

The Strategies of Development of New Non-Toxic Inhibitors of Amyloid Formation
 , , and
, , and
Abstract
:1. Introduction
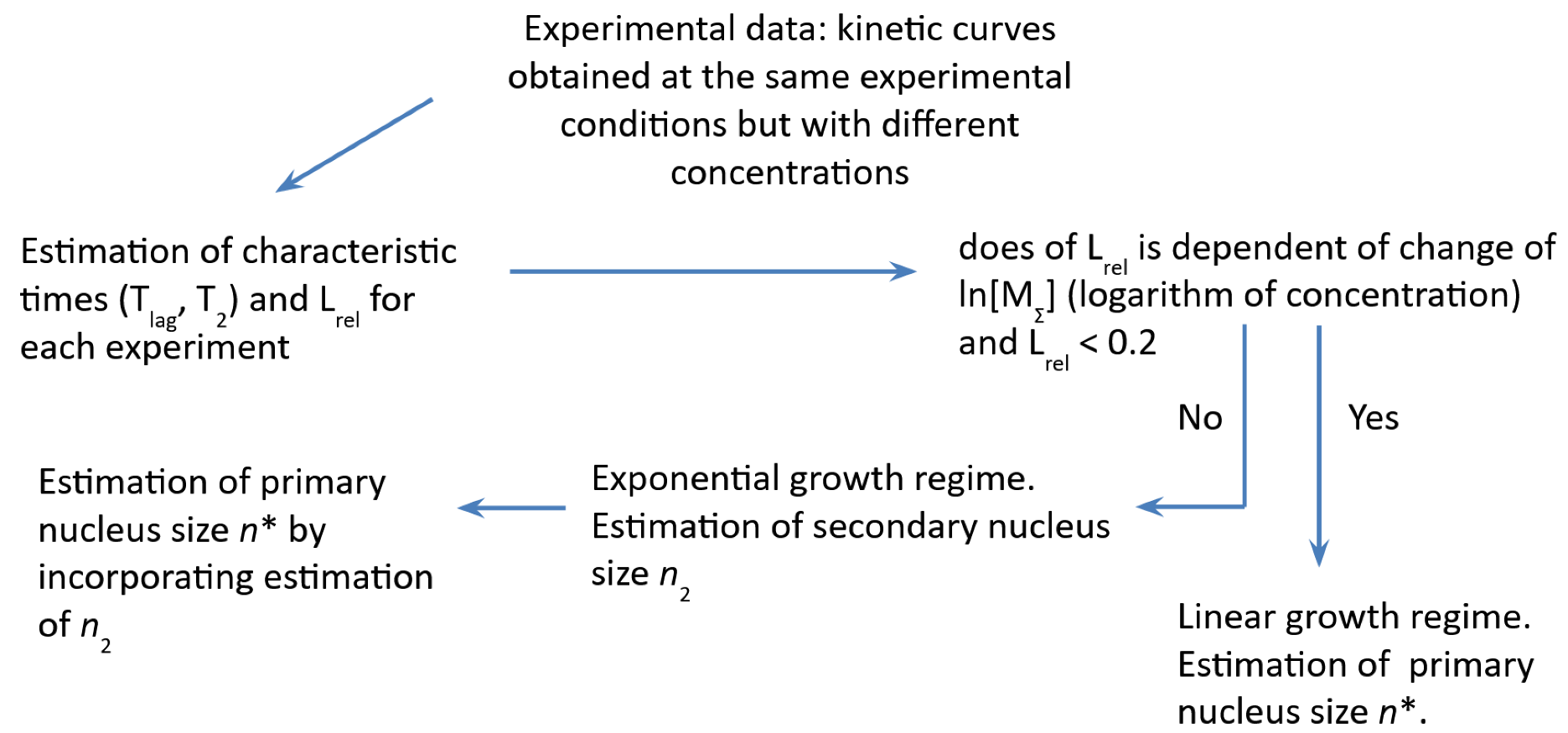
2. Amyloid Fibril Formation Mechanisms and Fibril Formation Nucleus Size Estimation
3. Protein Folding and Aggregation
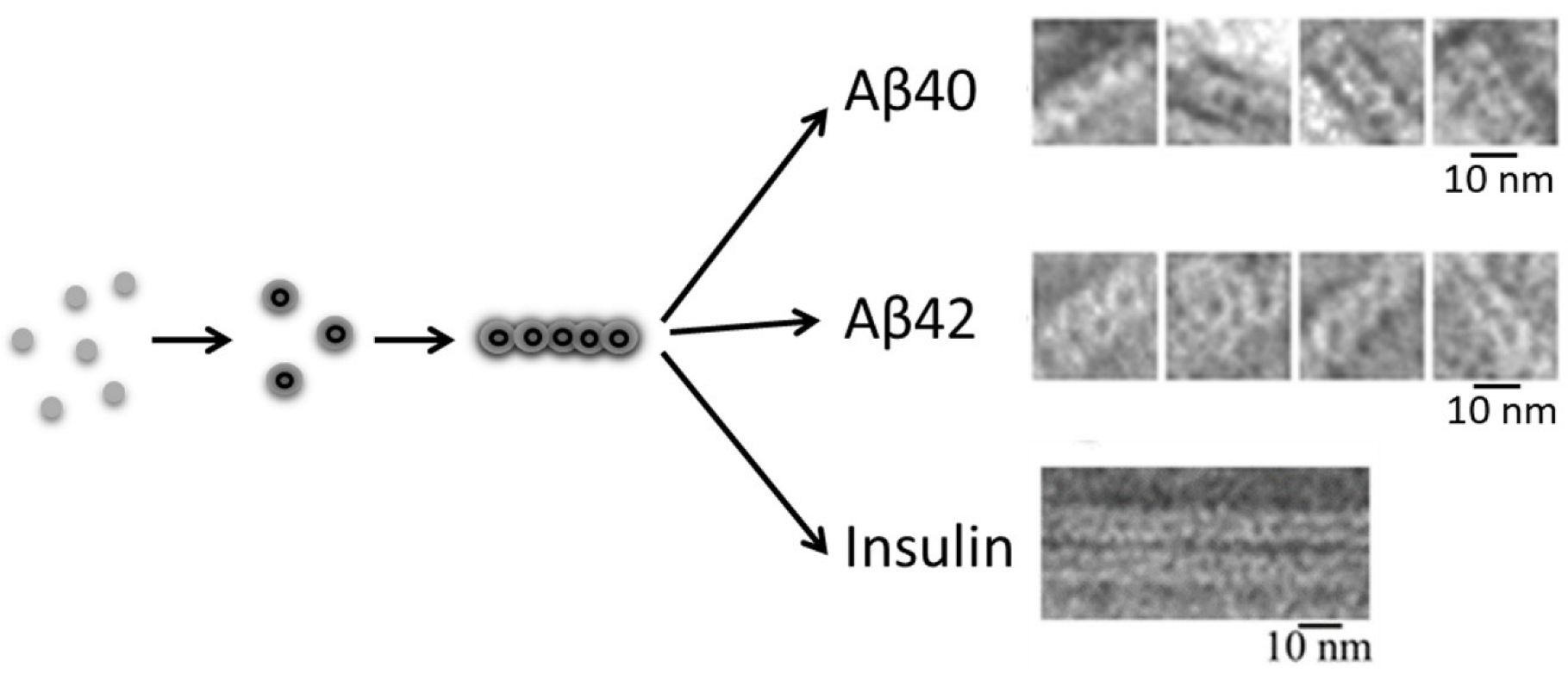
3.1. Aβ Peptide
3.2. α-Synuclein
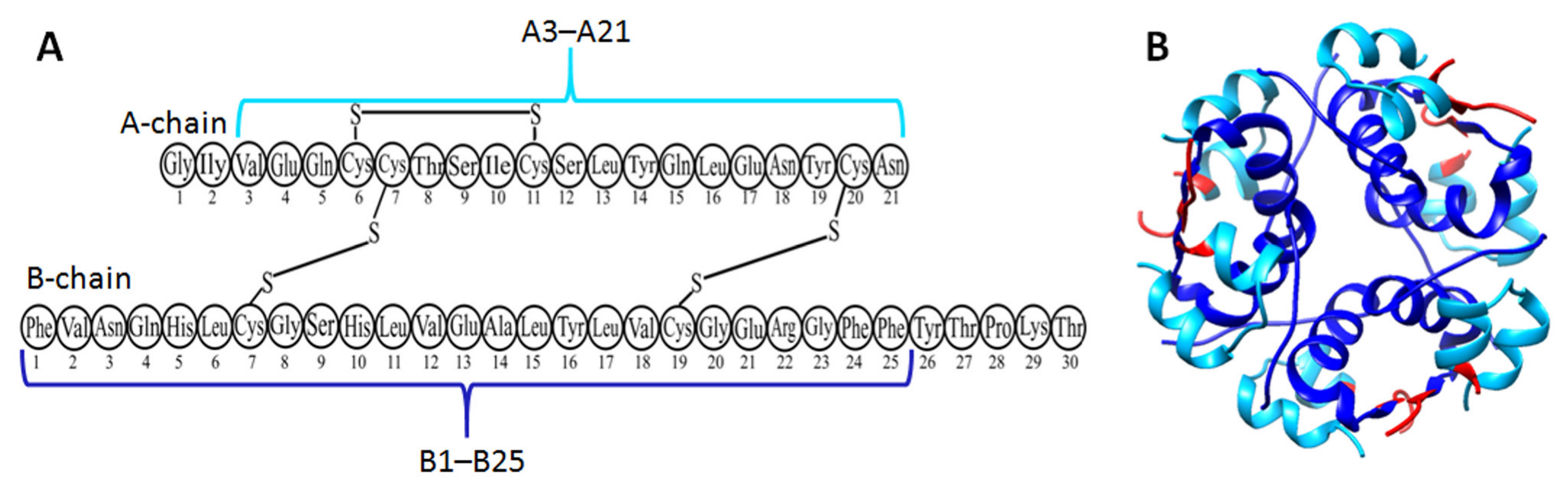
3.3. Insulin
4. Influence of Various Molecules on the Aggregation Process
4.1. Inhibitors for Aβ Fibril Formation
4.2. Inhibitors for α-Synuclein Fibril Formation
4.3. Inhibitors for Insulin Fibril Formation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
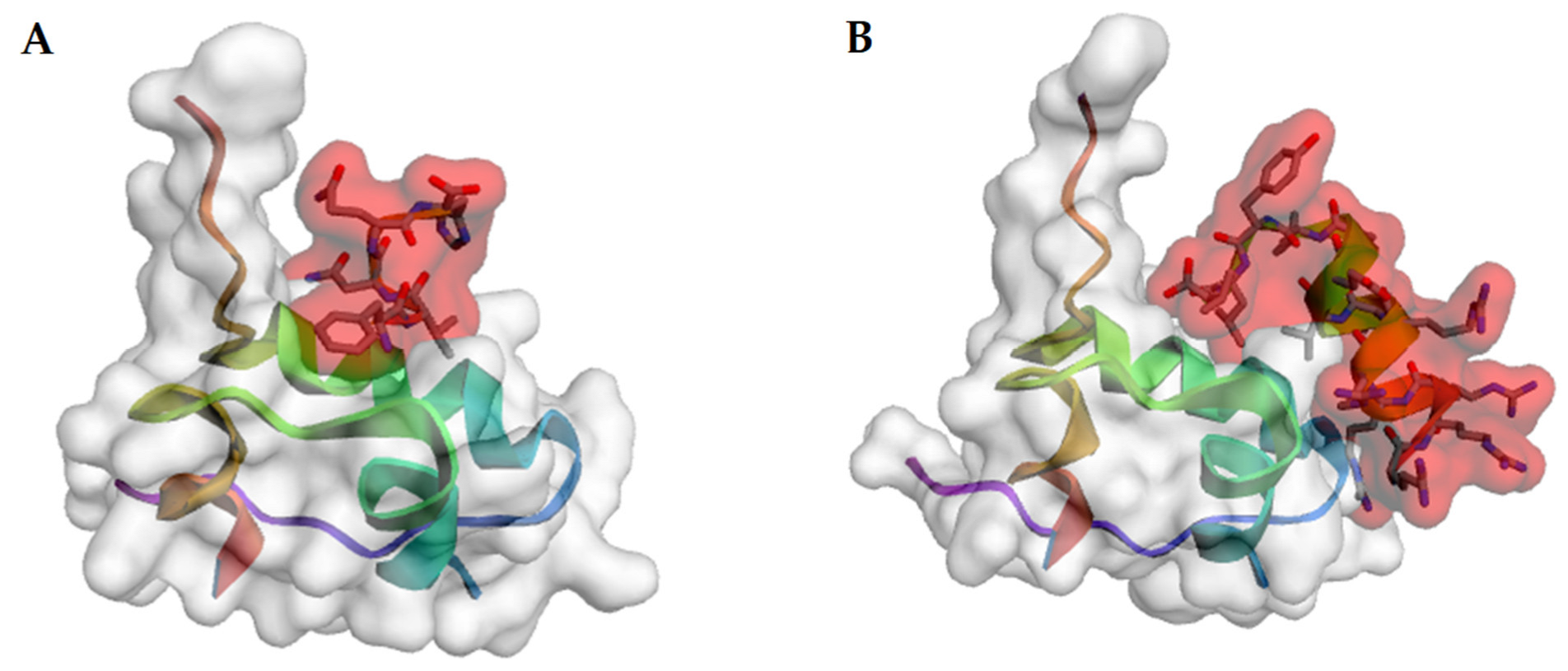{kind=link}
| Aβ | α-Synuclein | Insulin |
|---|---|---|
 | ||
| Myricetin | ||
| 10 µM, decreased of Aβ(1–42) fibril formation (10 µM Aβ solution) [189] | 200 µM, inhibited of α-synuclein fibril formation (200 µM α-synuclein solution) [190] | 400 µM, inhibited of insulin fibril formation (40 µM insulin solution) [191] |
 | ||
| Quercetin | ||
| 50 µM, decreased of Aβ(1–40) fibril formation (50 µM Aβ solution) [110] | 20 µM, inhibited of α-synuclein fibril formation (70 µM α-synuclein solution) [192] | 85 µM, inhibited of insulin fibril formation (170 µM insulin solution) [193] |
 | ||
| Epigallocatechin-3-gallate | ||
| 25 µM, inhibited of Aβ(1–42) fibril formation (2 µM Aβ solution) [194] | 100 µM, inhibited of α-synuclein fibril formation (100 µM α-synuclein solution) [185] | 25 µM, inhibited of insulin fibril formation (200 µM insulin solution) [185] |
 | ||
| Curcumin | ||
| 1 μM, disaggregated fibrils of Aβ(1–40) (11.6 µM Aβ solution) [195] | 72 μM, disaggregated fibrils of α-synuclein (48 µM α-synuclein solution) [138] | 4 μM, decreased of insulin fibril formation (3 µM insulin solution) [196] |
 | ||
| Gallic acid | ||
| 5 μM, reduced the fibril sizes of Aβ(1–42) particles (2.5 µM Aβ) [115] | 5 μM, inhibited of α-synuclein fibril formation (20 µM α-synuclein solution) [197] | 1700 μM, reduced insulin fibril formation (340 µM insulin solution) [198] |
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Uversky, V.N. The Protein Disorder Cycle. Biophys. Rev. 2021, 13, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Gulland, A. Number of People with Dementia Will Reach 65.7 Million by 2030, Says Report. BMJ 2012, 344, e2604. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Monge-García, V.; García-Ayllón, M.-S.; Sáez-Valero, J.; Sánchez-Payá, J.; Navarrete-Rueda, F.; Manzanares-Robles, J.; Gasparini-Berenguer, R.; Romero-Lorenzo, R.; Cortés-Gómez, M.A.; Monge-Argilés, J.-A. Relation between Alpha-Synuclein and Core CSF Biomarkers of Alzheimer’s Disease. Medicina 2021, 57, 954. [Google Scholar] [CrossRef]
- Brookes, A.J.; St Clair, D. Synuclein Proteins and Alzheimer’s Disease. Trends Neurosci. 1994, 17, 404–405. [Google Scholar] [CrossRef]
- Surguchov, A. Caveolin: A New Link Between Diabetes and AD. Cell. Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; Van Bronswijk, W.; Martins, R. Alzheimer’s Beta-Amyloid Peptides Compete for Insulin Binding to the Insulin Receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.-E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Dovidchenko, N.V.; Finkelstein, A.V.; Galzitskaya, O.V. How to Determine the Size of Folding Nuclei of Protofibrils from the Concentration Dependence of the Rate and Lag-Time of Aggregation. I. Modeling the Amyloid Protofibril Formation. J. Phys. Chem. B 2014, 118, 1189–1197. [Google Scholar] [CrossRef]
- Dovidchenko, N.V.; Galzitskaya, O.V. Computational Approaches to Identification of Aggregation Sites and the Mechanism of Amyloid Growth. Adv. Exp. Med. Biol. 2015, 855, 213–239. [Google Scholar] [CrossRef]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.A.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Differences in Nucleation Behavior Underlie the Contrasting Aggregation Kinetics of the Aβ40 and Aβ42 Peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef]
- Dovidchenko, N.V.; Glyakina, A.V.; Selivanova, O.M.; Grigorashvili, E.I.; Suvorina, M.Y.; Dzhus, U.F.; Mikhailina, A.O.; Shiliaev, N.G.; Marchenkov, V.V.; Surin, A.K.; et al. One of the Possible Mechanisms of Amyloid Fibrils Formation Based on the Sizes of Primary and Secondary Folding Nuclei of Aβ40 and Aβ42. J. Struct. Biol. 2016, 194, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Rajah, L.; Cohen, S.A.I.; Pfammatter, M.; Šarić, A.; Hellstrand, E.; Buell, A.K.; Aguzzi, A.; Linse, S.; Vendruscolo, M.; et al. Scaling Behaviour and Rate-Determining Steps in Filamentous Self-Assembly. Chem. Sci. 2017, 8, 7087–7097. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergström, A.-L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein Propagates from Mouse Brain to Grafted Dopaminergic Neurons and Seeds Aggregation in Cultured Human Cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Selivanova, O.M.; Surin, A.K.; Marchenkov, V.V.; Dzhus, U.F.; Grigorashvili, E.I.; Suvorina, M.Y.; Glyakina, A.V.; Dovidchenko, N.V.; Galzitskaya, O.V. The Mechanism Underlying Amyloid Polymorphism Is Opened for Alzheimer’s Disease Amyloid-β Peptide. J. Alzheimers Dis. 2016, 54, 821–830. [Google Scholar] [CrossRef]
- Pieri, L.; Madiona, K.; Melki, R. Structural and Functional Properties of Prefibrillar α-Synuclein Oligomers. Sci. Rep. 2016, 6, 24526. [Google Scholar] [CrossRef] [PubMed]
- Galzitskaya, O.V.; Selivanova, O.M. Rosetta Stone for Amyloid Fibrils: The Key Role of Ring-Like Oligomers in Amyloidogenesis. J. Alzheimers Dis. 2017, 59, 785–795. [Google Scholar] [CrossRef] [PubMed]
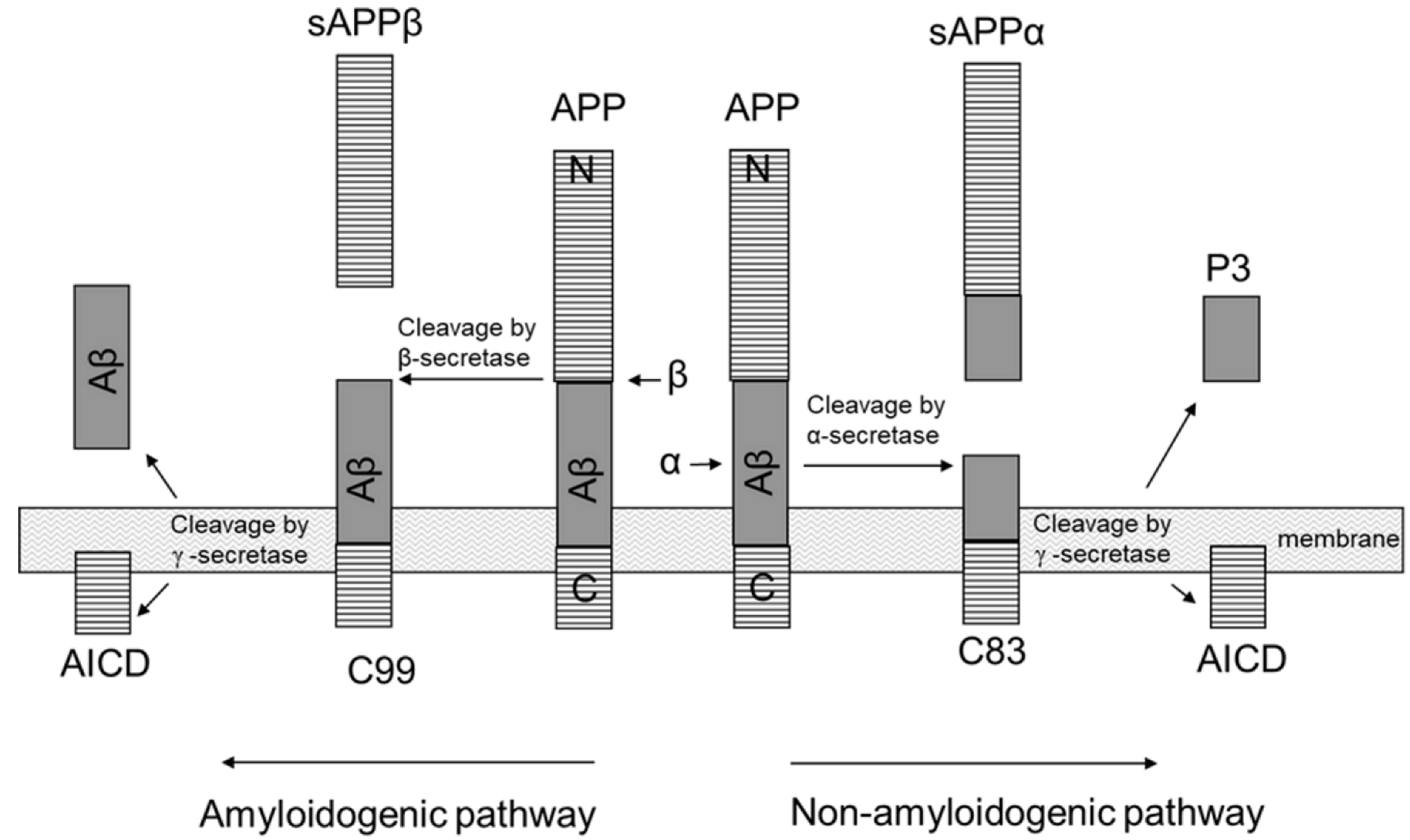
- Ling, Y.; Morgan, K.; Kalsheker, N. Amyloid Precursor Protein (APP) and the Biology of Proteolytic Processing: Relevance to Alzheimer’s Disease. Int. J. Biochem. Cell Biol. 2003, 35, 1505–1535. [Google Scholar] [CrossRef]
- Hardy, J. Amyloid, the Presenilins and Alzheimer’s Disease. Trends Neurosci. 1997, 20, 154–159. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The Cell Biology of Beta-Amyloid Precursor Protein and Presenilin in Alzheimer’s Disease. Trends Cell Biol. 1998, 8, 447–453. [Google Scholar] [CrossRef]
- Avila, J.; Pérez, M.; Lim, F.; Gómez-Ramos, A.; Hernández, F.; Lucas, J.J. Tau in Neurodegenerative Diseases: Tau Phosphorylation and Assembly. Neurotox. Res. 2004, 6, 477–482. [Google Scholar] [CrossRef]
- Avila, J.; Pérez, M.; Lucas, J.J.; Gómez-Ramos, A.; Santa María, I.; Moreno, F.; Smith, M.; Perry, G.; Hernández, F. Assembly in Vitro of Tau Protein and Its Implications in Alzheimer’s Disease. Curr. Alzheimer Res. 2004, 1, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ramos, A.; Smith, M.A.; Perry, G.; Avila, J. Tau Phosphorylation and Assembly. Acta NeuroBiol. Exp. 2004, 64, 33–39. [Google Scholar] [PubMed]
- Sambamurti, K.; Greig, N.H.; Lahiri, D.K. Advances in the Cellular and Molecular Biology of the Beta-Amyloid Protein in Alzheimer’s Disease. Neuromolecular Med. 2002, 1, 1–31. [Google Scholar] [CrossRef]
- Parvathy, S.; Hussain, I.; Karran, E.H.; Turner, A.J.; Hooper, N.M. Cleavage of Alzheimer’s Amyloid Precursor Protein by Alpha-Secretase Occurs at the Surface of Neuronal Cells. Biochemistry 1999, 38, 9728–9734. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of Amyloid-Β42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]
- Selivanova, O.M.; Surin, A.K.; Ryzhykau, Y.L.; Glyakina, A.V.; Suvorina, M.Y.; Kuklin, A.I.; Rogachevsky, V.V.; Galzitskaya, O.V. To Be Fibrils or To Be Nanofilms? Oligomers Are Building Blocks for Fibril and Nanofilm Formation of Fragments of Aβ Peptide. Langmuir 2018, 34, 2332–2343. [Google Scholar] [CrossRef]
- Galzitskaya, O.V.; Surin, A.K.; Glyakina, A.V.; Rogachevsky, V.V.; Selivanova, O.M. Should the Treatment of Amyloidosis Be Personified? Molecular Mechanism of Amyloid Formation by Aβ Peptide and Its Fragments. ADR 2018, 2, 181–199. [Google Scholar] [CrossRef] [Green Version]
- Selivanova, O.M.; Suvorina, M.Y.; Surin, A.K.; Dovidchenko, N.V.; Galzitskaya, O.V. Insulin and Lispro Insulin: What Is Common and Different in Their Behavior? Curr. Protein Pept. Sci. 2017, 18, 57–64. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril Structure of Amyloid-β(1–42) by Cryo–Electron Microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Markham, R.; Frey, S.; Hills, G.J. Methods for the Enhancement of Image Detail and Accentuation of Structure in Electron Microscopy. Virology 1963, 20, 88–102. [Google Scholar] [CrossRef]
- Inouye, H.; Fraser, P.E.; Kirschner, D.A. Structure of Beta-Crystallite Assemblies Formed by Alzheimer Beta-Amyloid Protein Analogues: Analysis by x-Ray Diffraction. Biophys. J. 1993, 64, 502–519. [Google Scholar] [CrossRef]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer’s Brain Tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, U.; Thurber, K.R.; Yau, W.-M.; Tycko, R. Molecular Structure of a Prevalent Amyloid-β Fibril Polymorph from Alzheimer’s Disease Brain Tissue. Proc. Natl. Acad. Sci. USA 2021, 118, e2023089118. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM Structures of Amyloid-β 42 Filaments from Human Brains. Science 2022, 375, 167–172. [Google Scholar] [CrossRef]
- Liu, D.; Wei, Q.; Xia, W.; He, C.; Zhang, Q.; Huang, L.; Wang, X.; Sun, Y.; Ma, Y.; Zhang, X.; et al. O-Glycosylation Induces Amyloid-β To Form New Fibril Polymorphs Vulnerable for Degradation. J. Am. Chem. Soc. 2021, 143, 20216–20223. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Yau, W.-M.; Tycko, R. Experimental Constraints on Quaternary Structure in Alzheimer’s β-Amyloid Fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.-M.; Tycko, R. Molecular Structural Basis for Polymorphism in Alzheimer’s β-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [Green Version]
- Qiang, W.; Yau, W.-M.; Luo, Y.; Mattson, M.P.; Tycko, R. Antiparallel β-Sheet Architecture in Iowa-Mutant β-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 4443–4448. [Google Scholar] [CrossRef]
- Lu, J.-X.; Qiang, W.; Yau, W.-M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef]
- Schütz, A.K.; Vagt, T.; Huber, M.; Ovchinnikova, O.Y.; Cadalbert, R.; Wall, J.; Güntert, P.; Böckmann, A.; Glockshuber, R.; Meier, B.H. Atomic-Resolution Three-Dimensional Structure of Amyloid β Fibrils Bearing the Osaka Mutation. Angew. Chem. Int. Ed. 2015, 54, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Sgourakis, N.G.; Yau, W.-M.; Qiang, W. Modeling an In-Register, Parallel “Iowa” Aβ Fibril Structure Using Solid-State NMR Data from Labeled Samples with Rosetta. Structure 2015, 23, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D Structure of Alzheimer’s Amyloid-β(1–42) Fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) Fibril Structure Illuminates Self-Recognition and Replication of Amyloid in Alzheimer’s Disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-Resolution Structure of a Disease-Relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ 42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Kyriukha, Y.A.; Afitska, K.; Kurochka, A.S.; Sachan, S.; Galkin, M.; Yushchenko, D.A.; Shvadchak, V.V. α-Synuclein Dimers as Potent Inhibitors of Fibrillization. J. Med. Chem. 2019, 62, 10342–10351. [Google Scholar] [CrossRef]
- Zhang, Y.; Hashemi, M.; Lv, Z.; Williams, B.; Popov, K.I.; Dokholyan, N.V.; Lyubchenko, Y.L. High-Speed Atomic Force Microscopy Reveals Structural Dynamics of α-Synuclein Monomers and Dimers. J. Chem. Phys. 2018, 148, 123322. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Mammi, S.; Bubacco, L. Structural Insights on Physiological Functions and Pathological Effects of Alpha-Synuclein. FASEB J. 2009, 23, 329–340. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Sokolovskiy, I.V.; Galzitskaya, O.V. IsUnstruct: Prediction of the Residue Status to Be Ordered or Disordered in the Protein Chain by a Method Based on the Ising Model. J. Biomol. Struct. Dyn. 2013, 31, 1034–1043. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, C.; Wang, S.; Yang, D.; Zhang, Y.; Xu, L.; Ma, L.; Zheng, J.; Petersen, R.B.; Zheng, L.; et al. Copper and Iron Ions Accelerate the Prion-like Propagation of α-Synuclein: A Vicious Cycle in Parkinson’s Disease. Int. J. Biol. Macromol. 2020, 163, 562–573. [Google Scholar] [CrossRef]
- Wu, K.-P.; Weinstock, D.S.; Narayanan, C.; Levy, R.M.; Baum, J. Structural Reorganization of Alpha-Synuclein at Low PH Observed by NMR and REMD Simulations. J. Mol. Biol. 2009, 391, 784–796. [Google Scholar] [CrossRef]
- Roberts, H.L.; Brown, D.R. Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ Is a Key Factor in α-Synuclein-Induced Neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a Partially Folded Intermediate in Alpha-Synuclein Fibril Formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic Reticulum Stress Is Important for the Manifestations of α-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef]
- Choi, D.-H.; Cristóvão, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.-S. NADPH Oxidase 1-Mediated Oxidative Stress Leads to Dopamine Neuron Death in Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Carranza, D.L.; Zhang, Y.; Guerrero-Muñoz, M.J.; Kayed, R.; Rincon-Limas, D.E.; Fernandez-Funez, P. Differential Activation of the ER Stress Factor XBP1 by Oligomeric Assemblies. Neurochem. Res. 2012, 37, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-Synuclein Oligomers: A New Hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, N.; Lemminger, L.; Pedersen, J.N.; Nielsen, S.B.; Otzen, D.E. The N-Terminus of α-Synuclein Is Essential for Both Monomeric and Oligomeric Interactions with Membranes. FEBS Lett. 2014, 588, 497–502. [Google Scholar] [CrossRef]
- Karpinar, D.P.; Balija, M.B.G.; Kügler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.-Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-Fibrillar Alpha-Synuclein Variants with Impaired Beta-Structure Increase Neurotoxicity in Parkinson’s Disease Models. EMBO J. 2009, 28, 3256–3268. [Google Scholar] [CrossRef]
- Van Raaij, M.E.; van Gestel, J.; Segers-Nolten, I.M.J.; de Leeuw, S.W.; Subramaniam, V. Concentration Dependence of Alpha-Synuclein Fibril Length Assessed by Quantitative Atomic Force Microscopy and Statistical-Mechanical Theory. Biophys. J. 2008, 95, 4871–4878. [Google Scholar] [CrossRef]
- Sanchez, S.E.; Whiten, D.R.; Meisl, G.; Ruggeri, F.S.; Hidari, E.; Klenerman, D. Alpha Synuclein Only Forms Fibrils In Vitro When Larger than Its Critical Size of 70 Monomers. Chembiochem 2021, 22, 2867–2871. [Google Scholar] [CrossRef]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-State NMR Structure of a Pathogenic Fibril of Full-Length Human α-Synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Pálmadóttir, T.; Malmendal, A.; Leiding, T.; Lund, M.; Linse, S. Charge Regulation during Amyloid Formation of α-Synuclein. J. Am. Chem. Soc. 2021, 143, 7777–7791. [Google Scholar] [CrossRef]
- Afitska, K.; Fucikova, A.; Shvadchak, V.V.; Yushchenko, D.A. α-Synuclein Aggregation at Low Concentrations. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Giasson, B.I. The Emerging Role of α-Synuclein Truncation in Aggregation and Disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-W.; Giasson, B.I.; Lewis, K.A.; Lee, V.M.; Demartino, G.N.; Thomas, P.J. A Precipitating Role for Truncated Alpha-Synuclein and the Proteasome in Alpha-Synuclein Aggregation: Implications for Pathogenesis of Parkinson Disease. J. Biol. Chem. 2005, 280, 22670–22678. [Google Scholar] [CrossRef] [PubMed]
- Vasili, E.; Dominguez-Meijide, A.; Outeiro, T.F. Spreading of α-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front. Mol. Neurosci. 2019, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Bisi, N.; Feni, L.; Peqini, K.; Pérez-Peña, H.; Ongeri, S.; Pieraccini, S.; Pellegrino, S. α-Synuclein: An All-Inclusive Trip Around Its Structure, Influencing Factors and Applied Techniques. Front Chem. 2021, 9, 666585. [Google Scholar] [CrossRef]
- Trexler, A.J.; Rhoades, E. N-Terminal Acetylation Is Critical for Forming α-Helical Oligomer of α-Synuclein. Protein Sci. 2012, 21, 601–605. [Google Scholar] [CrossRef]
- Bell, R.; Thrush, R.J.; Castellana-Cruz, M.; Oeller, M.; Staats, R.; Nene, A.; Flagmeier, P.; Xu, C.K.; Satapathy, S.; Galvagnion, C.; et al. N-Terminal Acetylation of α-Synuclein Slows down Its Aggregation Process and Alters the Morphology of the Resulting Aggregates. Biochemistry 2022, 61, 1743–1756. [Google Scholar] [CrossRef]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural Characterization of Toxic Oligomers That Are Kinetically Trapped during α-Synuclein Fibril Formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef]
- Bhak, G.; Lee, J.-H.; Hahn, J.-S.; Paik, S.R. Granular Assembly of α-Synuclein Leading to the Accelerated Amyloid Fibril Formation with Shear Stress. PLoS ONE 2009, 4, e4177. [Google Scholar] [CrossRef] [PubMed]
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.-M.; Raussens, V. Toxic Prefibrillar α-Synuclein Amyloid Oligomers Adopt a Distinctive Antiparallel β-Sheet Structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha Synuclein Aggregation Drives Ferroptosis: An Interplay of Iron, Calcium and Lipid Peroxidation. Cell Death Differ. 2020, 27, 2781–2796. [Google Scholar] [CrossRef]
- Lee, H.-J.; Suk, J.-E.; Patrick, C.; Bae, E.-J.; Cho, J.-H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.-J. Direct Transfer of α-Synuclein from Neuron to Astroglia Causes Inflammatory Responses in Synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [Green Version]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A Natural Product Inhibits the Initiation of α-Synuclein Aggregation and Suppresses Its Toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Guerrero-Muñoz, M.J.; Sengupta, U.; Gerson, J.E.; Kayed, R. α-Synuclein Oligomers Induce a Unique Toxic Tau Strain. Biol. Psychiatry 2018, 84, 499–508. [Google Scholar] [CrossRef]
- Gerson, J.E.; Farmer, K.M.; Henson, N.; Castillo-Carranza, D.L.; Carretero Murillo, M.; Sengupta, U.; Barrett, A.; Kayed, R. Tau Oligomers Mediate α-Synuclein Toxicity and Can Be Targeted by Immunotherapy. Mol. Neurodegener. 2018, 13, 13. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein Occurs Physiologically as a Helically Folded Tetramer That Resists Aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A Soluble α-Synuclein Construct Forms a Dynamic Tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Bhattacharya, S.; Thompson, D. Re-Designing the α-Synuclein Tetramer. Chem. Commun. 2018, 54, 8080–8083. [Google Scholar] [CrossRef]
- Nuber, S.; Rajsombath, M.; Minakaki, G.; Winkler, J.; Müller, C.P.; Ericsson, M.; Caldarone, B.; Dettmer, U.; Selkoe, D.J. Abrogating Native α-Synuclein Tetramers in Mice Causes a L-DOPA-Responsive Motor Syndrome Closely Resembling Parkinson’s Disease. Neuron 2018, 100, 75–90.e5. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.R. Insulin Amyloid at Injection Sites of Patients with Diabetes. Amyloid 2016, 23, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kesserwan, S.; Mulka, A.; Sharafieh, R.; Qiao, Y.; Wu, R.; Kreutzer, D.L.; Klueh, U. Advancing Continuous Subcutaneous Insulin Infusion In Vivo: New Insights into Tissue Challenges. J. Biomed. Mater. Res. A 2021, 109, 1065–1079. [Google Scholar] [CrossRef]
- Jaklin, M.; Hritz, J.; Hribar-Lee, B. A New Fibrillization Mechanism of β-Lactoglobulin in Glycine Solutions. Int. J. Biol. Macromol. 2022, 216, 414–425. [Google Scholar] [CrossRef]
- Sen, S.; Ali, R.; Onkar, A.; Ganesh, S.; Verma, S. Strategies for Interference of Insulin Fibrillogenesis: Challenges and Advances. ChemBioChem 2022, 23, e202100678. [Google Scholar] [CrossRef]
- Ansari, A.M.; Osmani, L.; Matsangos, A.E.; Li, Q.K. Current Insight in the Localized Insulin-Derived Amyloidosis (LIDA): Clinico-Pathological Characteristics and Differential Diagnosis. Pathol.-Res. Pract. 2017, 213, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, K.; Zako, T.; Fukunaga, J.; Sörgjerd, K.M.; Ogata, K.; Kogure, K.; Kosano, H.; Noritake, M.; Maeda, M.; Ando, Y.; et al. Toxicity of Insulin-Derived Amyloidosis: A Case Report. BMC Endocr. Disord. 2019, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- Qafary, M.; Rashno, F.; Khajeh, K.; Khaledi, M.; Moosavi-Movahedi, A.A. Insulin Fibrillation: Strategies for Inhibition. Prog. Biophys. Mol. Biol. 2022, 175, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Sharafdini, R.; Mosaddeghi, H. Inhibition of Insulin Amyloid Fibrillation by Salvianolic Acids and Calix[ n ]Arenes: Molecular Docking Insight. J. Comput. Biophys. Chem. 2021, 20, 539–555. [Google Scholar] [CrossRef]
- Yang, L.-F.; Zeng, C.-M. The Degradation Products of Ascorbic Acid Inhibit Amyloid Fibrillation of Insulin and Destabilize Preformed Fibrils. Molecules 2018, 23, 3121. [Google Scholar] [CrossRef] [PubMed]
- Gorai, B.; Vashisth, H. Progress in Simulation Studies of Insulin Structure and Function. Front. Endocrinol. 2022, 13, 908724. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, O.M.; Suvorina, M.Y.; Dovidchenko, N.V.; Eliseeva, I.A.; Surin, A.K.; Finkelstein, A.V.; Schmatchenko, V.V.; Galzitskaya, O.V. How to Determine the Size of Folding Nuclei of Protofibrils from the Concentration Dependence of the Rate and Lag-Time of Aggregation. II. Experimental Application for Insulin and LysPro Insulin: Aggregation Morphology, Kinetics, and Sizes of Nuclei. J. Phys. Chem. B 2014, 118, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Surin, A.K.; Grishin, S.Y.; Galzitskaya, O.V. Identification of Amyloidogenic Regions in the Spine of Insulin Fibrils. Biochem. (Mosc.) 2019, 84, 47–55. [Google Scholar] [CrossRef]
- Surin, A.K.; Grishin, S.Y.; Galzitskaya, O.V. Determination of Amyloid Core Regions of Insulin Analogues Fibrils. Prion 2020, 14, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Flavonols and Flavones as BACE-1 Inhibitors: Structure-Activity Relationship in Cell-Free, Cell-Based and in Silico Studies Reveal Novel Pharmacophore Features. Biochim. Biophys Acta 2008, 1780, 819–825. [Google Scholar] [CrossRef]
- John, S.; Thangapandian, S.; Sakkiah, S.; Lee, K.W. Potent BACE-1 Inhibitor Design Using Pharmacophore Modeling, in Silico Screening and Molecular Docking Studies. BMC Bioinform. 2011, 12 (Suppl. S1), S28. [Google Scholar] [CrossRef]
- Huang, D.; Lüthi, U.; Kolb, P.; Edler, K.; Cecchini, M.; Audetat, S.; Barberis, A.; Caflisch, A. Discovery of Cell-Permeable Non-Peptide Inhibitors of Beta-Secretase by High-Throughput Docking and Continuum Electrostatics Calculations. J. Med. Chem. 2005, 48, 5108–5111. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, K.; Singh, K.D.; Chinnasamy, S.; Nagamani, S.; Krishnasamy, G.; Thiyagarajan, C.; Premkumar, P.; Anusuyadevi, M. High Throughput Virtual Screening and E-Pharmacophore Filtering in the Discovery of New BACE-1 Inhibitors. Interdiscip. Sci. 2013, 5, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Vassar, R. Targeting the β Secretase BACE1 for Alzheimer’s Disease Therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Minter, L.M.; Turley, D.M.; Das, P.; Shin, H.M.; Joshi, I.; Lawlor, R.G.; Cho, O.H.; Palaga, T.; Gottipati, S.; Telfer, J.C.; et al. Inhibitors of Gamma-Secretase Block In Vivo and In Vitro T Helper Type 1 Polarization by Preventing Notch Upregulation of Tbx21. Nat. Immunol. 2005, 6, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular Prion Protein Regulates Beta-Secretase Cleavage of the Alzheimer’s Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef]
- Hooper, N.M.; Turner, A.J. A New Take on Prions: Preventing Alzheimer’s Disease. Trends Biochem. Sci. 2008, 33, 151–155. [Google Scholar] [CrossRef]
- Fu, Z.; Aucoin, D.; Ahmed, M.; Ziliox, M.; Van Nostrand, W.E.; Smith, S.O. Capping of Aβ42 Oligomers by Small Molecule Inhibitors. Biochemistry 2014, 53, 7893–7903. [Google Scholar] [CrossRef]
- Alghamdi, A.; Birch, D.J.S.; Vyshemirsky, V.; Rolinski, O.J. Impact of the Flavonoid Quercetin on β-Amyloid Aggregation Revealed by Intrinsic Fluorescence. J. Phys. Chem. B 2022, 126, 7229–7237. [Google Scholar] [CrossRef] [PubMed]
- Cuccioloni, M.; Cecarini, V.; Bonfili, L.; Pettinari, R.; Tombesi, A.; Pagliaricci, N.; Petetta, L.; Angeletti, M.; Eleuteri, A.M. Enhancing the Amyloid-β Anti-Aggregation Properties of Curcumin via Arene-Ruthenium(II) Derivatization. IJMS 2022, 23, 8710. [Google Scholar] [CrossRef]
- Gautieri, A.; Beeg, M.; Gobbi, M.; Rigoldi, F.; Colombo, L.; Salmona, M. The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42. IJMS 2019, 20, 4641. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG Remodels Mature α-Synuclein and Amyloid-β Fibrils and Reduces Cellular Toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef]
- Tavanti, F.; Pedone, A.; Menziani, M.C. Insights into the Effect of Curcumin and (–)-Epigallocatechin-3-Gallate on the Aggregation of Aβ(1–40) Monomers by Means of Molecular Dynamics. IJMS 2020, 21, 5462. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, X.; Liu, J.; Ma, Q.; Zhuo, Z.; Chen, H.; Zhou, L.; Yang, S.; Zheng, L.; Ning, C.; et al. Gallic Acid Disruption of Aβ1–42 Aggregation Rescues Cognitive Decline of APP/PS1 Double Transgenic Mouse. Neurobiol. Dis. 2019, 124, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.; Wu, L.; Zhang, C.; Davis, R.M.; Xu, B. Natural Product-Based Nanomedicine: Recent Advances and Issues. Int. J. Nanomed. 2015, 10, 6055–6074. [Google Scholar] [CrossRef]
- Basnet, P.; Skalko-Basnet, N. Curcumin: An Anti-Inflammatory Molecule from a Curry Spice on the Path to Cancer Treatment. Molecules 2011, 16, 4567–4598. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, S.; Greco, C.; Tortora, P.; Aprile, F. Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms. IJMS 2018, 19, 2677. [Google Scholar] [CrossRef]
- Ghosh, N.; Kundu, L.M. Breaker Peptides against Amyloid-β Aggregation: A Potential Therapeutic Strategy for Alzheimer’s Disease. Future Med. Chem. 2021, 13, 1767–1794. [Google Scholar] [CrossRef] [PubMed]
- Catania, M.; Colombo, L.; Sorrentino, S.; Cagnotto, A.; Lucchetti, J.; Barbagallo, M.C.; Vannetiello, I.; Vecchi, E.R.; Favagrossa, M.; Costanza, M.; et al. A Novel Bio-Inspired Strategy to Prevent Amyloidogenesis and Synaptic Damage in Alzheimer’s Disease. Mol. Psychiatry 2022, 27, 5227–5234. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Shuaib, S.; Mann, S.; Goyal, B. Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-β (Aβ) Aggregation: Potential Therapeutics of Alzheimer’s Disease. ACS Comb. Sci. 2017, 19, 55–80. [Google Scholar] [CrossRef]
- Xiong, N.; Zhao, Y.; Dong, X.; Zheng, J.; Sun, Y. Design of a Molecular Hybrid of Dual Peptide Inhibitors Coupled on AuNPs for Enhanced Inhibition of Amyloid Β-Protein Aggregation and Cytotoxicity. Small 2017, 13, 1601666. [Google Scholar] [CrossRef] [PubMed]
- Stark, T.; Lieblein, T.; Pohland, M.; Kalden, E.; Freund, P.; Zangl, R.; Grewal, R.; Heilemann, M.; Eckert, G.P.; Morgner, N.; et al. Peptidomimetics That Inhibit and Partially Reverse the Aggregation of Aβ1–42. Biochemistry 2017, 56, 4840–4849. [Google Scholar] [CrossRef]
- Russ, H.; Mazzanti, M.; Parsons, C.; Riemann, K.; Gebauer, A.; Rammes, G. The Small Molecule GAL-201 Efficiently Detoxifies Soluble Amyloid β Oligomers: New Approach towards Oral Disease-Modifying Treatment of Alzheimer’s Disease. IJMS 2022, 23, 5794. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.; Lou, J.; Mehrazma, B.; Rauk, A.; Beazely, M.; Leonenko, Z. Pseudopeptide Amyloid Aggregation Inhibitors: In Silico, Single Molecule and Cell Viability Studies. IJMS 2021, 22, 1051. [Google Scholar] [CrossRef] [PubMed]
- Taş, K.; Volta, B.D.; Lindner, C.; El Bounkari, O.; Hille, K.; Tian, Y.; Puig-Bosch, X.; Ballmann, M.; Hornung, S.; Ortner, M.; et al. Designed Peptides as Nanomolar Cross-Amyloid Inhibitors Acting via Supramolecular Nanofiber Co-Assembly. Nat. Commun. 2022, 13, 5004. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Bera, A.; Bhadury, P.; De, P. From Small Molecules to Synthesized Polymers: Potential Role in Combating Amyloidogenic Disorders. ACS Chem. Neurosci. 2021, 12, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Dean, D.N.; Fogel, A.L.; Liu, F.; Abel, B.A.; McCormick, C.L.; Kharlampieva, E.; Rangachari, V.; Morgan, S.E. Aqueous RAFT Synthesis of Glycopolymers for Determination of Saccharide Structure and Concentration Effects on Amyloid β Aggregation. Biomacromolecules 2017, 18, 3359–3366. [Google Scholar] [CrossRef]
- Evgrafova, Z.; Voigt, B.; Roos, A.H.; Hause, G.; Hinderberger, D.; Balbach, J.; Binder, W.H. Modulation of Amyloid β Peptide Aggregation by Hydrophilic Polymers. Phys. Chem. Chem. Phys. 2019, 21, 20999–21006. [Google Scholar] [CrossRef] [PubMed]
- Sehar, U.; Rawat, P.; Reddy, A.P.; Kopel, J.; Reddy, P.H. Amyloid Beta in Aging and Alzheimer’s Disease. IJMS 2022, 23, 12924. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S. Bad News and Good News in AD, and How to Reconcile Them. Nat. Rev. Neurol. 2019, 15, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-T.; Lin, D.-H.; Luo, X.-Y.; Zhang, F.; Ji, L.-N.; Du, H.-N.; Song, G.-Q.; Hu, J.; Zhou, J.-W.; Hu, H.-Y. Inhibition of Alpha-Synuclein Fibrillization by Dopamine Analogs via Reaction with the Amino Groups of Alpha-Synuclein. Implication for Dopaminergic Neurodegeneration. FEBS J. 2005, 272, 3661–3672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Gallagher, A.; Hong, D.-P.; Long, C.; Fink, A.L.; Uversky, V.N. At Low Concentrations, 3,4-Dihydroxyphenylacetic Acid (DOPAC) Binds Non-Covalently to Alpha-Synuclein and Prevents Its Fibrillation. J. Mol. Biol. 2009, 388, 597–610. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Manning-Bog, A.B.; Di Monte, D.A.; Fink, A.L. Dopamine and L-Dopa Disaggregate Amyloid Fibrils: Implications for Parkinson’s and Alzheimer’s Disease. FASEB J. 2004, 18, 962–964. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, S.; Shi, D.; Bai, Q.; Liu, H.; Yao, X. Influence of EGCG on α-Synuclein (AS) Aggregation and Identification of Their Possible Binding Mode: A Computational Study Using Molecular Dynamics Simulation. Chem. Biol. Drug Des. 2018, 91, 162–171. [Google Scholar] [CrossRef]
- Grønnemose, A.L.; Østerlund, E.C.; Otzen, D.E.; Jørgensen, T.J.D. EGCG Has Dual and Opposing Effects on the N-Terminal Region of Self-Associating α-Synuclein Oligomers. J. Mol. Biol. 2022, 434, 167855. [Google Scholar] [CrossRef]
- Sternke-Hoffmann, R.; Peduzzo, A.; Bolakhrif, N.; Haas, R.; Buell, A.K. The Aggregation Conditions Define Whether EGCG Is an Inhibitor or Enhancer of α-Synuclein Amyloid Fibril Formation. Int. J. Mol. Sci. 2020, 21, 61995. [Google Scholar] [CrossRef]
- Ahmad, B.; Lapidus, L.J. Curcumin Prevents Aggregation in α-Synuclein by Increasing Reconfiguration Rate. J. Biol. Chem. 2012, 287, 9193–9199. [Google Scholar] [CrossRef]
- Xu, B.; Chen, J.; Liu, Y. Curcumin Interacts with α-Synuclein Condensates to Inhibit Amyloid Aggregation under Phase Separation. ACS Omega 2022, 7, 30281–30290. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nehru, B. Curcumin Affords Neuroprotection and Inhibits α-Synuclein Aggregation in Lipopolysaccharide-Induced Parkinson’s Disease Model. Inflammopharmacology 2018, 26, 349–360. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Medvedeva, M.; Barinova, K.; Melnikova, A.; Semenyuk, P.; Kolmogorov, V.; Gorelkin, P.; Erofeev, A.; Muronetz, V. Naturally Occurring Cinnamic Acid Derivatives Prevent Amyloid Transformation of Alpha-Synuclein. Biochimie 2020, 170, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, M.; Kitsilovskaya, N.; Stroylova, Y.; Sevostyanova, I.; Saboury, A.A.; Muronetz, V. Hydroxycinnamic Acid Derivatives from Coffee Extracts Prevent Amyloid Transformation of Alpha-Synuclein. Biomedicines 2022, 10, 2255. [Google Scholar] [CrossRef] [PubMed]
- Muronetz, V.I.; Barinova, K.; Kudryavtseva, S.; Medvedeva, M.; Melnikova, A.; Sevostyanova, I.; Semenyuk, P.; Stroylova, Y.; Sova, M. Natural and Synthetic Derivatives of Hydroxycinnamic Acid Modulating the Pathological Transformation of Amyloidogenic Proteins. Molecules 2020, 25, 4647. [Google Scholar] [CrossRef]
- Caruana, M.; Högen, T.; Levin, J.; Hillmer, A.; Giese, A.; Vassallo, N. Inhibition and Disaggregation of α-Synuclein Oligomers by Natural Polyphenolic Compounds. FEBS Lett. 2011, 585, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.R.; Tay, K.C.; Su, Y.X.; Wong, C.K.; Tan, W.N.; Khaw, K.Y. Potential of Naturally Derived Alkaloids as Multi-Targeted Therapeutic Agents for Neurodegenerative Diseases. Molecules 2021, 26, 728. [Google Scholar] [CrossRef] [PubMed]
- Kardani, J.; Roy, I. Understanding Caffeine’s Role in Attenuating the Toxicity of α-Synuclein Aggregates: Implications for Risk of Parkinson’s Disease. ACS Chem. Neurosci. 2015, 6, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, Q.; Zhang, L.; Wang, Q.; Yang, Z.; Liu, J.; Feng, L. Caffeic Acid Reduces A53T α-Synuclein by Activating JNK/Bcl-2-Mediated Autophagy in Vitro and Improves Behaviour and Protects Dopaminergic Neurons in a Mouse Model of Parkinson’s Disease. Pharmacol. Res. 2019, 150, 104538. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, S.S.; Fayed, H.S.; Zhu, Q.; Lu, J.-H.; Vaikath, N.N.; Ponraj, J.; Mansour, S.; El-Agnaf, O.M.A. Natural Alkaloid Compounds as Inhibitors for Alpha-Synuclein Seeded Fibril Formation and Toxicity. Molecules 2021, 26, 3736. [Google Scholar] [CrossRef] [PubMed]
- Pujols, J.; Peña-Díaz, S.; Lázaro, D.F.; Peccati, F.; Pinheiro, F.; González, D.; Carija, A.; Navarro, S.; Conde-Giménez, M.; García, J.; et al. Small Molecule Inhibits α-Synuclein Aggregation, Disrupts Amyloid Fibrils, and Prevents Degeneration of Dopaminergic Neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef] [Green Version]
- Peña-Díaz, S.; Pujols, J.; Vasili, E.; Pinheiro, F.; Santos, J.; Manglano-Artuñedo, Z.; Outeiro, T.F.; Ventura, S. The Small Aromatic Compound SynuClean-D Inhibits the Aggregation and Seeded Polymerization of Multiple α-Synuclein Strains. J. Biol. Chem. 2022, 298, 101902. [Google Scholar] [CrossRef] [PubMed]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; Lopes da Fonseca, T.; Koch, J.C.; Becker, S.; Tönges, L.; Bähr, M.; et al. Fasudil Attenuates Aggregation of α-Synuclein in Models of Parkinson’s Disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef]
- Pena-DIaz, S.; Ventura, S. One Ring Is Sufficient to Inhibit α-Synuclein Aggregation. Neural Regen. Res. 2022, 17, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhao, Y.; Xue, F.; Zheng, Y.; Huang, H.; Wang, W.; Chang, Y.; Yang, H.; Zhang, J. Exosomal DNA Aptamer Targeting α-Synuclein Aggregates Reduced Neuropathological Deficits in a Mouse Parkinson’s Disease Model. Mol. Nucleic Acids 2019, 17, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.H.; Saha, R.; Blanco, C.; Bagchi, D.; Chen, I.A. Modulation of α-Synuclein Aggregation In Vitro by a DNA Aptamer. Biochemistry 2022, 61, 1757–1765. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Duffuler, P.; Bhullar, K.S.; de Campos Zani, S.C.; Wu, J. Bioactive Peptides: From Basic Research to Clinical Trials and Commercialization. J. Agric. Food Chem. 2022, 70, 3585–3595. [Google Scholar] [CrossRef]
- Heinemann, L.; Krinelke, L. Insulin Infusion Set: The Achilles Heel of Continuous Subcutaneous Insulin Infusion. J. Diabetes Sci. Technol. 2012, 6, 954–964. [Google Scholar] [CrossRef]
- Weber, C.; Kammerer, D.; Streit, B.; Licht, A.H. Phenolic Excipients of Insulin Formulations Induce Cell Death, pro-Inflammatory Signaling and MCP-1 Release. Toxicol. Rep. 2015, 2, 194–202. [Google Scholar] [CrossRef]
- Paiva, T.O.; Bastos, A.E.P.; Marquês, J.T.; Viana, A.S.; Lima, P.A.; de Almeida, R.F.M. M-Cresol Affects the Lipid Bilayer in Membrane Models and Living Neurons. RSC Adv. 2016, 6, 105699–105712. [Google Scholar] [CrossRef]
- Das, A.; Gangarde, Y.M.; Pariary, R.; Bhunia, A.; Saraogi, I. An Amphiphilic Small Molecule Drives Insulin Aggregation Inhibition and Amyloid Disintegration. Int. J. Biol. Macromol. 2022, 218, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Channuwong, P.; Salae, K.; Chongruchiroj, S.; Cheng, H.; Suantawee, T.; Thilavech, T.; Adisakwattana, S. Dietary Anthocyanins Inhibit Insulin Fibril Formation and Cytotoxicity in 3T3-L1 Preadipocytes. Int. J. Biol. Macromol. 2022, 223, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Ratha, B.N.; Ghosh, A.; Brender, J.R.; Gayen, N.; Ilyas, H.; Neeraja, C.; Das, K.P.; Mandal, A.K.; Bhunia, A. Inhibition of Insulin Amyloid Fibrillation by a Novel Amphipathic Heptapeptide. J. Biol. Chem. 2016, 291, 23545–23556. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, M.; Yousefi, R.; Farjadian, F.; Uversky, V.N. Insulin Fibrillation: Toward Strategies for Attenuating the Process. Chem. Commun. 2020, 56, 11354–11373. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Shah, M.; Saraogi, I. Molecular Aspects of Insulin Aggregation and Various Therapeutic Interventions. ACS Bio Med. Chem. Au. 2022, 2, 205–221. [Google Scholar] [CrossRef]
- Maity, D. Inhibition of Amyloid Protein Aggregation Using Selected Peptidomimetics. ChemMedChem 2023, 18, e202200499. [Google Scholar] [CrossRef]
- Zhou, A.; Xie, J.; Han, H.; Chen, Y.; Zhao, C.; Li, J. Supramolecular Nanoparticles of Insulin and Pentapeptide for Inhibition of Fibrillation and Controlled Release. J. Biomed. Nanotechnol. 2018, 14, 959–967. [Google Scholar] [CrossRef]
- Siddiqi, M.K.; Majid, N.; Alam, P.; Malik, S.; Alam, A.; Rajan, S.; Ajmal, M.R.; Khan, R.H. Both Beta Sheet Breaker and Alpha Helix Forming Pentapeptide Inhibits Protein Fibrillation: Implication for the Treatment of Amyloid Disorders. Int. J. Biol. Macromol. 2020, 143, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Roy Chowdhury, S.; Mondal, S.; Iyer, P.K. Blocking Oligomeric Insulin Amyloid Fibrillation via Perylenebisimides Containing Dipeptide Tentacles. ACS Biomater. Sci. Eng. 2018, 4, 4076–4083. [Google Scholar] [CrossRef]
- Gibson, T.J.; Murphy, R.M. Inhibition of Insulin Fibrillogenesis with Targeted Peptides. Protein Sci. 2006, 15, 1133–1141. [Google Scholar] [CrossRef]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. FoldAmyloid: A Method of Prediction of Amyloidogenic Regions from Protein Sequence. Bioinformatics 2010, 26, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Trovato, A.; Seno, F.; Tosatto, S.C.E. The PASTA Server for Protein Aggregation Prediction. Protein Eng. Des. Sel. 2007, 20, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez de la Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the Sequence Determinants of Amyloid Structure Using Position-Specific Scoring Matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A Server for the Prediction and Evaluation of “Hot Spots” of Aggregation in Polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the Amylome, Proteins Capable of Forming Amyloid-like Fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Tan, K.H.; Chin, S.P.; Heh, C.H. Automated in Silico EGFR Peptide Inhibitor Elongation Using Self-Evolving Peptide Algorithm. Curr. Comput.-Aided Drug Des. 2022, 18, 150–158. [Google Scholar] [CrossRef]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-Dock Web Server for the Flexible Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Landreh, M.; Stukenborg, J.-B.; Willander, H.; Söder, O.; Johansson, J.; Jörnvall, H. Proinsulin C-Peptide Interferes with Insulin Fibril Formation. Biochem. Biophys. Res. Commun. 2012, 418, 489–493. [Google Scholar] [CrossRef]
- Kovalska, V.B.; Losytskyy, M.Y.; Varzatskii, O.A.; Cherepanov, V.V.; Voloshin, Y.Z.; Mokhir, A.A.; Yarmoluk, S.M.; Volkov, S. V Study of Anti-Fibrillogenic Activity of Iron(II) Clathrochelates. Bioorganic Med. Chem. 2014, 22, 1883–1888. [Google Scholar] [CrossRef]
- Nusrat, S.; Zaman, M.; Masroor, A.; Siddqi, M.K.; Zaidi, N.; Neelofar, K.; Abdelhameed, A.S.; Khan, R.H. Deciphering the Enhanced Inhibitory, Disaggregating and Cytoprotective Potential of Promethazine towards Amyloid Fibrillation. Int. J. Biol. Macromol. 2018, 106, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Khan, M.V.; Zakariya, S.M.; Nusrat, S.; Meeran, S.M.; Alam, P.; Ajmal, M.R.; Wahiduzzaman, W.; Shahein, Y.E.; Abouelella, A.M.; et al. Amino Group of Salicylic Acid Exhibits Enhanced Inhibitory Potential against Insulin Amyloid Fibrillation with Protective Aptitude toward Amyloid Induced Cytotoxicity. J. Cell. Biochem. 2018, 119, 3945–3956. [Google Scholar] [CrossRef] [PubMed]
- Kurpe, S.; Grishin, S.; Surin, A.; Selivanova, O.; Fadeev, R.; Dzhus, U.; Gorbunova, E.; Mustaeva, L.; Azev, V.; Galzitskaya, O. Antimicrobial and Amyloidogenic Activity of Peptides Synthesized on the Basis of the Ribosomal S1 Protein from Thermus Thermophilus . Int. J. Mol. Sci. 2020, 21, 76382. [Google Scholar] [CrossRef]
- Kravchenko, S.V.; Domnin, P.A.; Grishin, S.Y.; Panfilov, A.V.; Azev, V.N.; Mustaeva, L.G.; Gorbunova, E.Y.; Kobyakova, M.I.; Surin, A.K.; Glyakina, A.V.; et al. Multiple Antimicrobial Effects of Hybrid Peptides Synthesized Based on the Sequence of Ribosomal S1 Protein from Staphylococcus Aureus . Int. J. Mol. Sci. 2022, 23, 10524. [Google Scholar] [CrossRef]
- Grishin, S.Y.; Domnin, P.A.; Kravchenko, S.V.; Azev, V.N.; Mustaeva, L.G.; Gorbunova, E.Y.; Kobyakova, M.I.; Surin, A.K.; Makarova, M.A.; Kurpe, S.R.; et al. Is It Possible to Create Antimicrobial Peptides Based on the Amyloidogenic Sequence of Ribosomal S1 Protein of P. Aeruginosa? Int. J. Mol. Sci. 2021, 22, 89776. [Google Scholar] [CrossRef] [PubMed]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blázquez-Castro, A. Tetrazolium Salts and Formazan Products in Cell Biology: Viability Assessment, Fluorescence Imaging, and Labeling Perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Scieuzo, C.; Petrone, A.M.; Salvia, R.; Manniello, M.D.; Franco, A.; Lucchetti, D.; Vassallo, A.; Vogel, H.; Sgambato, A.; et al. Antimicrobial Peptides: A New Hope in Biomedical and Pharmaceutical Fields. Front. Cell. Infect. Microbiol. 2021, 11, 668632. [Google Scholar] [CrossRef]
- Mishra, N.K.; Joshi, K.B.; Verma, S. Inhibition of Human and Bovine Insulin Fibril Formation by Designed Peptide Conjugates. Mol. Pharm. 2013, 10, 3903–3912. [Google Scholar] [CrossRef] [PubMed]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Multifunction of Myricetin on Aβ: Neuroprotection via a Conformational Change of Aβ and Reduction of Aβ via the Interference of Secretases. J. Neurosci. Res. 2008, 86, 368–377. [Google Scholar] [CrossRef]
- Xu, B.; Mo, X.; Chen, J.; Yu, H.; Liu, Y. Myricetin Inhibits A-Synuclein Amyloid Aggregation by Delaying the Liquid-to-Solid Phase Transition. ChemBioChem 2022, 23, e202200216. [Google Scholar] [CrossRef]
- Prajapati, K.P.; Singh, A.P.; Dubey, K.; Ansari, M.; Temgire, M.; Anand, B.G.; Kar, K. Myricetin Inhibits Amyloid Fibril Formation of Globular Proteins by Stabilizing the Native Structures. Colloids Surf. B Biointerfaces 2020, 186, 110640. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Han, S.; Fink, A.L. Oxidized Quercetin Inhibits α-Synuclein Fibrillization. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 2872–2881. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-B.; Wang, Y.-M.; Zeng, C.-M. Quercetin Inhibits Amyloid Fibrillation of Bovine Insulin and Destabilizes Preformed Fibrils. Biochem. Biophys. Res. Commun. 2011, 415, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Ziaunys, M.; Mikalauskaite, K.; Sakalauskas, A.; Smirnovas, V. Interplay between Epigallocatechin-3-Gallate and Ionic Strength during Amyloid Aggregation. PeerJ 2021, 9, e12381. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin Inhibits Formation of Amyloid β Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid In Vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Rabiee, A.; Ebrahim-Habibi, A.; Ghasemi, A.; Nemat-Gorgani, M. How Curcumin Affords Effective Protection against Amyloid Fibrillation in Insulin. Food Funct. 2013, 4, 1474. [Google Scholar] [CrossRef]
- Liu, Y.; Carver, J.A.; Calabrese, A.N.; Pukala, T.L. Gallic Acid Interacts with α-Synuclein to Prevent the Structural Collapse Necessary for Its Aggregation. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2014, 1844, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Jayamani, J.; Shanmugam, G. Gallic Acid, One of the Components in Many Plant Tissues, Is a Potential Inhibitor for Insulin Amyloid Fibril Formation. Eur. J. Med. Chem. 2014, 85, 352–358. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galzitskaya, O.V.; Grishin, S.Y.; Glyakina, A.V.; Dovidchenko, N.V.; Konstantinova, A.V.; Kravchenko, S.V.; Surin, A.K. The Strategies of Development of New Non-Toxic Inhibitors of Amyloid Formation. Int. J. Mol. Sci. 2023, 24, 3781. https://doi.org/10.3390/ijms24043781
Galzitskaya OV, Grishin SY, Glyakina AV, Dovidchenko NV, Konstantinova AV, Kravchenko SV, Surin AK. The Strategies of Development of New Non-Toxic Inhibitors of Amyloid Formation. International Journal of Molecular Sciences. 2023; 24(4):3781. https://doi.org/10.3390/ijms24043781
Chicago/Turabian StyleGalzitskaya, Oxana V., Sergei Y. Grishin, Anna V. Glyakina, Nikita V. Dovidchenko, Anastasiia V. Konstantinova, Sergey V. Kravchenko, and Alexey K. Surin. 2023. "The Strategies of Development of New Non-Toxic Inhibitors of Amyloid Formation" International Journal of Molecular Sciences 24, no. 4: 3781. https://doi.org/10.3390/ijms24043781