
Alzheimer’s Disease: A Brief History of Immunotherapies Targeting Amyloid β
 and
and
Abstract
:1. Introduction
2. Disease Mechanism and Pathogenesis
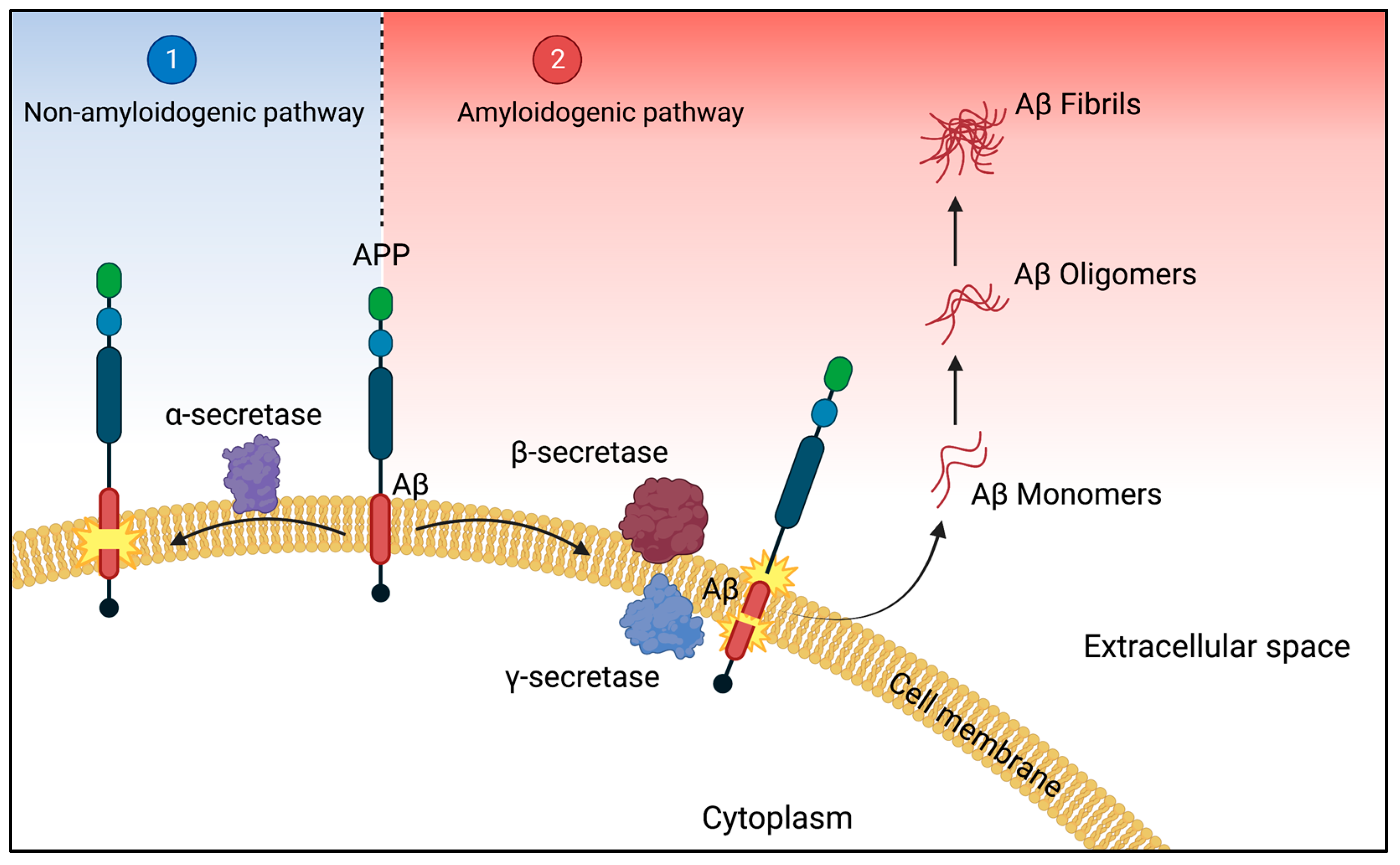
2.1. The Amyloid Pathway
2.2. Pathogenesis and Clinical Stages
3. Current Treatment Strategies Targeting Aβ
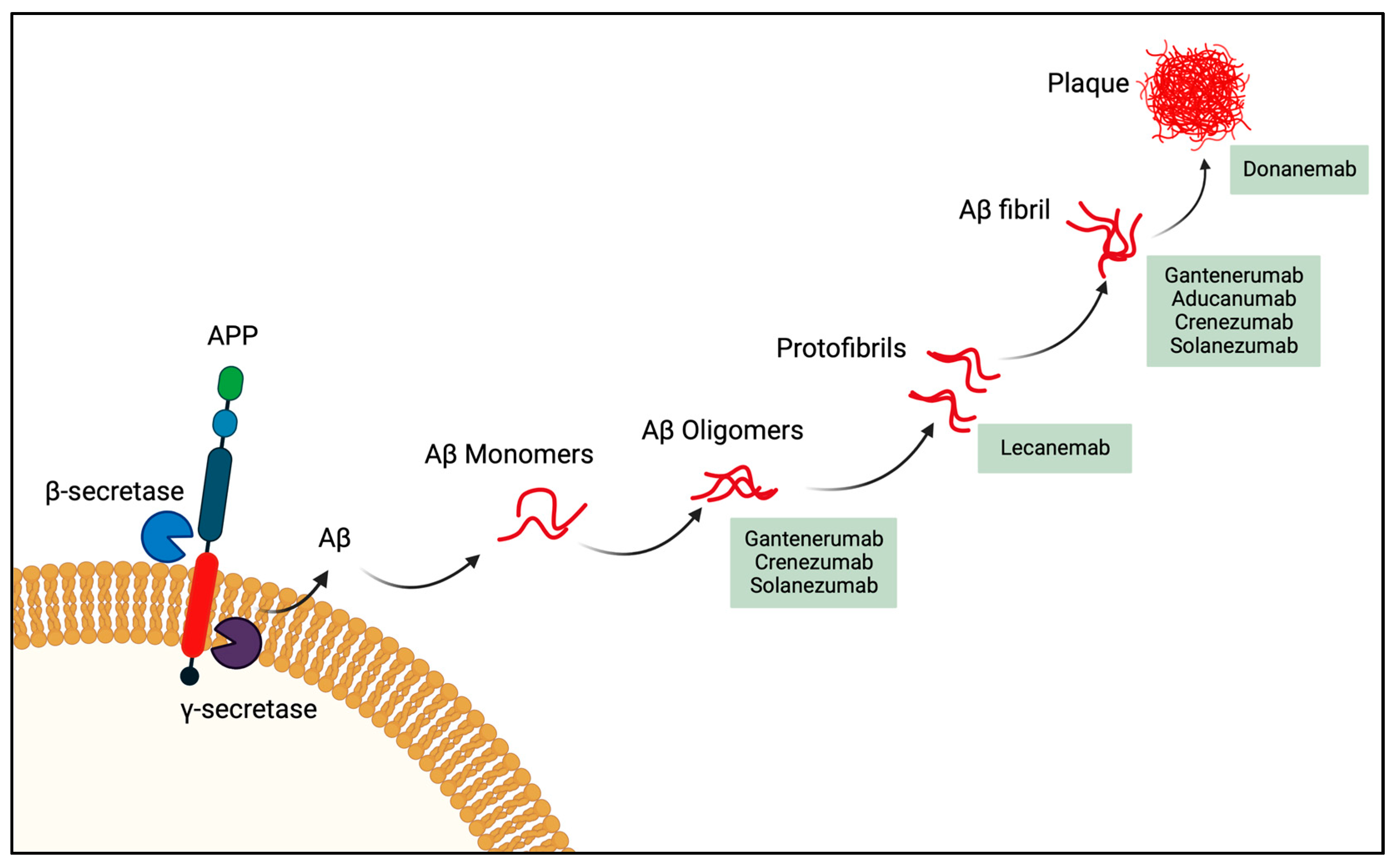
3.1. Passive Immunotherapy Phase III Clinical Agents
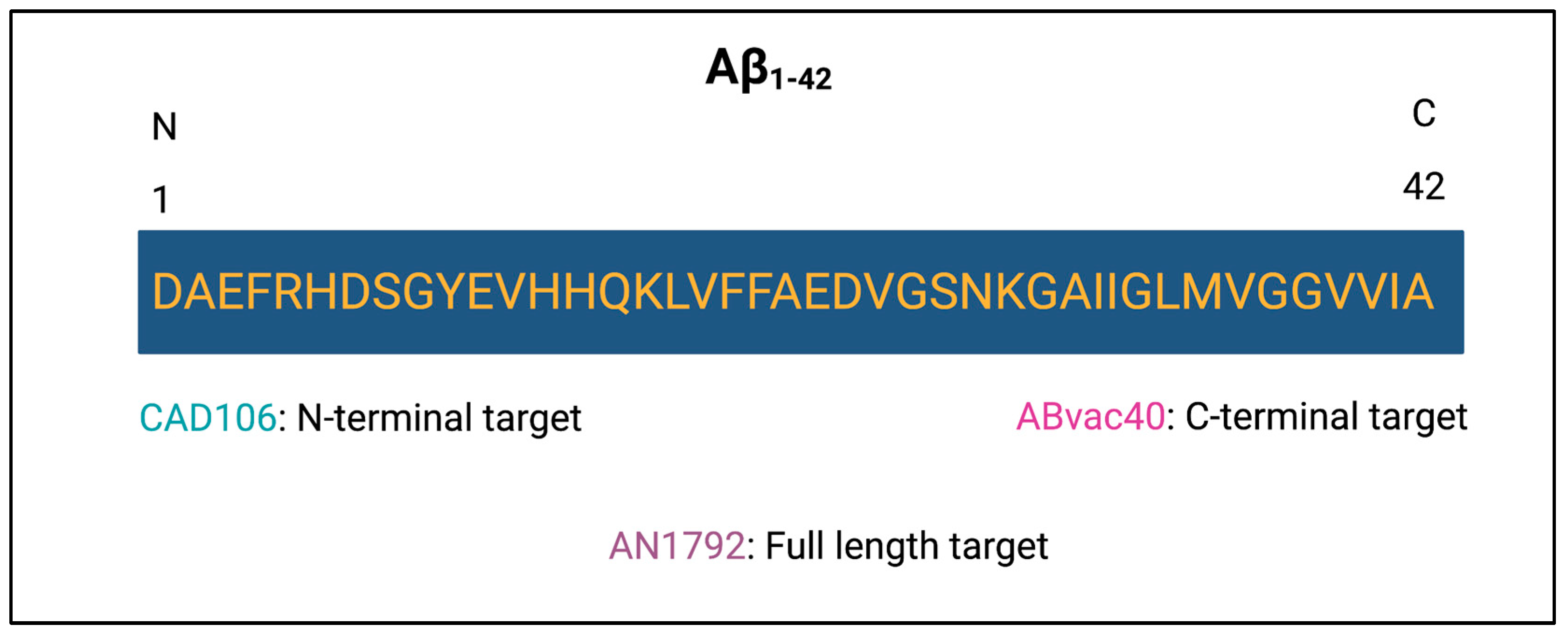
3.2. Active Immunotherapy
4. Alzheimer’s Disease Risk Factors
5. Summary and Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-β protein |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| mAb | Monoclonal antibody |
| VLP | Virus-like particle |
| LMIC | Low and middle-income countries |
| NFT | Neurofibrillary tangles |
References
- Hartl, F.U. Protein Misfolding Diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Terio, K.A.; O’Brien, T.; Lamberski, N.; Famula, T.R.; Munson, L. Amyloidosis in Black-Footed Cats (Felis nigripes). Vet. Pathol. 2004, 45, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermark, P.; Lundmark, K.; Westermark, G.T. Fibrils from Designed Non-Amyloid-Related Synthetic Peptides Induce AA-Amyloidosis during Inflammation in an Animal Model. PLoS ONE 2009, 4, e6041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjernberg, L.O.; Rising, A.; Johansson, J.; Jaudzems, K.; Westermark, P. Transmissible Amyloid. J. Intern. Med. 2016, 280, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galzitskaya, O.V.; Galushko, E.I.; Selivanova, O.M. Studies of the Process of Amyloid Formation by Aβ Peptide. Biochemistry 2018, 83, 62–80. [Google Scholar] [CrossRef]
- Fish, P.V.; Steadman, D.; Bayle, E.D.; Whiting, P. New Approaches for the Treatment of Alzheimer’s Disease. Bioorg. Med. Chem. Lett. 2019, 29, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.-C.; Crawford, F.; Houlden, H.; Warren, A.; HUghes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.; et al. Early-Onset Alzheimer’s Disease Caused by Mutations at Codon 717 of the /J-Amyloid Precursor Protein Gene. Nature 1991, 353, 844–846. [Google Scholar] [CrossRef]
- Chapleau, M.; Iaccarino, L.; Soleimani-Meigooni, D.; Rabinovici, G.D. The Role of Amyloid PET in Imaging Neurodegenerative Disorders: A Review. J. Nucl. Med. 2022, 63, 13–19. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef]
- Rajan, K.B.; Wilson, R.S.; Weuve, J.; Barnes, L.L.; Evans, D.A. Cognitive Impairment 18 Years before Clinical Diagnosis of Alzheimer Disease Dementia. Neurology 2015, 85, 898–904. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2020, 16, 391–460. [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Marasco, R.A. Current and Evolving Treatment Strategies for the Alzheimer Disease Continuum. Am. J. Manag. Care 2020, 26, S171–S183. [Google Scholar] [CrossRef]
- Shi, J.; Sabbagh, M.N.; Vellas, B. Alzheimer’s Disease beyond Amyloid: Strategies for Future Therapeutic Interventions. BMJ 2020, 371, m3684. [Google Scholar] [CrossRef]
- 2021 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef]
- Fillit, H.; Knopman, D.; Cummings, J.; Appel, F.; Schwartz, H.L. Opportunities for Improving Managed Care for Individuals with Dementia: Part 1-The Issues From The Institute for the Study of Aging. Am. J. Manag. Care 1999, 5, 309–315. [Google Scholar]
- Maciejewska, K.; Czarnecka, K.; Szymański, P. A Review of the Mechanisms Underlying Selected Comorbidities in Alzheimer’s Disease. Pharmacol. Rep. 2021, 73, 1565–1581. [Google Scholar] [CrossRef]
- Song, C.; Shi, J.; Zhang, P.; Zhang, Y.; Xu, J.; Zhao, L.; Zhang, R.; Wang, H.; Chen, H. Immunotherapy for Alzheimer’s Disease: Targeting β-Amyloid and Beyond. Transl. Neurodegener. 2022, 11, 18. [Google Scholar] [CrossRef]
- van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.J.; Bioi; Mackinnon, R.; Yellen, G.; Timpe, L.C. Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer’s Disease. Lett. Nat. 1991, 60, 704–706. [Google Scholar] [CrossRef]
- Murphy, M.P.; Levine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP Processing in Alzheimer’s Disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Frozza, R.L.; Lourenco, M.V.; de Felice, F.G. Challenges for Alzheimer’s Disease Therapy: Insights from Novel Mechanisms beyond Memory Defects. Front. Neurosci. 2018, 12, 37. [Google Scholar] [CrossRef]
- Patterson, C.; Feightner, J.W.; Garcia, A.; Hsiung, G.Y.R.; MacKnight, C.; Sadovnick, A.D. Diagnosis and Treatment of Dementia: 1. Risk Assessment and Primary Prevention of Alzheimer Disease. CMAJ Can. Med. Assoc. J. 2008, 178, 548–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A Recessive Mutation in the APP Gene with Dominant-Negative Effect on Amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Vassar, R.; de Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; de Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Brunkan, A.L.; Goate, A.M. Presenilin Function and γ-Secretase Activity. J. Neurochem. 2005, 93, 769–792. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yatest, J.; Cotmans, C.; Glabell, C. Assembly and Aggregation Properties of Synthetic Alzheimer’s A4/β Amyloid Peptide Analogs. J. Biol. Chem. 1992, 267, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Ashe, K.H.; Aguzzi, A. Prions, Prionoids and Pathogenic Proteins in Alzheimer Disease. Prion 2013, 7, 55–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Hefti, F.; Goure, W.F.; Jerecic, J.; Iverson, K.S.; Walicke, P.A.; Krafft, G.A. The Case for Soluble Aβ Oligomers as a Drug Target in Alzheimer’s Disease. Trends Pharmacol. Sci. 2013, 34, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Mandelkow, E.; Selkoe, D.J. Alzheimer Disease in 2020. Cold Spring Harb. Perspect. Med. 2012, 2, a011585. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s Disease: Pathogenesis, Diagnostics, and Therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Tarawneh, R.; Holtzman, D.M. The Clinical Problem of Symptomatic Alzheimer Disease and Mild Cognitive Impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s Disease: Definition, Natural History, and Diagnostic Criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Lopez, O.L.; Dekosky, S.T. Clinical Symptoms in Alzheimer’s Disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 89, pp. 207–216. [Google Scholar]
- Wattmo, C.; Minthon, L.; Wallin, Å.K. Mild versus Moderate Stages of Alzheimer’s Disease: Three-Year Outcomes in a Routine Clinical Setting of Cholinesterase Inhibitor Therapy. Alzheimers Res. Ther. 2016, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J. Alzheimer Disease; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Apostolova, L.G. Alzheimer Disease. Continuum 2016, 22, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Zhang, Y.; Zhang, X.; Tian, W.; Zhang, W.; Jia, Y.; Zhang, L. Preparation and in Vitro Activity of Single Chain Antibodies against Alzheimer’s Disease. Immunol. Lett. 2020, 227, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brody, D.L.; Holtzman, D.M. Active and Passive Immunotherapy for Neurodegenerative Disorders. Annu. Rev. Neurosci. 2008, 31, 175–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical Effects of A Immunization (AN1792) in Patients with AD in an Interrupted Trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Lemere, C.A. Immunotherapy for Alzheimer’s Disease: Hoops and Hurdles. Mol. Neurodegener. 2013, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Lee, G.; Nahed, P.; Kambar, M.E.Z.N.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s Disease Drug Development Pipeline: 2022. Alzheimers Dement. Transl. Res. Clin. Interv. 2022, 8, e12179. [Google Scholar] [CrossRef]
- Pardridge, W.M. Treatment of Alzheimer’s Disease and Blood–Brain Barrier Drug Delivery. Pharmaceuticals 2020, 13, 394. [Google Scholar] [CrossRef]
- Uhlmann, R.E.; Rother, C.; Rasmussen, J.; Schelle, J.; Bergmann, C.; Ullrich Gavilanes, E.M.; Fritschi, S.K.; Buehler, A.; Baumann, F.; Skodras, A.; et al. Acute Targeting of Pre-Amyloid Seeds in Transgenic Mice Reduces Alzheimer-like Pathology Later in Life. Nat. Neurosci. 2020, 23, 1580–1588. [Google Scholar] [CrossRef]
- Banks, W.A. From Blood-Brain Barrier to Blood-Brain Interface: New Opportunities for CNS Drug Delivery. Nat. Rev. Drug. Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally Administered Antibodies against Amyloid β-Peptide Enter the Central Nervous System and Reduce Pathology in a Mouse Model of Alzheimer Disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An Effector-Reduced Anti-β-Amyloid (Aβ) Antibody with Unique Aβ Binding Properties Promotes Neuroprotection and Glial Engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9689. [Google Scholar] [CrossRef]
- Solomon, B.; Koppel, R.; Hanan, E.; Katzav, T. Monoclonal Antibodies Inhibit in Vitro Fibrillar Aggregation of the Alzheimer B-Amyloid Peptide. Proc. Natl. Acad. Sci. USA 1996, 93, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Chu, F.; Zhu, F.; Zhu, J. Impact of Anti-Amyloid-β Monoclonal Antibodies on the Pathology and Clinical Profile of Alzheimer’s Disease: A Focus on Aducanumab and Lecanemab. Front. Aging Neurosci. 2022, 14, 870517. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.; Stein, P.; Cavazzoni, P. Approval of Aducanumab for Alzheimer Disease—The FDA’s Perspective. JAMA Intern. Med. 2021, 181, 1277–1278. [Google Scholar] [CrossRef]
- Ebell, M.H.; Barry, H.C. Why Physicians Should Not Prescribe for Alzheimer Disease. Am. Fam. Physician 2022, 105, 353–354. [Google Scholar] [PubMed]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A Novel Human Anti-Aβ Antibody Demonstrates Sustained Cerebral Amyloid-β Binding and Elicits Cell-Mediated Removal of Human Amyloid-β. J. Alzheimers Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Media & Investor Release Ad Hoc Announcement Pursuant to Art. 53 LR Roche Provides Update on Phase III GRADUATE Programme Evaluating Gantenerumab in Early Alzheimer’s Disease; F. Hoffmann-La Roche Ltd.: Basel, Switzerland, 2022.
- Salloway, S.; Honigberg, L.A.; Cho, W.; Ward, M.; Friesenhahn, M.; Brunstein, F.; Quartino, A.; Clayton, D.; Mortensen, D.; Bittner, T.; et al. Amyloid Positron Emission Tomography and Cerebrospinal Fluid Results from a Crenezumab Anti-Amyloid-β Antibody Double-Blind, Placebo-Controlled, Randomized Phase II Study in Mild-to-Moderate Alzheimer’s Disease (BLAZE). Alzheimers Res. Ther. 2018, 10, 96. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Bittner, T.; Sink, K.M.; Mackey, H.; Rabe, C.; Honig, L.S.; Cassetta, E.; Woodward, M.; Boada, M.; van Dyck, C.H.; et al. Evaluating the Safety and Efficacy of Crenezumab vs Placebo in Adults with Early Alzheimer Disease: Two Phase 3 Randomized Placebo-Controlled Trials. JAMA Neurol. 2022, 79, 1113–1121. [Google Scholar] [CrossRef]
- Hardy, J.; Bogdanovic, N.; Winblad, B.; Portelius, E.; Andreasen, N.; Cedazo-Minguez, A.; Zetterberg, H. Pathways to Alzheimer’s Disease. J. Intern. Med. 2014, 275, 296–303. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; DeLong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A Plaque-Specific Antibody Clears Existing β-Amyloid Plaques in Alzheimer’s Disease Mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [Green Version]
- Bouter, Y.; Liekefeld, H.; Pichlo, S.; Westhoff, A.C.; Fenn, L.; Bakrania, P.; Bayer, T.A. Donanemab Detects a Minor Fraction of Amyloid-β Plaques in Post-Mortem Brain Tissue of Patients with Alzheimer’s Disease and Down Syndrome. Acta Neuropathol. 2022, 143, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.; Lo, A.; Evans, C.; Wessels, A.; Ardayfio, P.; Andersen, S.; Shcherbinin, S.; Sparks, J.; Sims, J.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Olloquequi, J.; Ettcheto, M.; Cano, A.; Sanchez-López, E.; Carrasco, M.; Espinosa, T.; Beas-Zarate, C.; Gudiño-Cabrera, G.; Ureña-Guerrero, M.E.; Verdaguer, E.; et al. Impact of New Drugs for Therapeutic Intervention in Alzheimer’s Disease. Front. Biosci. Landmark 2022, 27, 146. [Google Scholar] [CrossRef] [PubMed]
- Parrocha, C.M.T.; Nowick, J.S. Current Peptide Vaccine and Immunotherapy Approaches against Alzheimer’s Disease. Pept. Sci. 2023, 115, e24289. [Google Scholar] [CrossRef] [PubMed]
- Lecanemab Confirmatory Phase 3 Clarity Ad Study Met Primary Endpoint, Showing Highly Statistically Significant Reduction of Clinical Decline in Large Global Clinical Study of 1795 Participants with Early Alzheimer’s Disease. Available online: https://investors.biogen.com/news-releases/news-release-details/lecanemab-confirmatory-phase-3-clarity-ad-study-met-primary (accessed on 19 December 2022).
- vander Zanden, C.M.; Chi, E.Y. Passive Immunotherapies Targeting Amyloid β and Tau Oligomers in Alzheimer’s Disease. J. Pharm. Sci. 2020, 109, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The Murine Version of BAN2401 (MAb158) Selectively Reduces Amyloid-β Protofibrils in Brain and Cerebrospinal Fluid of Tg-ArcSwe Mice. J. Alzheimers Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef]
- Söllvander, S.; Nikitidou, E.; Gallasch, L.; Zyśk, M.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. The Aβ Protofibril Selective Antibody MAb158 Prevents Accumulation of Aβ in Astrocytes and Rescues Neurons from Aβ-Induced Cell Death. J. Neuroinflamm. 2018, 15, 98. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A Randomized, Double-Blind, Phase 2b Proof-of-Concept Clinical Trial in Early Alzheimer’s Disease with Lecanemab, an Anti-Aβ Protofibril Antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2022, 388, 9–21. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 9 January 2023).
- Armstrong, A. Facing a Familiar Side Effect Problem, Eisai Makes the Case for Its next Alzheimer’s Drug after Patient Deaths. Available online: https://www.fiercebiotech.com/biotech/eisai-biogen-lecanemab-alzheimers-aria-patient-deaths-adverse-events (accessed on 9 December 2022).
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Illouz, T.; Madar, R.; Hirsh, T.; Biragyn, A.; Okun, E. Induction of an Effective Anti-Amyloid-β Humoral Response in Aged Mice. Vaccine 2021, 39, 4817–4829. [Google Scholar] [CrossRef] [PubMed]
- Davtyan, H.; Ghochikyan, A.; Petrushina, I.; Hovakimyan, A.; Davtyan, A.; Poghosyan, A.; Marleau, A.M.; Movsesyan, N.; Kiyatkin, A.; Rasool, S.; et al. Immunogenicity, Efficacy, Safety, and Mechanism of Action of Epitope Vaccine (Lu AF20513) for Alzheimer’s Disease: Prelude to a Clinical Trial. J. Neurosci. 2013, 33, 4923–4934. [Google Scholar] [CrossRef] [Green Version]
- Orgogozo, J.-M.; Gilman, S.; Dartigues, J.-F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute Meningoencephalitis in a Subset of Patients with AD after A42 Immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.; Seabrook, T.J.; Lazo, N.D.; Jiang, L.; Das, P.; Janus, C.; Lemere, C.A. Short Amyloid-β (Aβ) Immunogens Reduce Cerebral Aβ Load and Learning Deficits in an Alzheimer’s Disease Mouse Model in the Absence of an Aβ-Specific Cellular Immune Response. J. Neurosci. 2006, 26, 4717–4728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiessner, C.; Wiederhold, K.H.; Tissot, A.C.; Frey, P.; Danner, S.; Jacobson, L.H.; Jennings, G.T.; Lüönd, R.; Ortmann, R.; Reichwald, J.; et al. The Second-Generation Active Aβ Immunotherapy CAD106 Reduces Amyloid Accumulation in APP Transgenic Mice While Minimizing Potential Side Effects. J. Neurosci. 2011, 31, 9323–9331. [Google Scholar] [CrossRef]
- Lambracht-Washington, D.; Rosenberg, R.N. Advances in the Development of Vaccines for Alzheimer’s Disease. Discov. Med. 2013, 15, 319–326. [Google Scholar]
- Bachmann, M.F.; Jennings, G.T.; Vogel, M. A Vaccine against Alzheimer`s Disease: Anything Left but Faith? Expert. Opin. Biol. Ther. 2019, 19, 73–78. [Google Scholar] [CrossRef]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, Tolerability, and Antibody Response of Active Abeta Immunotherapy with CAD106 in Patients with Alzheimer’s Disease: Randomised, Double-Blind, Placebo-Controlled, First-in-Human Study. LancetNeurol 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Aβ Immunotherapy CAD106 in Alzheimer’s Disease: A Phase 2b Study. Alzheimers Dement. Transl. Res. Clin. Interv. 2017, 3, 10–22. [Google Scholar] [CrossRef]
- Riviere, M.; Turner, R.S.; Sui, Y.; Laurent, N.; Langbaum, J.B.; Cazorla, P.; Ricart, J.; Seneca, N.; Caputo, A.; Reiman, E.M.; et al. API Generation Program: Active Immunotherapy CAD106 Slows Amyloid Deposition in Cognitively Unimpaired APOE4 Homozygotes. Alzheimers Dement. 2021, 17, e051287. [Google Scholar] [CrossRef]
- Huang, X. Alzheimer’s Disease: Drug Discovery; Exon Publications: Brisbane, Australia, 2020; ISBN-13 978-0-6450017-0-9. [Google Scholar]
- Lacosta, A.M.; Pascual-Lucas, M.; Pesini, P.; Casabona, D.; Pérez-Grijalba, V.; Marcos-Campos, I.; Sarasa, L.; Canudas, J.; Badi, H.; Monleón, I.; et al. Safety, Tolerability and Immunogenicity of an Active Anti-Aβ 40 Vaccine (ABvac40) in Patients with Alzheimer’s Disease: A Randomised, Double-Blind, Placebo-Controlled, Phase i Trial. Alzheimers Res. Ther. 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Fettelschoss, A.; Zabel, F.; Bachmann, M.F. Vaccination against Alzheimer Disease: An Update on Future Strategies. Hum. Vaccin. Immunother. 2014, 10, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. How Plausible Is an Alzheimer’s Disease Vaccine? Expert Opin. Drug Discov. 2020, 15, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, C.J. Sex and the Development of Alzheimer’s Disease. J. Neurosci. Res. 2017, 95, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, R.A. Risk Factors for Alzheimer’s Disease. Folia. Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [Green Version]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s Disease. Lancet 2011, 377, 1019–1050. [Google Scholar] [CrossRef]
- van Cauwenberghe, C.; van Broeckhoven, C.; Sleegers, K. The Genetic Landscape of Alzheimer Disease: Clinical Implications and Perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Li, N.M.; Liu, K.F.; Qiu, Y.J.; Zhang, H.H.; Nakanishi, H.; Qing, H. Mutations of β-Amyloid Precursor Protein Alter the Consequence of Alzheimer’s Disease Pathogenesis. Neural Regen. Res. 2019, 14, 658–665. [Google Scholar] [CrossRef]
- de Strooper, B. Loss-of-Function Presenilin Mutations in Alzheimer Disease. Talking Point on the Role of Presenilin Mutations in Alzheimer Disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.V.F.; Loures, C.D.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.D.G. Alzheimer’s Disease: Risk Factors and Potentially Protective Measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [Green Version]
- Cahill, L. Why Sex Matters for Neuroscience. Nat. Rev. Neurosci. 2006, 7, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Buckley, R.F.; Mormino, E.C.; Amariglio, R.E.; Properzi, M.J.; Rabin, J.S.; Lim, Y.Y.; Papp, K.V.; Jacobs, H.I.L.; Burnham, S.; Hanseeuw, B.J.; et al. Sex, Amyloid, and APOE Ε4 and Risk of Cognitive Decline in Preclinical Alzheimer’s Disease: Findings from Three Well-Characterized Cohorts. Alzheimers Dement. 2018, 14, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, D.; Leng, S.X. Link between Type 2 Diabetes and Alzheimer’s Disease: From Epidemiology to Mechanism and Treatment. Clin. Interv. Aging 2015, 10, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef] [PubMed]
- Uwishema, O.; Mahmoud, A.; Sun, J.; Correia, I.F.S.; Bejjani, N.; Alwan, M.; Nicholas, A.; Oluyemisi, A.; Dost, B. Is Alzheimer’s Disease an Infectious Neurological Disease? A Review of the Literature. Brain Behav. 2022, 12, e2728. [Google Scholar] [CrossRef]
- Vaz, M.; Silvestre, S. Alzheimer’s Disease: Recent Treatment Strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- 2019 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2019, 15, 321–387. [CrossRef]
- Lemere, C.A.; Masliah, E. Can Alzheimer Disease Be Prevented by Amyloid-Β Immunotherapy? Nat. Rev. Neurol. 2010, 6, 108–119. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Drug | Sponsor | Status of Clinical Trial | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| Aβ aggregates | Aducanumab | Biogen | Phase III | NCT04241068 |
| Aβ fibrils | Gantenerumab | Roche | Phase III | NCT03443973 |
| Aβ aggregates | Crenezumab | Genentech | Phase III | NCT03114657 |
| Amyloid β | Solanezumab | Eli Lilly | Phase II/III | NCT01900665 |
| Aβ aggregates | Donanemab | Eli Lilly | Phase III | NCT04437511 |
| Aβ Protofibrils | Lecanemab | Eisai | Phase III | NCT04468659 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogt, A.-C.S.; Jennings, G.T.; Mohsen, M.O.; Vogel, M.; Bachmann, M.F. Alzheimer’s Disease: A Brief History of Immunotherapies Targeting Amyloid β. Int. J. Mol. Sci. 2023, 24, 3895. https://doi.org/10.3390/ijms24043895
Vogt A-CS, Jennings GT, Mohsen MO, Vogel M, Bachmann MF. Alzheimer’s Disease: A Brief History of Immunotherapies Targeting Amyloid β. International Journal of Molecular Sciences. 2023; 24(4):3895. https://doi.org/10.3390/ijms24043895
Chicago/Turabian StyleVogt, Anne-Cathrine S., Gary T. Jennings, Mona O. Mohsen, Monique Vogel, and Martin F. Bachmann. 2023. "Alzheimer’s Disease: A Brief History of Immunotherapies Targeting Amyloid β" International Journal of Molecular Sciences 24, no. 4: 3895. https://doi.org/10.3390/ijms24043895