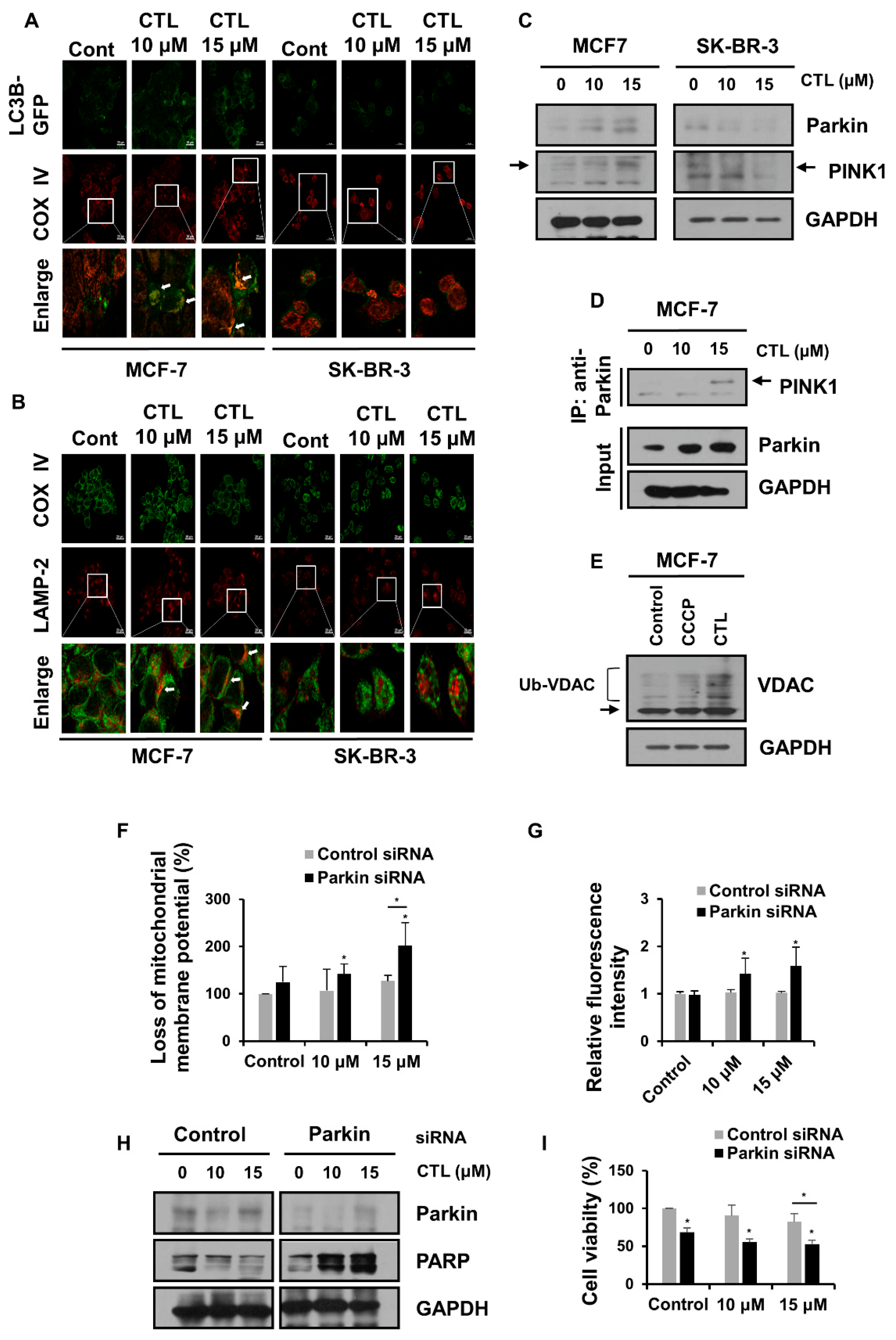
2.4. Mitophagy Protects MCF-7 Cells from CTL-Induced Mitochondrial Damage and Cell Death
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and Cell Viability Assay
4.3. Western Blot Analysis
4.4. Cell Fractionation
4.5. Apoptosis Analysis
4.6. Intracellular ROS Production
4.7. Loss of Mitochondrial Membrane Potential (MMP)
4.8. Acridine Orange (AO) Staining
4.9. Plasmid and Transfection
4.10. Confocal Microscopy
4.11. Immunoprecipitation (IP) Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.J.; Han, W. A Review of the Epidemiology of Breast Cancer in Asia: Focus on Risk Factors. Asian Pac. J. Cancer Prev. 2020, 21, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The, L. Breast cancer targeted therapy: Successes and challenges. Lancet 2017, 389, 2350. [Google Scholar]
- Ju, J.; Zhu, A.J.; Yuan, P. Progress in targeted therapy for breast cancer. Chronic Dis. Transl. Med. 2018, 4, 164–175. [Google Scholar]
- Fehm, T.N.; Stickeler, E.; Fasching, P.A.; Janni, W.; Kolberg-Liedtke, C.; Kolberg, H.C.; Luftner, D.; Muller, V.; Schutz, F.; Thomssen, C.; et al. Update Breast Cancer 2021 Part 3-Current Developments in the Treatment of Early Breast Cancer: Review and Assessment of Specialised Treatment Scenarios by an International Expert Panel. Geburtshilfe Frauenheilkd. 2021, 81, 654–665. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar]
- Kang, J.; Pervaiz, S. Mitochondria: Redox metabolism and dysfunction. Biochem. Res. Int. 2012, 2012, 896751. [Google Scholar]
- Zablocka, A.; Janusz, M. The two faces of reactive oxygen species. Postepy. Hig. Med. Dosw. 2008, 62, 118–124. [Google Scholar]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Osbild, S.; Brault, L.; Battaglia, E.; Bagrel, D. Resistance to cisplatin and adriamycin is associated with the inhibition of glutathione efflux in MCF-7-derived cells. Anticancer Res. 2006, 26, 3595–3600. [Google Scholar] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [PubMed]
- Dadsena, S.; Jenner, A.; Garcia-Saez, A.J. Mitochondrial outer membrane permeabilization at the single molecule level. Cell. Mol. Life Sci. 2021, 78, 3777–3790. [Google Scholar] [PubMed]
- Pena-Blanco, A.; Garcia-Saez, A.J. Bax, Bak and beyond-mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Tu, H.C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar]
- Tan, M.; Liu, W.; Liu, F.; Zhang, W.; Gao, H.; Cheng, J.; Chen, Y.; Wang, Z.; Cao, Y.; Ran, H. Silk Fibroin-Coated Nanoagents for Acidic Lysosome Targeting by a Functional Preservation Strategy in Cancer Chemotherapy. Theranostics 2019, 9, 961–973. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Antitumor activity and mechanism of costunolide and dehydrocostus lactone: Two natural sesquiterpene lactones from the Asteraceae family. Biomed Pharm. 2020, 125, 109955. [Google Scholar]
- Fang, Y.; Li, J.; Wu, Y.; Gui, J.; Shen, Y. Costunolide Inhibits the Growth of OAW42-A Multidrug-Resistant Human Ovarian Cancer Cells by Activating Apoptotic and Autophagic Pathways, Production of Reactive Oxygen Species (ROS), Cleaved Caspase-3 and Cleaved Caspase-9. Med. Sci. Monit. 2019, 25, 3231–3237. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Wu, D.; Cheng, W.; Gao, J.; Zhang, Z.; Ge, J.; Zhou, W.; Xu, Z. Costunolide Induces Autophagy and Apoptosis by Activating ROS/MAPK Signaling Pathways in Renal Cell Carcinoma. Front Oncol. 2020, 10, 582273. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Huang, X.; Lei, X.; Jin, Z.; Wu, M.; Liu, X.; Huang, Y.; Zhao, X.; Xiong, Y.; Sun, J.; et al. Costunolide-Induced Apoptosis via Promoting the Reactive Oxygen Species and Inhibiting AKT/GSK3beta Pathway and Activating Autophagy in Gastric Cancer. Front. Cell Dev. Biol. 2021, 9, 722734. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Park, S.; Zhang, H.; Park, S.; Kwon, W.; Kim, E.; Zhang, X.; Jang, S.; Yoon, D.; Choi, S.K.; et al. Targeting AKT with costunolide suppresses the growth of colorectal cancer cells and induces apoptosis in vitro and in vivo. J. Exp. Clin. Cancer Res. 2021, 40, 114. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, Y.; Fan, J.; Lin, X.; Liu, C.; Xu, Y.; Ji, W.; Yan, C.; Su, C. Costunolide and dehydrocostuslactone combination treatment inhibit breast cancer by inducing cell cycle arrest and apoptosis through c-Myc/p53 and AKT/14-3-3 pathway. Sci. Rep. 2017, 7, 41254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitchai, D.; Roy, A.; Banu, S. In vitro and in silico evaluation of NF-kappaB targeted costunolide action on estrogen receptor-negative breast cancer cells--a comparison with normal breast cells. Phytother Res. 2014, 28, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Manikkam, R. Cytotoxic Impact of Costunolide Isolated from Costus speciosus on Breast Cancer via Differential Regulation of Cell Cycle-An In-vitro and In-silico Approach. Phytother Res. 2015, 29, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, T.; Wang, G.D.; Ma, C.; Zhou, Y.F. Costunolide, an active sesquiterpene lactone, induced apoptosis via ROS-mediated ER stress and JNK pathway in human U2OS cells. Biomed Pharm. 2016, 80, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Liu, Y.; Hu, Y.; Liu, X.; Jiang, H.; Yang, S.; Shao, Z.; Xia, Y.; Xiong, L. ROS-mediated lysosomal membrane permeabilization is involved in bupivacaine-induced death of rabbit intervertebral disc cells. Redox Biol. 2018, 18, 65–76. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Stroikin, Y.; Mild, H.; Johansson, U.; Roberg, K.; Ollinger, K. Lysosome-targeted stress reveals increased stability of lipofuscin-containing lysosomes. Age 2008, 30, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sorensen, T.; Rafn, B.; Bottzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jaattela, M. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Zhao, L.P.; Ye, J.Y.; Yang, L.; Huang, Y.; Jiang, X.P.; Zhang, Q.; Jia, J.Z.; Zhang, D.X.; Huang, Y. The Lysosomal Membrane Protein Lamp2 Alleviates Lysosomal Cell Death by Promoting Autophagic Flux in Ischemic Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelqvist, H.; Johansson, A.C.; Linderoth, E.; Johansson, U.; Antonsson, B.; Steinfeld, R.; Kagedal, K.; Ollinger, K. Lysosome-mediated apoptosis is associated with cathepsin D-specific processing of bid at Phe24, Trp48, and Phe183. Ann. Clin. Lab. Sci. 2012, 42, 231–242. [Google Scholar] [PubMed]
- Johansson, A.C.; Steen, H.; Ollinger, K.; Roberg, K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003, 10, 1253–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emert-Sedlak, L.; Shangary, S.; Rabinovitz, A.; Miranda, M.B.; Delach, S.M.; Johnson, D.E. Involvement of cathepsin D in chemotherapy-induced cytochrome c release, caspase activation, and cell death. Mol. Cancer Ther. 2005, 4, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Scaduto, R.C., Jr.; Grotyohann, L.W. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Zhang, H.T.; Mi, L.; Wang, T.; Yuan, L.; Li, X.H.; Dong, L.S.; Zhao, P.; Fu, J.L.; Yao, B.Y.; Zhou, Z.C. PINK1/Parkin-mediated mitophagy play a protective role in manganese induced apoptosis in SH-SY5Y cells. Toxicol. Vitr. 2016, 34, 212–219. [Google Scholar] [CrossRef]
- Cui, Y.; Li, B.; Du, J.; Huo, S.; Song, M.; Shao, B.; Wang, B.; Li, Y. Dibutyl phthalate causes MC3T3-E1 cell damage by increasing ROS to promote the PINK1/Parkin-mediated mitophagy. Environ. Toxicol. 2022, 37, 2341–2353. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, (3), 794–798. [Google Scholar]
- Choi, Y.K.; Seo, H.S.; Choi, H.S.; Choi, H.S.; Kim, S.R.; Shin, Y.C.; Ko, S.G. Induction of Fas-mediated extrinsic apoptosis, p21WAF1-related G2/M cell cycle arrest and ROS generation by costunolide in estrogen receptor-negative breast cancer cells, MDA-MB-231. Mol. Cell Biochem. 2012, 363, 119–128. [Google Scholar] [CrossRef]
- Yan, Z.; Xu, T.; An, Z.; Hu, Y.; Chen, W.; Ma, J.; Shao, C.; Zhu, F. Costunolide induces mitochondria-mediated apoptosis in human gastric adenocarcinoma BGC-823 cells. BMC Complement. Altern. Med. 2019, 19, 151. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Yi, J.K.; Lim, S.G.; Park, S.; Zhang, H.; Kim, E.; Jang, S.; Lee, M.H.; Liu, K.; Kim, K.R.; et al. Costunolide Induces Apoptosis via the Reactive Oxygen Species and Protein Kinase B Pathway in Oral Cancer Cells. Int. J. Mol. Sci. 2021, 22, 7509. [Google Scholar] [CrossRef]
- Zhuge, W.; Chen, R.; Vladimir, K.; Dong, X.; Zia, K.; Sun, X.; Dai, X.; Bao, M.; Shen, X.; Liang, G. Costunolide specifically binds and inhibits thioredoxin reductase 1 to induce apoptosis in colon cancer. Cancer Lett. 2018, 412, 46–58. [Google Scholar] [CrossRef]
- Chen, J.; Chen, B.; Zou, Z.; Li, W.; Zhang, Y.; Xie, J.; Liu, C. Costunolide enhances doxorubicin-induced apoptosis in prostate cancer cells via activated mitogen-activated protein kinases and generation of reactive oxygen species. Oncotarget 2017, 8, 107701–107715. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Cho, S.G.; Woo, S.M.; Yun, Y.J.; Jo, J.; Kim, W.; Shin, Y.C.; Ko, S.G. Saussurea lappa Clarke-Derived Costunolide Prevents TNF alpha -Induced Breast Cancer Cell Migration and Invasion by Inhibiting NF- kappa B Activity. Evid. Based Complement. Altern. Med. 2013, 2013, 936257. [Google Scholar] [CrossRef] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: Involvement of reactive oxygen species and cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Repnik, U.; Turk, B. Lysosomal-mitochondrial cross-talk during cell death. Mitochondrion 2010, 10, 662–669. [Google Scholar] [CrossRef]
- Vianello, C.; Cocetta, V.; Catanzaro, D.; Dorn, G.W., 2nd; De Milito, A.; Rizzolio, F.; Canzonieri, V.; Cecchin, E.; Roncato, R.; Toffoli, G.; et al. Cisplatin resistance can be curtailed by blunting Bnip3-mediated mitochondrial autophagy. Cell Death Dis. 2022, 13, 398. [Google Scholar] [CrossRef]
- Yao, N.; Wang, C.; Hu, N.; Li, Y.; Liu, M.; Lei, Y.; Chen, M.; Chen, L.; Chen, C.; Lan, P.; et al. Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis. 2019, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wang, T.; Liu, Y.; Li, X.; Xu, S.; Wu, C.; Zou, H.; Cao, M.; Jin, G.; Lang, J.; et al. Mitophagy promotes sorafenib resistance through hypoxia-inducible ATAD3A dependent Axis. J. Exp. Clin. Cancer Res. 2020, 39, 274. [Google Scholar] [CrossRef]
- Bahar, E.; Han, S.Y.; Kim, J.Y.; Yoon, H. Chemotherapy Resistance: Role of Mitochondrial and Autophagic Components. Cancers 2022, 14, 1462. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.-J.; Choi, Y.K.; Ko, S.-G.; Cheon, C.; Kim, T.Y. Investigation of Molecular Mechanisms Involved in Sensitivity to the Anti-Cancer Activity of Costunolide in Breast Cancer Cells. Int. J. Mol. Sci. 2023, 24, 4009. https://doi.org/10.3390/ijms24044009
Choi Y-J, Choi YK, Ko S-G, Cheon C, Kim TY. Investigation of Molecular Mechanisms Involved in Sensitivity to the Anti-Cancer Activity of Costunolide in Breast Cancer Cells. International Journal of Molecular Sciences. 2023; 24(4):4009. https://doi.org/10.3390/ijms24044009
Chicago/Turabian StyleChoi, Yu-Jeong, Youn Kyung Choi, Seong-Gyu Ko, Chunhoo Cheon, and Tai Young Kim. 2023. "Investigation of Molecular Mechanisms Involved in Sensitivity to the Anti-Cancer Activity of Costunolide in Breast Cancer Cells" International Journal of Molecular Sciences 24, no. 4: 4009. https://doi.org/10.3390/ijms24044009