

Advanced Microscopy Techniques for Molecular Biophysics

Abstract
:1. Introduction
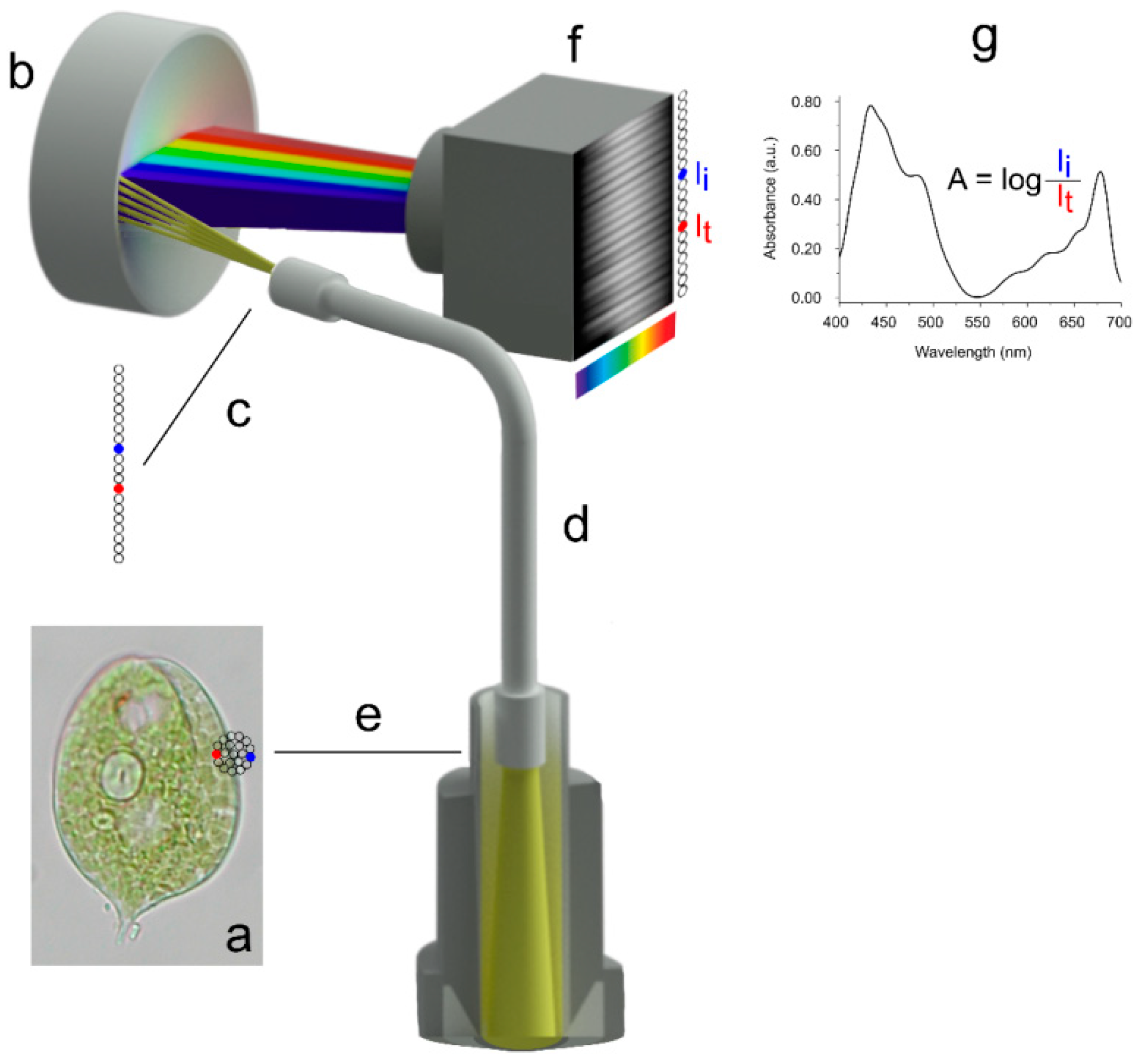
2. Microspectrophotometry
2.1. General Description
2.2. Theory
- (a)
- SR, the spectral radiance of the lamp, expressed in W cm−2 sr−1 nm−1, i.e., the radiant flux per unit area, solid angle and spectral bandwidth;
- (b)
- SB, the spectral bandwidth, expressed in nm, for each measuring wavelength;
- (c)
- OF, the optical flux of the optical system, i.e., the ability of the optical system to transfer light energy. OF is expressed in cm² sr and is a purely geometric quantity applied to the volume through which the light is transferred. In wide-field microscopy (WFM) the OF is about 10−3 cm2 sr, and the smallest optical flux giving a reliable measurement value is about 10−8 cm2 sr.
- (d)
- TR, the total transmittance of the optical system, is a measure of the percentage of light that remains after loss by absorption, reflection and diffraction of optical components (i.e., lens, mirrors, ...);
- (e)
- ILS, the interactions of the light with the sample, is related to the absorption cross section of absorbing molecules and their number, and it is a measure of the percentage of light that remains after the sample absorption.
2.3. A Working Set-Up
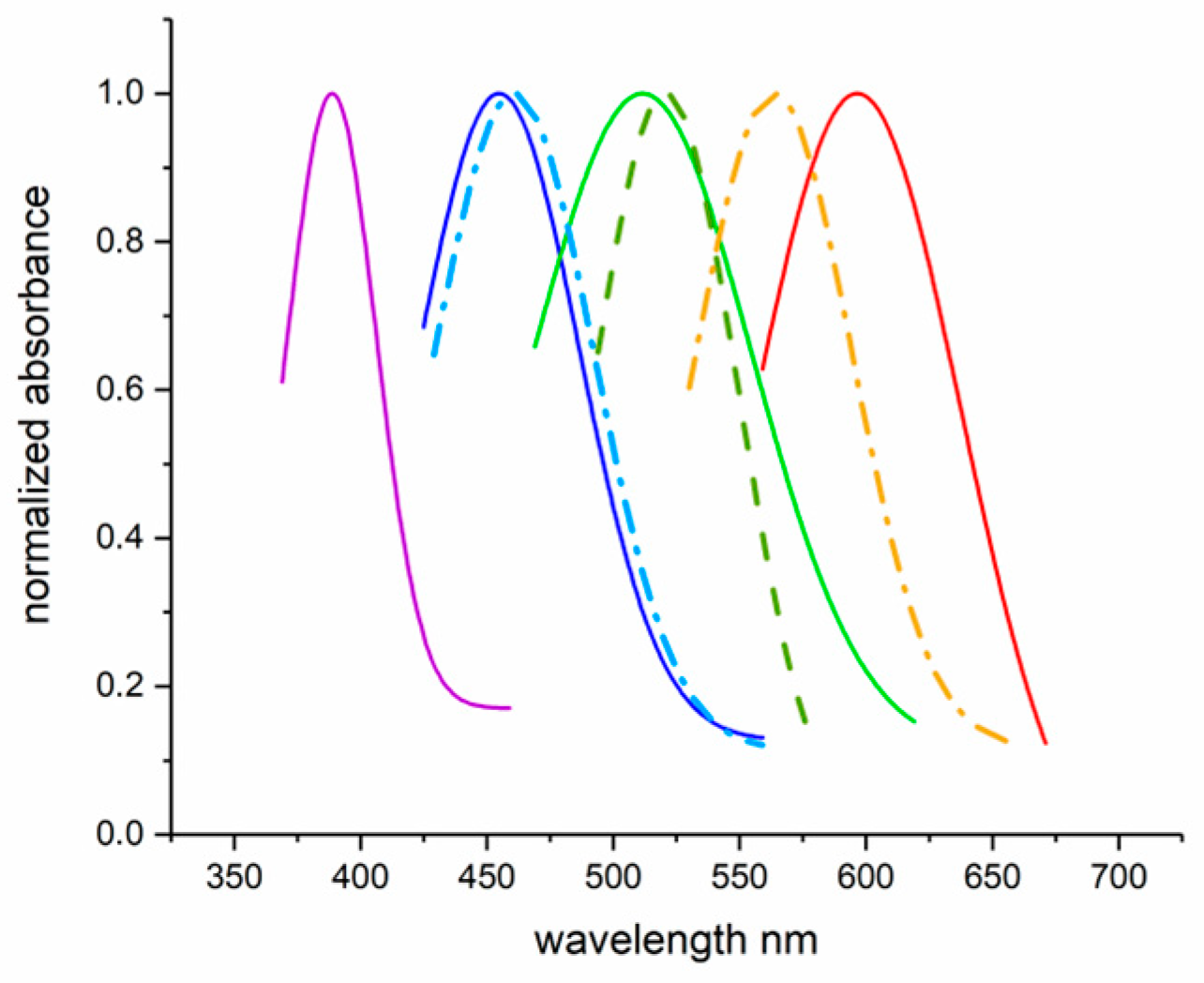
2.4. Experimental Example 1: MSP on Retinal and Extra-Retinal Photoreceptors of Teleost Fish
2.5. Experimental Example 2: Which Is the Prey?
3. Super-Resolution Localization Microscopy
3.1. General Description
3.2. Theory
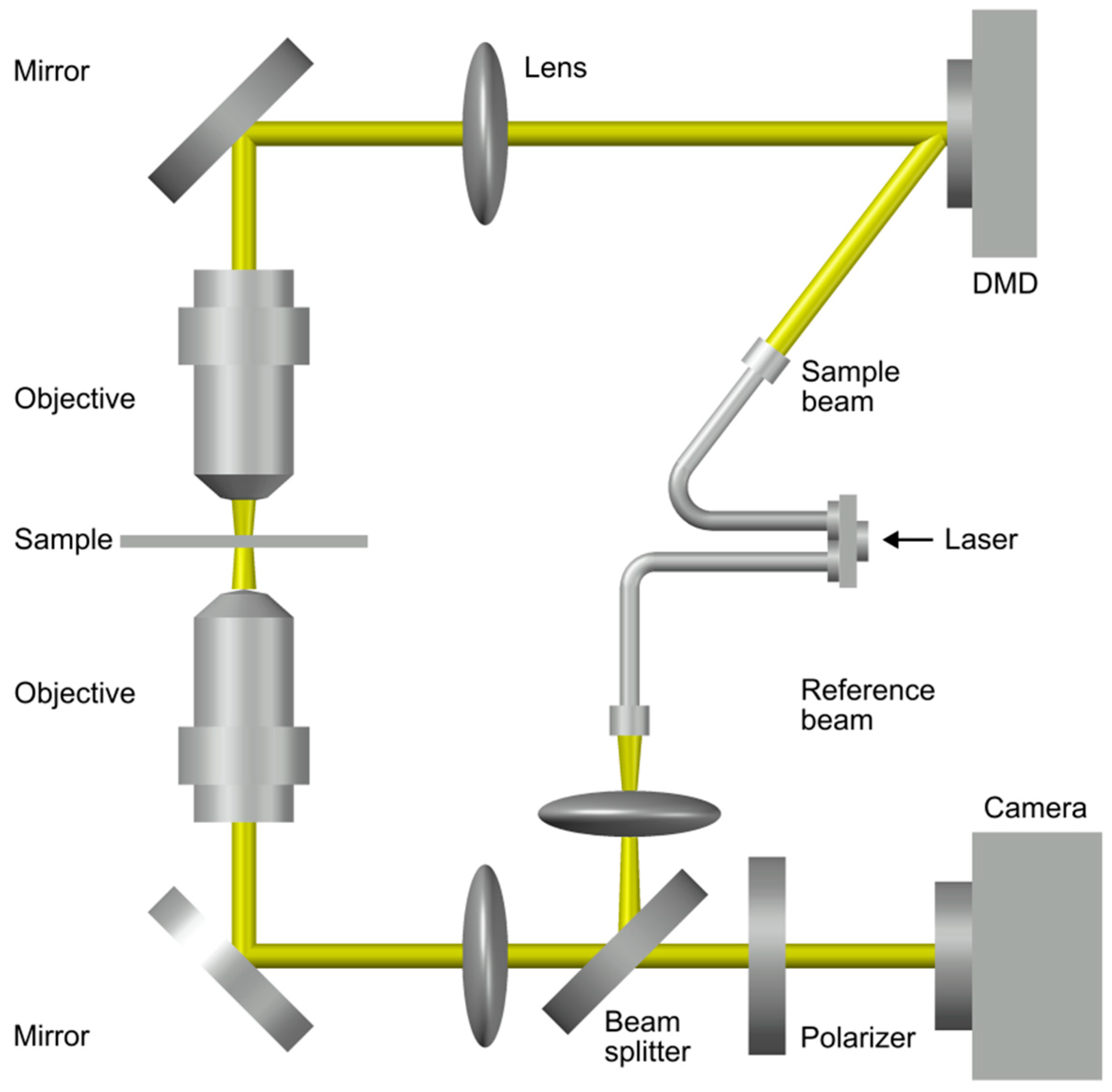
3.3. A Working Set-Up
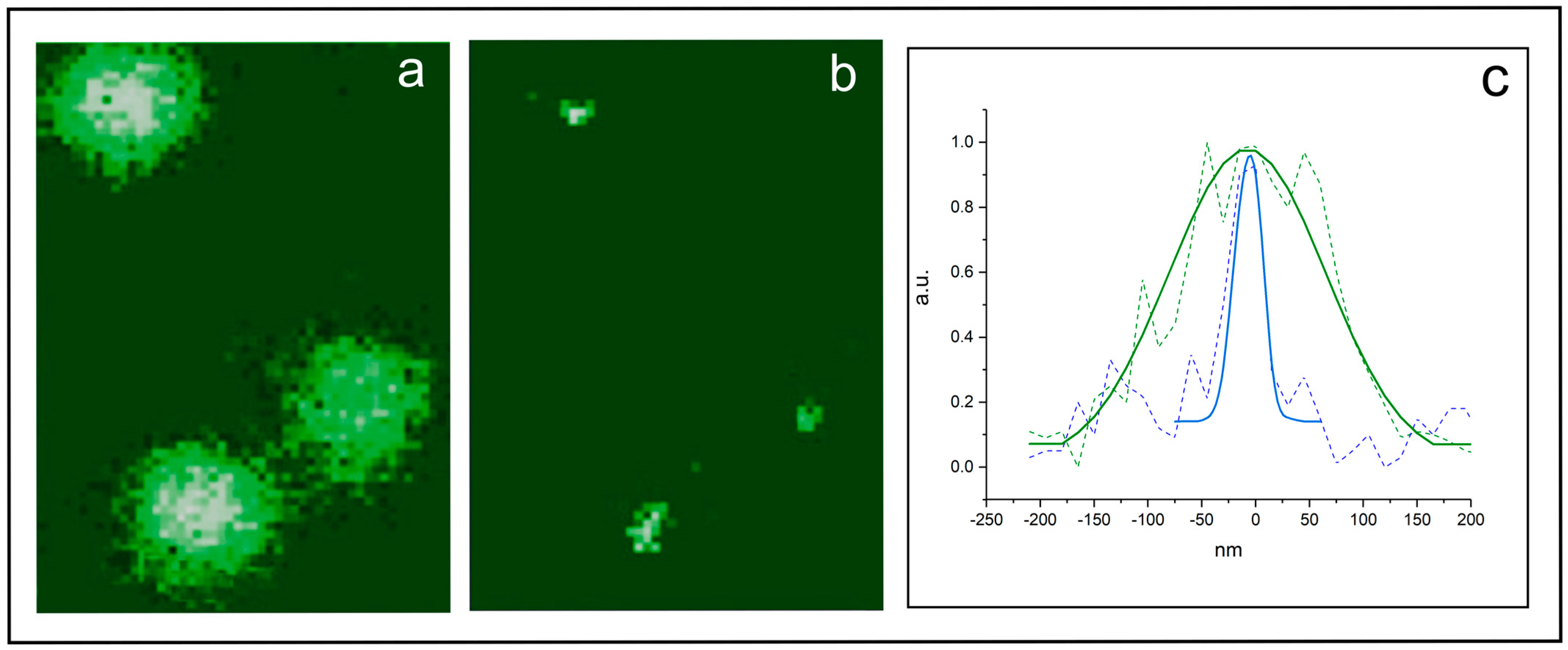
3.4. Experimental Example 1: Single-Molecule Tracking and Imaging
3.5. Experimental Example 2: STED or Confocal Microscopy, Which Is the Best?
4. Holotomography
4.1. General Description
4.2. Theory
4.3. A Working Set-Up
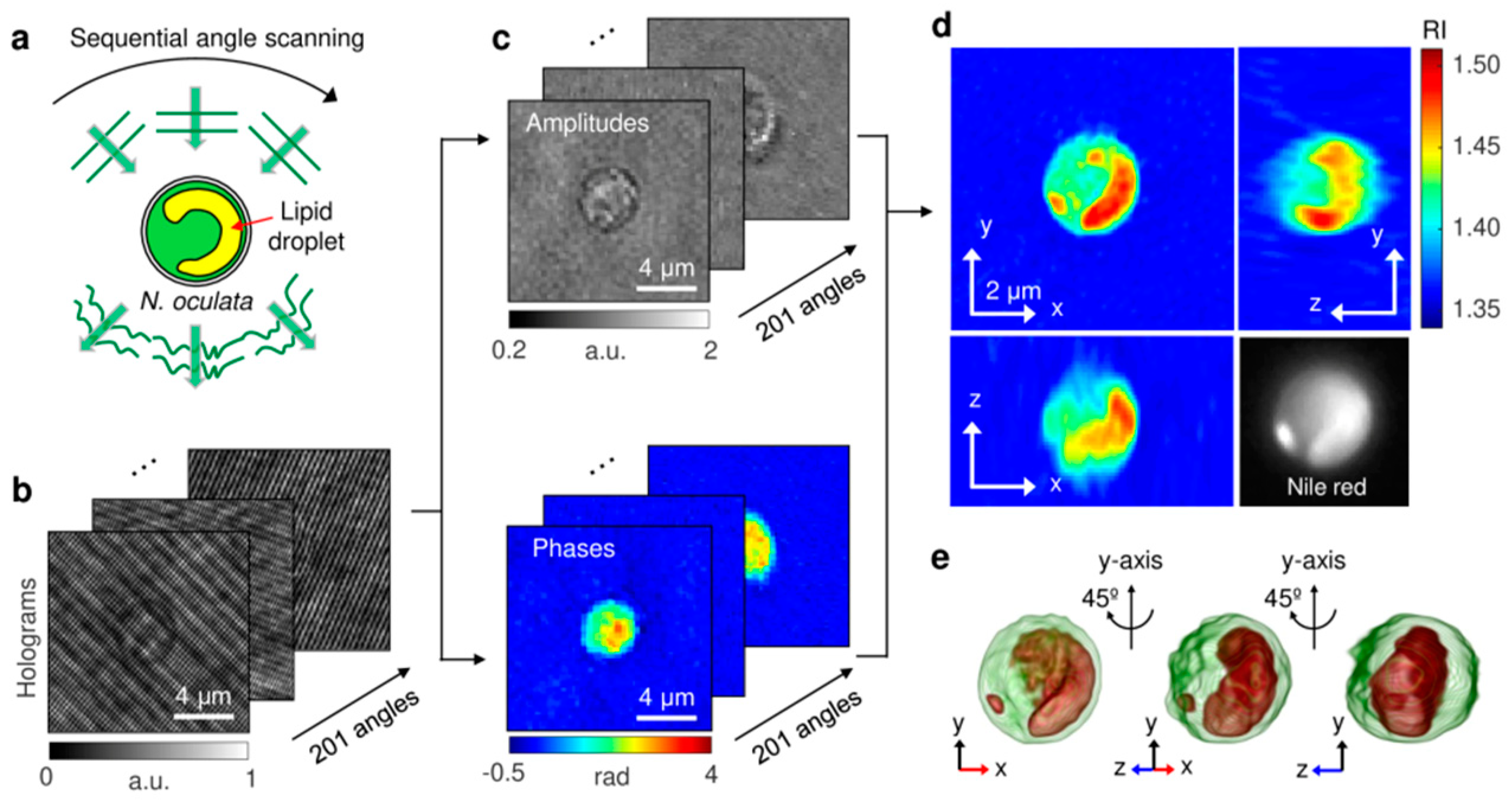
4.4. Experimental Example 1: Quantitative Measurement of Lipid Contents in Individual Microalgal Cells
4.5. Experimental Example 2: The 3D Structure of Euglena Gracilis Photoreceptor
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gualtieri, P. Molecular biology in living cells by means of digital optical microscopy. Micron Microsc. Acta 1992, 23, 239–257. [Google Scholar] [CrossRef]
- Gualtieri, P. Microspectroscopy of photoreceptor pigments in flagellated algae. Crit. Rev. Plant Sci. 1991, 9, 475–495. [Google Scholar] [CrossRef]
- Wannemacher, R. Confocal Laser Scanning Microscopy. In Encyclopedia of Nanotechnology; Bhushan, B., Ed.; Springer: Dordrecht, The Netherlands, 2015. [Google Scholar]
- Yu, L.; Lei, Y.; Ma, Y.; Liu, M.; Zheng, J.; Dan, D.; Gao, P. A Comprehensive Review of Fluorescence Correlation Spectroscopy. Front. Phys. 2021, 9, 644450. [Google Scholar] [CrossRef]
- Broussard, J.A.; Green, K.J. Research Techniques Made Simple: Methodology and Applications of Förster Resonance Energy Transfer (FRET) Microscopy. J. Investig. Dermatol. 2017, 137, e185–e191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercatelli, R.; Quercioli, F.; Barsanti, L.; Evangelista, V.; Coltelli, P.; Passarelli, V.; Frassanito, A.M.; Gualtieri, P. Intramolecular photo-switching and intermolecular energy transfer as primary photo-events in photoreceptive processes: The case of Euglena gracilis. Biochem. Biophys. Res. Commun. 2009, 385, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Heintzmann, R.; Huser, T. Super-Resolution Structured Illumination Microscopy. Chem. Rev. 2017, 117, 13890–13908. [Google Scholar] [CrossRef]
- Szymborska, A.; de Marco, A.; Daigle, N.; Cordes, V.C.; Briggs, J.A.; Ellenberg, J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science 2013, 341, 655–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertocchi, C.; Wang, Y.; Ravasio, A.; Hara, Y.; Wu, Y.; Sailov, T.; Baird, M.A.; Davidson, M.W.; Zaidel-Bar, R.; Toyama, Y.; et al. Nanoscale architecture of cadherin-based cell adhesions. Nat. Cell Biol. 2017, 19, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Gualtieri, P.; Passarelli, V.; Barsanti, L. A simple instrument to perform in vivo absorption spectra of pigmented cedllular organelles. Micron Microsc. Acta 1989, 20, 107–110. [Google Scholar] [CrossRef]
- Barsanti, L.; Passarelli, V.; Walne, P.L.; Gualtieri, P. In Vivo Photocycle of the Euglena gracilis Photoreceptor. Biophys. J. 1997, 72, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Barsanti, L.; Coltelli, P.; Evangelista, V.; Frassanito, A.M.; Vesentini, N.; Santoro, F.; Gualtieri, P. In Vivo Absorption Spectra of the Two Stable States of the Euglena Photoreceptor photocycle. Photochem. Photobiol. 2009, 85, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Barsanti, L.; Evangelista, V.; Frassanito, A.M.; Gualtieri, P. Fundamental questions and concepts about photoreception and the case of Euglena gracilis. Integr. Biol. 2012, 4, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.C.; Barsanti, L.; Evangelista, E.; Conforti, V.; Gualtieri, P. Effects of chromium on photosynthetic and photoreceptive apparatus of the alga Chlamydomonas reinhardtii. Environ. Res. 2007, 105, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Juarez, A.B.; Barsanti, L.; Passarelli, V.; Evangelista, V.; Vesentini, N.; Conforti, V.; Gualtieri, P. In vivo microspectroscopy monitoring of chromium effects on the photosynthetic and photoreceptive apparatus of Eudorina unicocca and Chlorella kessleri. J. Environ. Monit. 2008, 10, 1313–1318. [Google Scholar] [CrossRef]
- Coltelli, P.; Barsanti, L.; Evangelista, V.; Frassanito, A.M.; Gualtieri, P. Reconstruction of the absorption spectrum of an object spot from the colour values of the corresponding pixel(s) in its digital image: The challenge of algal colours. J. Microsc. 2016, 264, 311–320. [Google Scholar] [CrossRef]
- Gualtieri, P.; Barsanti, L.; Passarelli, V. Absorption spectrum of a single isolated paraflagellar swelling of Euglena gracilis. Biochem. Biophys. Acta 1989, 993, 293–296. [Google Scholar] [CrossRef]
- Croce, A.C.; Bottiroli, G. Autofluorescence spectroscopy and imaging: A tool for biomedical research and diagnosis. Eur. J. Histochem. 2014, 12, 2461. [Google Scholar] [CrossRef] [Green Version]
- Barsanti, L.; Gualtieri, P. Algae: Anatomy, Biochemistry, and Biotechnology, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2023. [Google Scholar]
- Evangelista, V.; Frassanito, A.M.; Barsanti, L.; Gualtieri, P. Microspectroscopy of the Photosynthetic Compartment of Algae. Photochem. Photobiol. 2006, 82, 1039–1046. [Google Scholar] [CrossRef]
- Barsanti, L.; Evangelista, V.; Frassanito, A.M.; Vesentini, N.; Passarelli, V.; Gualtieri, P. Absorption microspectroscopy, theory and applications in the case of the photosynthetic compartment. Micron 2007, 38, 197–213. [Google Scholar] [CrossRef]
- Evangelista, V.; Evangelisti, M.; Barsanti, L.; Frassanito, A.M.; Passarelli, V.; Gualtieri, P. A polychromator-based microspectrophotometer. Int. J. Biol. Sci. 2007, 3, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Kusmic, C.; Barsanti, L.; Passarelli, V.; Gualtieri, P. Photoreceptor morphology and visual pigment content in the pineal organ and in the retina of juvenile and adult trout, Salmo irideus. Micron 1993, 24, 279–286. [Google Scholar] [CrossRef]
- Kondrashev, S.L.; Lamash, N.E. Unusual A1/A2–visual pigment conversion during light/dark adaptation in marine fish. Comp. Biochem. Physiol. Part A 2019, 238, 110560. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, H.H.; Hansen, P.J.; Larsen, J. Growth and grazing responses of two chloroplast-retaining dinoflagellates: Effect of irradiance and prey species. Mar. Ecol. Prog. Ser. 2000, 201, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Javornicky, P. Taxonomic notes on some freshwater planktonic Cryptophyceae based on light microscopy. Hydrobiologia 2003, 502, 271–283. [Google Scholar] [CrossRef]
- Novarino, G. A companion to the identification of cryptomonads flagellates (Cryptophyceae=Cryptomonadea). Hydrobiologia 2003, 502, 225–270. [Google Scholar] [CrossRef]
- Barsanti, L.; Evangelista, V.; Passarelli, V.; Frassanito, A.M.; Coltelli, P.; Gualtieri, P. Microspectrophotometry as a method to identify kleptoplastids in the naked freshwater dinoflagellate Gymnodinium acidotum. J. Phycol. 2009, 45, 1304–1309. [Google Scholar] [CrossRef]
- Mockl, L.; Lamb, D.C.; Bruchle, C. Super-resolved Fluorescence Microscopy: Nobel Prize in Chemistry 2014 for Eric Betzig, Stefan Hell, and William E. Moerner. Angew. Chem. Int. Ed. 2014, 53, 13972–13977. [Google Scholar] [CrossRef]
- Moerner, W.E.; Kador, L. Optical detection and spectroscopy of single molecules in a solid. Phys. Rev. Lett. 1989, 62, 2535–2538. [Google Scholar] [CrossRef] [Green Version]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [Green Version]
- Vicidomini, G.; Bianchini, P.; Diaspro, A. STED super-resolved microscopy. Nat. Methods 2018, 15, 173–182. [Google Scholar] [CrossRef]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzig, E. Proposed method for molecular optical imaging. Opt. Lett. 1995, 20, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [Green Version]
- Heilemann, M.; Van De Linde, S.; Schüttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. 2008, 47, 6172–6176. [Google Scholar] [CrossRef]
- Van de Linde, S.; Löschberger, A.; Klein, T.; Heidbreder, M.; Wolter, S.; Heilemann, M.; Sauer, M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011, 6, 991–1009. [Google Scholar] [CrossRef]
- Löschberger, A.; van de Linde, S.; Dabauvalle, M.-C.; Rieger, B.; Heilemann, M.; Krohne, G.; Sauer, M. Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution. J. Cell Sci. 2012, 125, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Ravalia, A.S.; Lau, J.; Barron, J.C.; Purchase, S.L.M.; Southwell, A.L.; Hayden, M.R.; Nafar, F.; Parsons, M.P. Super-resolution imaging reveals extrastriatal synaptic dysfunction in presymptomatic Huntington disease mice. Neurobiol. Dis. 2021, 152, 105293. [Google Scholar] [CrossRef]
- Wang, M.; Marr, J.M.; Davanco, M.; Gilman, J.W.; Liddle, J.A. Nanoscale deformation in polymers revealed by single-molecule super-resolution location—Orientation microscopy. Mater. Horiz. 2019, 6, 817–825. [Google Scholar] [CrossRef]
- Walker, J.G. Optical imaging with resolution exceeding the Rayleigh criterion. Opt. Acta 1983, 30, 1197–1202. [Google Scholar] [CrossRef]
- Vangindertael, J.; Camacho, R.; Sempels, W.; Mizuno, H.; Dedecker, P.; Janssen, K.P.F. An introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl. Fluoresc. 2018, 6, 022003. [Google Scholar] [CrossRef]
- Busch, P.; Heinonen, T.; Lahti, P. Heisenberg’s uncertainty principle. Phys. Rep. 2007, 452, 155–176. [Google Scholar] [CrossRef] [Green Version]
- Unternährer, M.; Bessire, B.; Gasparini, L.; Perenzoni, M.; Stefanov, A. Super-Resolution Quantum Imaging at the Heisenberg Limit. Optica 2018, 5, 1150–1154. [Google Scholar] [CrossRef] [Green Version]
- Bertero, M.; Pike, E.R. Resolution in diffraction limited imaging, a singular value analysis. I the case of coherent illumination. Opt. Acta 1982, 29, 727–746. [Google Scholar] [CrossRef] [Green Version]
- Bertero, M.; Boccacci, P.; Pike, E.R. Resolution in diffraction limited imaging, a singular value analysis. II the case of incoherent illumination. Opt. Acta 1982, 29, 1599–1611. [Google Scholar] [CrossRef]
- Frieden, B.R. Probability, Statistical Optics, and Data Testing; Springer Series in Information Sciences; Springer: Berlin, Germany, 1991; Volume 10, 444p. [Google Scholar]
- Tressler, C.; Stolle, M.; Fradin, C. Fluorescence correlation spectroscopy with a doughnut-shaped excitation profile as a characterization tool in STED microscopy. Opt. Express 2014, 22, 31154–31166. [Google Scholar] [CrossRef] [PubMed]
- Coto Hernández, I.; Buttafava, M.; Boso, G.; Diaspro, A.; Tosi, A.; Vicidomini, G. Gated STED microscopy with time-gated single-photon avalanche diode. Biomed. Opt. Express 2015, 6, 2258–2267. [Google Scholar] [CrossRef] [Green Version]
- Vicidomini, G.; Schönle, A.; Ta, H.; Han, K.Y.; Moneron, G.; Eggeling, C.; Hell, S.W. STED nanoscopy with time-gated detection: Theoretical and experimental aspects. PLoS ONE 2013, 8, e54421. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Thiruthakkathevan, S.; Prabhakar, A. Active Fibre Mode-locked Lasers in Synchronization for STED Microscopy. In Optics, Photonics and Laser Technology; Springer Series in Optical, Sciences; Ribeiro, P., Andrews, D., Raposo, M., Eds.; Springer: Cham, Switzerland, 2017; Volume 222. [Google Scholar]
- Xie, H.; Liu, Y.; Jin, D.; Santangelo, P.J.; Xi, P. Analytical description of high-aperture STED resolution with 0-2π vortex phase modulation. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2013, 30, 1640–1645. [Google Scholar] [CrossRef] [Green Version]
- Ishii, H.; Otomo, K.; Hung, J.-H.; Tsutsumi, M.; Yokoyama, H.; Nemoto, T. Two-photon STED nanoscopy realizing 100-nm spatial resolution utilizing high-peak-power sub-nanosecond 655-nm pulses. Biomed. Opt. Express 2019, 10, 3104–3113. [Google Scholar] [CrossRef]
- Kolmakov, K.; Winter, F.R.; Sednev, M.V.; Ghosh, S.; Borisov, S.M.; Nizovtsev, A.V. Everlasting rhodamine dyes and true deciding factors in their STED microscopy performance. Photochem. Photobiol. Sci. 2020, 19, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Gahlmann, A.; Moerner, W.E. Exploring bacterial cell biology with single-molecule tracking and super-resolution imaging. Nat. Rev. Microbiol. 2014, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khater, I.M.; Nabi, I.R.; Hamarneh, G. A Review of Super-Resolution Single-Molecule Localization Microscopy Cluster Analysis and Quantification Methods. Patterns 2020, 1, 100038. [Google Scholar] [CrossRef]
- Donnert, G.; Keller, J.; Medda, R.; Andrei, M.A.; Rizzoli, S.O.; Luhrmann, R.; Jahn, R.; Eggeling, C.; Hell, S.W. Macromolecular-scale resolution in biological fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 11440–11445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Yoon, J.; Shin, S.; Lee, S.; Yang, S.-A.; Park, Y. Optical diffraction tomography techniques for the study of cell pathophysiology. J. Biomed. Photonics Eng. 2016, 2, 020201-1. [Google Scholar] [CrossRef] [Green Version]
- Haeberlé, O.; Belkebir, K.; Giovaninni, H.; Sentenac, A. Tomographic Diffractive Microscopy: Basics, techniques and perspectives. J. Mod. Opt. 2010, 57, 686–699. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Lee, S.; Lee, M.; Oh, J.; Yang, S.A.; Park, Y. Holotomography: Refractive Index as an Intrinsic Imaging Contrast for 3-D Label-Free Live Cell Imaging. Adv. Exp. Med. Biol. 2021, 1310, 211–238. [Google Scholar]
- Kim, Y.; Shim, H.; Kim, K.; Park, H.; Jang, S.; Park, Y. Profiling individual human red blood cells using common-path diffraction optical tomography. Sci. Rep. 2014, 4, 6659. [Google Scholar] [CrossRef] [Green Version]
- Rappaz, B.; Cano, E.; Colomb, T.; Kühn, J.; Depeursinge, C.; Simanis, V.; Magistretti, P.J.; Marquet, P. Noninvasive characterization of the fission yeast cell cycle by monitoring dry mass with digital holographic microscopy. J. Biomed. Opt. 2009, 14, 034049. [Google Scholar] [CrossRef]
- Bennet, M.; Gur, D.; Yoon, J.; Park, Y.; Faivre, D. A Bacteria-Based Remotely Tunable Photonic Device. Adv. Opt. Mater. 2016, 5, 1600617. [Google Scholar] [CrossRef]
- Kim, T.; Zhou, R.; Mir, M.; Babacan, S.D.; Carney, P.S.; Goddard, L.L.; Popescu, G. White-light diffraction tomography of unlabelled live cells. Nat. Photonics 2014, 8, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, K.; Mubarok, A.; Panduwirawan, A.; Lee, K.; Lee, S.; Park, H.; Park, Y. High-resolution 3-D refractive index tomography and 2-D synthetic aperture imaging of live phytoplankton. J. Opt. Soc. Korea 2014, 18, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Hong, S.J.; Kim, H.B.; Kim, G.; Lee, M.; Shin, S.; Lee, S.; Kim, D.J.; Lee, C.G.; Park, Y. Label-free non-invasive quantitative measurement of lipid contents in individual microalgal cells using refractive index tomography. Sci. Rep. 2018, 8, 6524. [Google Scholar] [CrossRef] [PubMed]
- Kuś, A.; Dudek, M.; Kemper, B.; Kujawińska, M.; Vollmer, A. Tomographic phase microscopy of living three-dimensional cell cultures. J. Biomed. Opt. 2014, 19, 046009. [Google Scholar] [CrossRef]
- Su, J.W.; Hsu, W.C.; Chou, C.Y.; Chang, C.H.; Sung, K.B. Digital holographic microtomography for high-resolution refractive index mapping of live cells. J. Biophotonics 2013, 6, 416–424. [Google Scholar] [CrossRef]
- Takeda, M.; Ina, H.; Kobayashi, S. Fourier-transform method of fringe-pattern analysis for computer-based topography and interferometry. J. Opt. Soc. Am. 1982, 72, 156–160. [Google Scholar] [CrossRef]
- Kim, K.; Yoon, H.; Diez-Silva, M.; Dao, M.; Dasari, R.R.; Park, Y. High-resolution three-dimensional imaging of red blood cells parasitized by Plasmodium falciparum and in situ hemozoin crystals using optical diffraction tomography. J. Biomed. Opt. 2014, 19, 011005. [Google Scholar] [CrossRef] [Green Version]
- Wolf, E. Three-dimensional structure determination of semi-transparent objects from holographic data. Opt. Commun. 1969, 1, 153–156. [Google Scholar] [CrossRef]
- Kim, T.; Zhou, R.; Goddard, L.L.; Popescu, G. Solving inverse scattering problems in biological samples by quantitative phase imaging. Laser Photonics Rev. 2016, 10, 13–39. [Google Scholar] [CrossRef]
- Ghiglia, D.C.; Pritt, M.D. Two-Dimensional Phase Unwrapping—Theory, Algorithms, and Software; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Le Clerc, F.; Collot, L.; Gross, M. Numerical heterodyne holography with two-dimensional photodetector arrays. Opt. Lett. 2000, 25, 716–718. [Google Scholar] [CrossRef] [Green Version]
- Le Clerc, F.; Gross, M.; Collot, L. Synthetic-aperture experiment in the visible with on-axis digital heterodyne holography. Opt. Lett. 2001, 26, 1550–1552. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Lee, K.; Jin, K.H.; Shin, S.; Lee, S.; Park, Y.; Ye, J.C. Comparative study of iterative reconstruction algorithms for missing cone problems in optical diffraction tomography. Opt. Express 2015, 23, 16933–16948. [Google Scholar] [CrossRef] [Green Version]
- Sung, Y.; Choi, W.; Fang-Yen, C.; Badizadegan, K.; Dasari, R.R.; Feld, M.S. Optical diffraction tomography for high resolution live cell imaging. Opt. Express 2009, 17, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, P.A. Nitrogen in the Marine Environment; Carpenter, E.J., Capone, D.G., Eds.; Academic Press: Cambridge, MA, USA, 1983; Volume 309. [Google Scholar]
- Li, Y.; Horsman, M.; Wang, B.; Wu, N.; Lan, C.Q. Effects of nitrogen sources on cell growth and lipid accumulation of green alga Neochloris oleoabundans. Appl. Microbiol. Biotechnol. 2008, 81, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Firestone, D. Physical and Chemical Characteristics of Oils, Fats, and Waxes, 2nd ed.; AOCS Press: Champaign, IL, USA, 1999. [Google Scholar]
- Popescu, G.; Park, Y.; Lue, N.; Best-Popescu, C.; Deflores, L.; Dasari, R.R.; Feld, M.S.; Badizadegan, K. Optical imaging of cell mass and growth dynamics. Am. J. Physiol. Cell Physiol. 2008, 295, C538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, Y.; Hubbard, J.; Holzer, G.; Tornabene, T. Total lipid production of the green alga Nannochloropsis sp. QII under different nitrogen regimes. J. Phycol. 1987, 23, 289–296. [Google Scholar] [CrossRef]
- Rodolfi, L.; Chini Zittelli, G.; Bassi, N.; Padovani, G.; Biondi, N.; Bonini, G.; Tredici, M.R. Microalgae for oil: Strain selection, induction of lipid synthesis and outdoor mass cultivation in a low-cost photobioreactor. Biotechnol. Bioeng. 2009, 102, 100–112. [Google Scholar] [CrossRef]
- Rosati, G.; Verni, F.; Barsanti, L.; Passarelli, V.; Gualtieri, P. Ultrastructure of the apical zone of Euglena gracilis. Electron Microsc. Rev. 1991, 4, 319–342. [Google Scholar] [CrossRef]
- Walne, P.L.; Passarelli, V.; Lenzi, P.; Barsanti, L.; Gualtieri, P. Isolation of the Flagellar Swelling and Identification of Retinal in the Phototactic Flagellate, Ochromonas danica (Chrysophyceae). J. Eukaryot. Microbiol. 1995, 42, 7–11. [Google Scholar] [CrossRef]
- Barsanti, L.; Passarelli, V.; Walne, P.L.; Gualtieri, P. The photoreceptor protein of Euglena gracilis. FEBS Lett. 2000, 482, 247–251. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lateral Resolution (nm) | Axial Resolution (nm) | Methods of Illumination | Methods of Image Generation | Fluorescent Probe | Five Aspects of Microscopy | Merits | Limitations | |
|---|---|---|---|---|---|---|---|---|
| WFM | 250 | 700 | Lamp | Direct visualization | Conventional |  | Perfect for living cell Very easy to use | Low axial and lateral resolution |
| CLSM | 180–250 | 500 | Pinhole | Scanning excitation beam | Conventional |  | High axial resolution 3D imaging capability Reduced photobleaching | Limited field of view Slow processing |
| 2D-STED | 40–60 | 600 | Hardware shaped excitation beam | Scanning excitation beam | Photo switchable |  | Resolution directly improved with PSF to ~50 nm 2 colour imaging (more if combined with other advanced techniques) No post-processing Lateral resolution 20–70 nm 20 micron depth penetration | Photobleaching, Phototoxic Requires very stable dyes Difficult for live cells Requires complex alignment Low dynamic range |
| SIM | 100–125 | 2D 500–700 3D 250–350 | Patterned wide field | Multiple images combining Fourier space | Conventional |  | Live cell imaging Multicolour imaging No special sample preparation Simple to use Fast | Require sensitive cameras Photobleaching/toxicity Subject to artifacts Limited sample thickness |
| PALM/STORM | 10–50 | 500–700 | Stochastic fluorophore excitation | Multiple single molecules frames | Photoactivable |  | Single molecule sensitivity Highest potential resolution Multichannel imaging Low illumination power | Specific fluorophores required Shallow depth-limited Long acquisition times Impractical for live cell |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barsanti, L.; Birindelli, L.; Sbrana, F.; Lombardi, G.; Gualtieri, P. Advanced Microscopy Techniques for Molecular Biophysics. Int. J. Mol. Sci. 2023, 24, 9973. https://doi.org/10.3390/ijms24129973
Barsanti L, Birindelli L, Sbrana F, Lombardi G, Gualtieri P. Advanced Microscopy Techniques for Molecular Biophysics. International Journal of Molecular Sciences. 2023; 24(12):9973. https://doi.org/10.3390/ijms24129973
Chicago/Turabian StyleBarsanti, Laura, Lorenzo Birindelli, Francesca Sbrana, Giovanni Lombardi, and Paolo Gualtieri. 2023. "Advanced Microscopy Techniques for Molecular Biophysics" International Journal of Molecular Sciences 24, no. 12: 9973. https://doi.org/10.3390/ijms24129973