
Survey of Protein Sequence Embedding Models


Abstract
:1. Introduction
2. Results
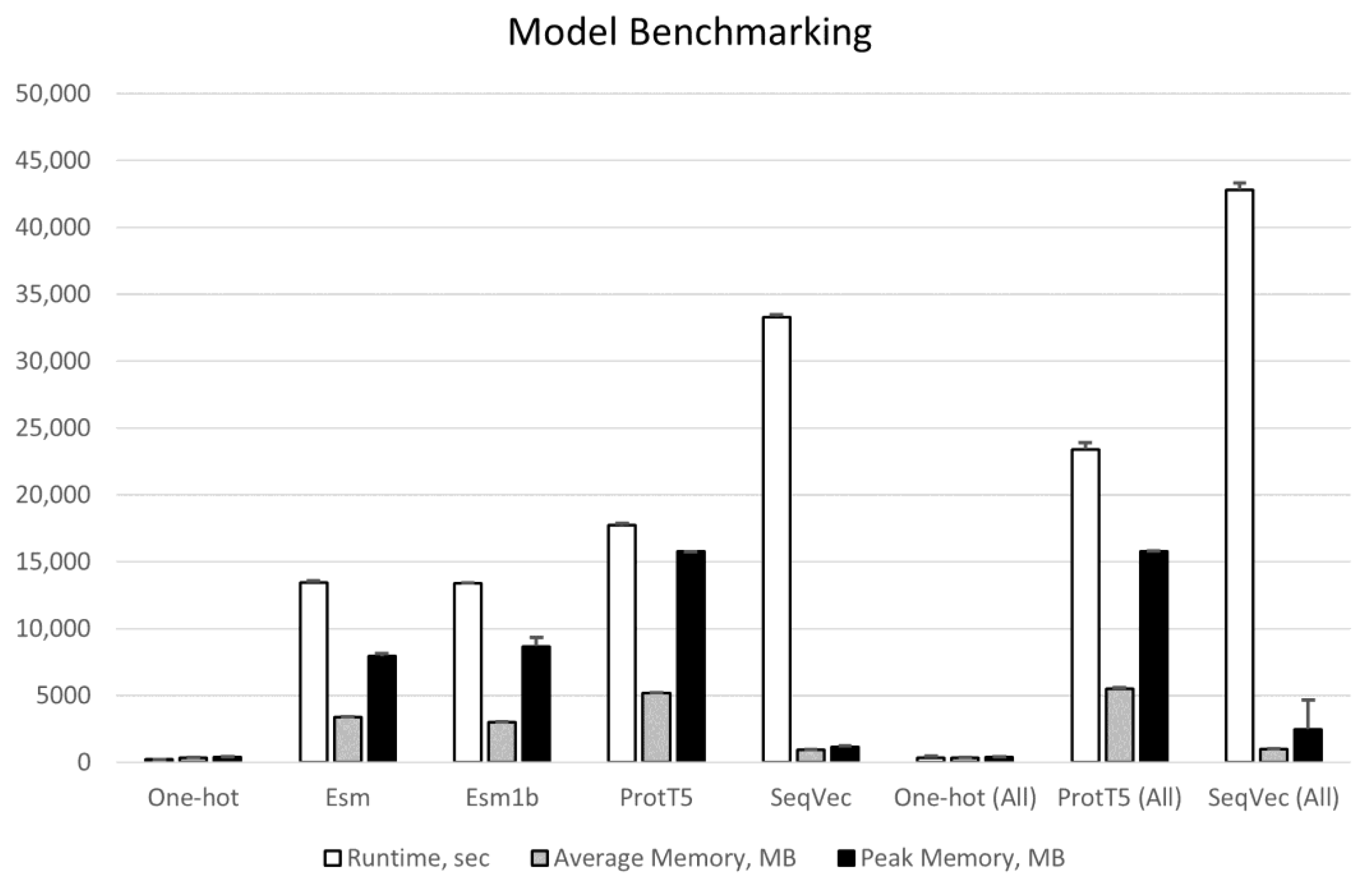
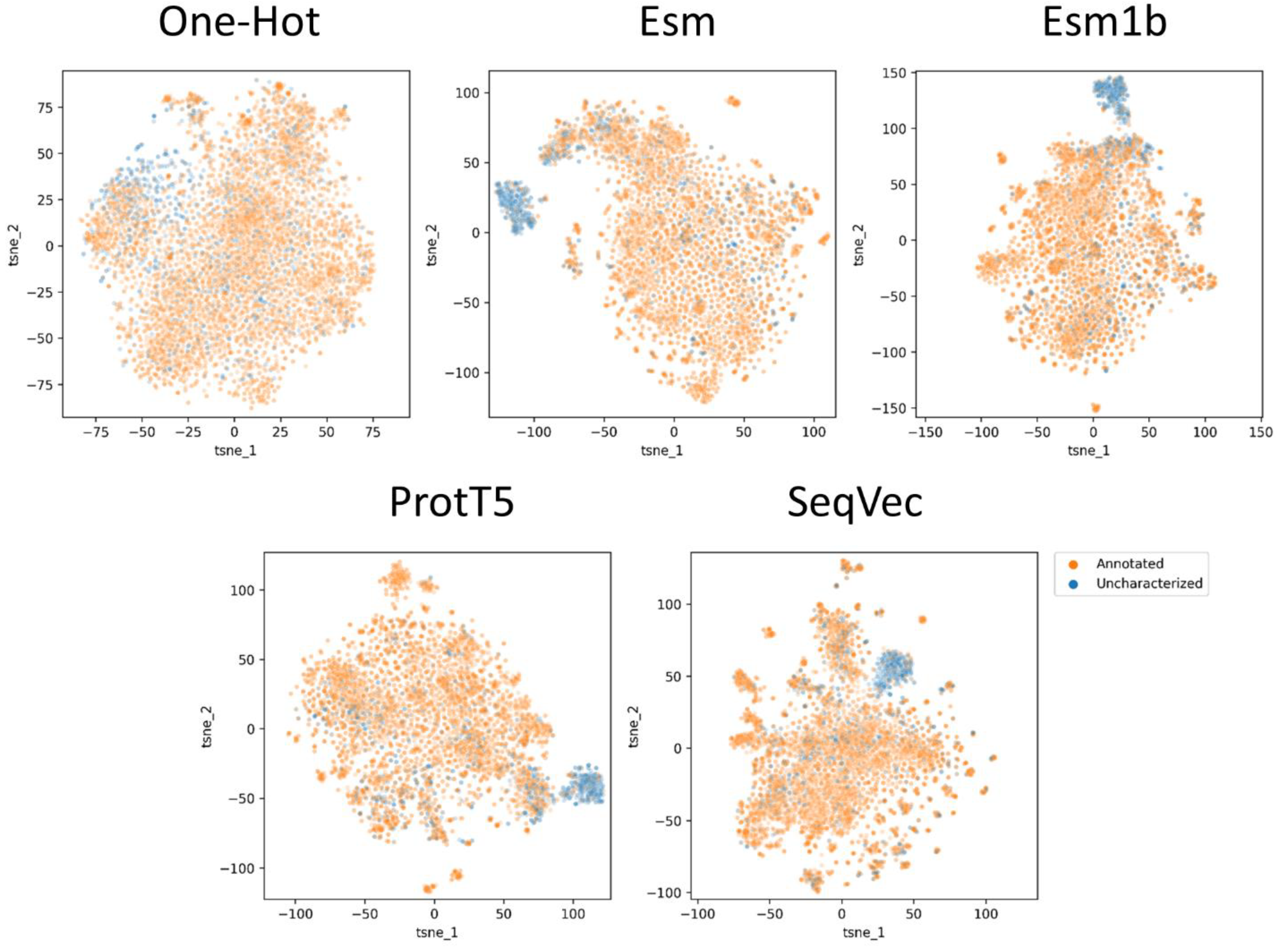
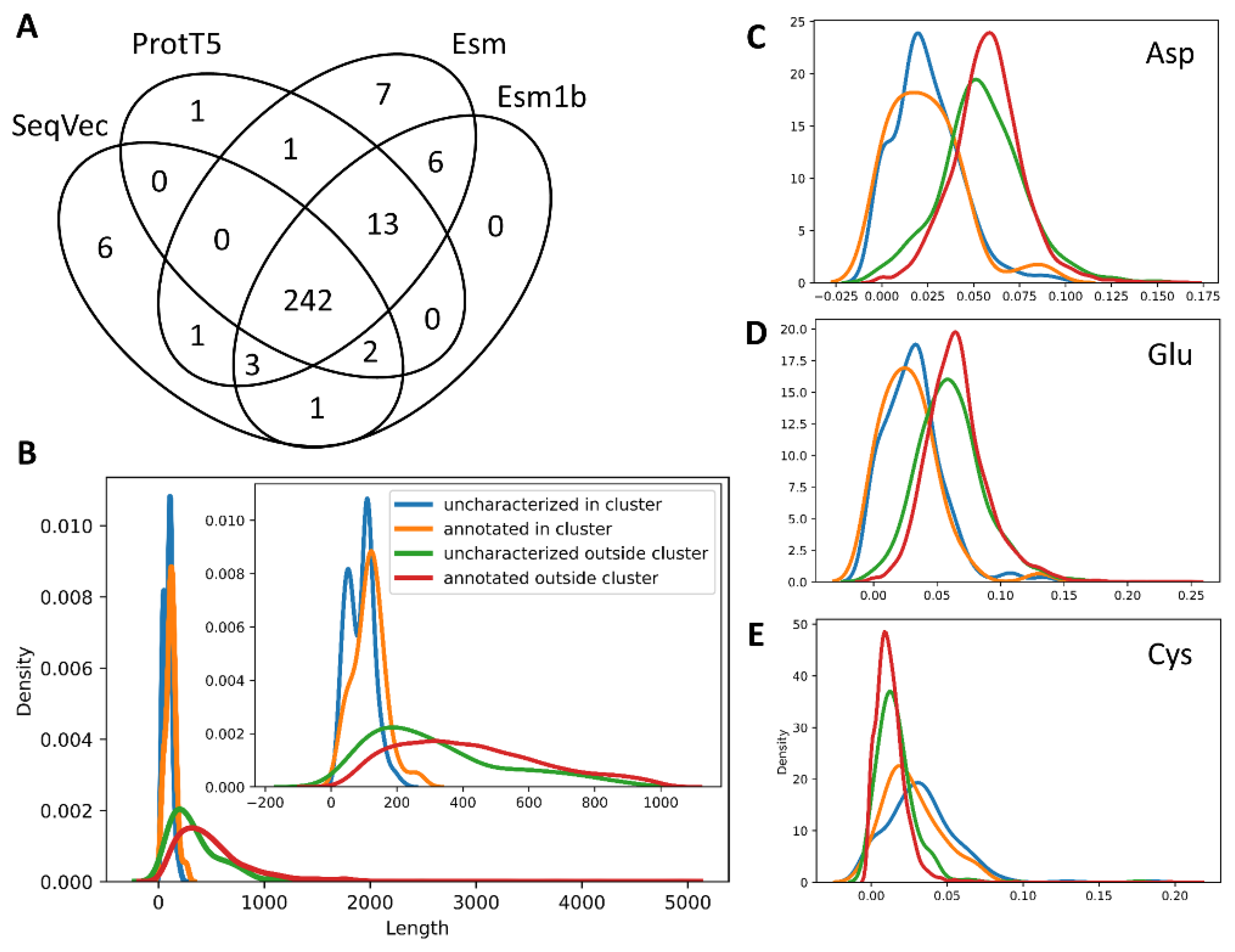
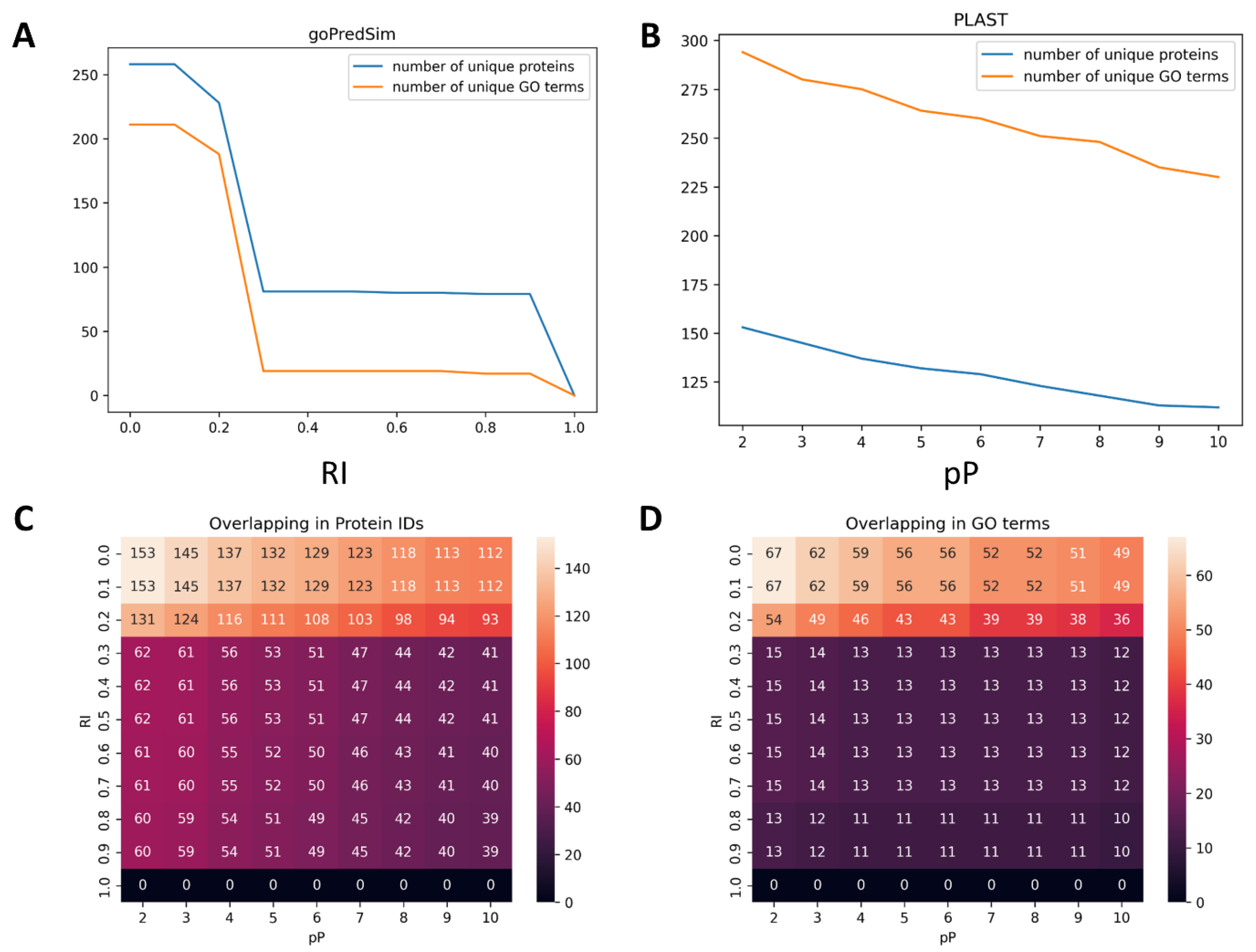
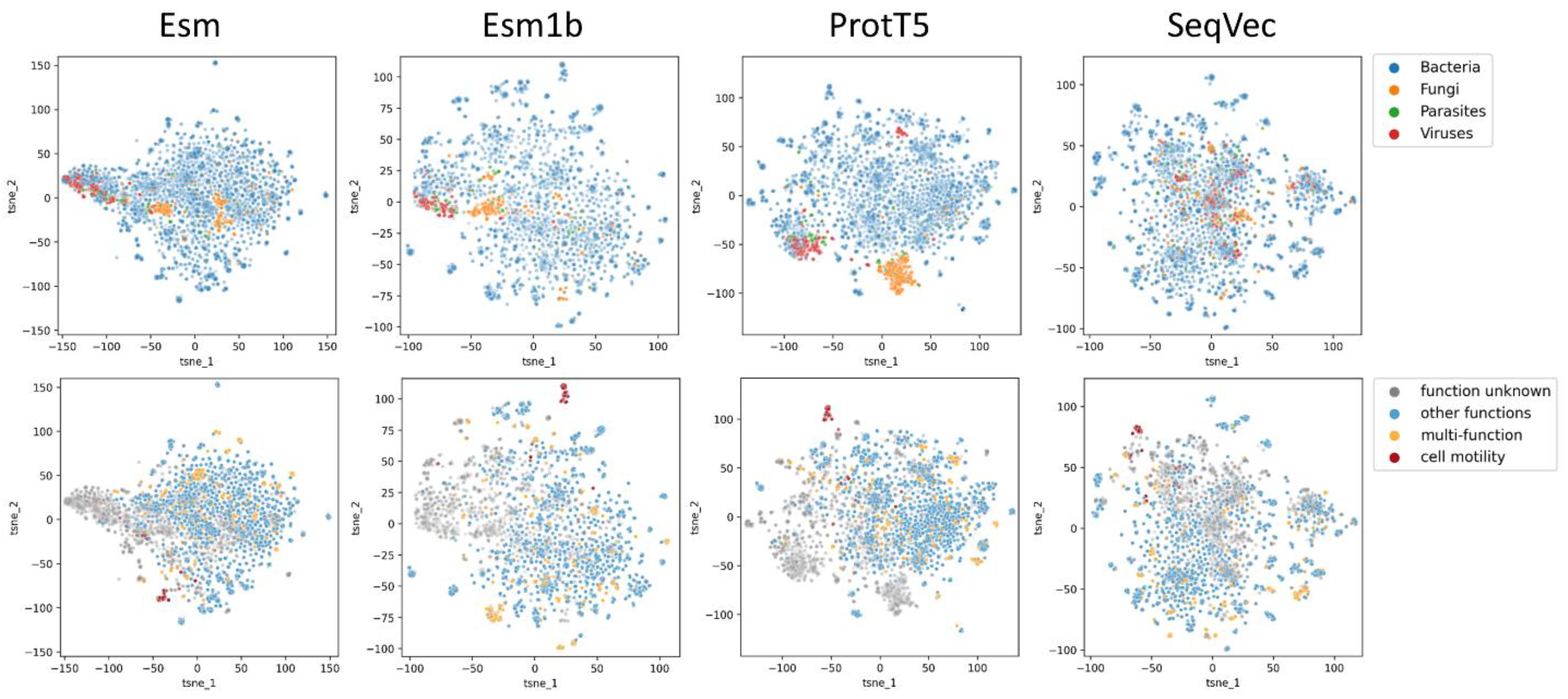
2.1. Proteome Wide Benchmarking
2.2. Human Protein Variants and Diseases
2.3. TEM-1 Variants and Antimicrobial Resistance
2.4. Mating Factors
2.5. Virulence Factors
3. Discussion
4. Materials and Methods
Protein Sequence Embedding Models
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: Nlp, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar] [PubMed]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural. Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 6000–6010. [Google Scholar]
- Heinzinger, M.; Elnaggar, A.; Wang, Y.; Dallago, C.; Nechaev, D.; Matthes, F.; Rost, B. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinform. 2019, 20, 723. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Morcillo, A.; Makrodimitris, S.; van Ham, R.; Gomez, A.M.; Sanchez, V.; Reinders, M.J.T. Unsupervised protein embeddings outperform hand-crafted sequence and structure features at predicting molecular function. Bioinformatics 2021, 37, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Sonderby, C.K.; Sonderby, S.K.; Nielsen, H.; Winther, O. Deeploc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, H.; Zeng, T.; Li, Z.; Chen, L.; Huang, T.; Cai, Y.D. Identification of protein subcellular localization with network and functional embeddings. Front. Genet. 2020, 11, 626500. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Casadio, R. Deepsig: Deep learning improves signal peptide detection in proteins. Bioinformatics 2018, 34, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [PubMed]
- Littmann, M.; Heinzinger, M.; Dallago, C.; Olenyi, T.; Rost, B. Embeddings from deep learning transfer go annotations beyond homology. Sci. Rep. 2021, 11, 1160. [Google Scholar] [CrossRef]
- Saar, K.L.; Morgunov, A.S.; Qi, R.; Arter, W.E.; Krainer, G.; Lee, A.A.; Knowles, T.P.J. Learning the molecular grammar of protein condensates from sequence determinants and embeddings. Proc. Natl. Acad. Sci. USA 2021, 118, e2019053118. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. ; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Littmann, M.; Heinzinger, M.; Dallago, C.; Weissenow, K.; Rost, B. Protein embeddings and deep learning predict binding residues for various ligand classes. Sci. Rep. 2021, 11, 23916. [Google Scholar] [CrossRef] [PubMed]
- Marquet, C.; Heinzinger, M.; Olenyi, T.; Dallago, C.; Erckert, K.; Bernhofer, M.; Nechaev, D.; Rost, B. Embeddings from protein language models predict conservation and variant effects. Hum. Genet. 2022, 141, 1629–1647. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. Proteinbert: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Kilinc, M.; Jia, K.; Jernigan, R.L. Protein language model performs efficient homology detection. bioRxiv 2022. [Google Scholar] [CrossRef]
- Jacquier, H.; Birgy, A.; Le Nagard, H.; Mechulam, Y.; Schmitt, E.; Glodt, J.; Bercot, B.; Petit, E.; Poulain, J.; Barnaud, G.; et al. Capturing the mutational landscape of the beta-lactamase tem-1. Proc. Natl. Acad. Sci. USA 2013, 110, 13067–13072. [Google Scholar] [CrossRef]
- Martin, S.H.; Wingfield, B.D.; Wingfield, M.J.; Steenkamp, E.T. Causes and consequences of variability in peptide mating pheromones of ascomycete fungi. Mol. Biol. Evol. 2011, 28, 1987–2003. [Google Scholar] [CrossRef]
- Sayers, S.; Li, L.; Ong, E.; Deng, S.; Fu, G.; Lin, Y.; Yang, B.; Zhang, S.; Fa, Z.; Zhao, B.; et al. Victors: A web-based knowledge base of virulence factors in human and animal pathogens. Nucleic. Acids. Res. 2019, 47, D693–D700. [Google Scholar] [CrossRef] [PubMed]
- Dallago, C.; Schutze, K.; Heinzinger, M.; Olenyi, T.; Littmann, M.; Lu, A.X.; Yang, K.K.; Min, S.; Yoon, S.; Morton, J.T.; et al. Learned embeddings from deep learning to visualize and predict protein sets. Curr. Protoc. 2021, 1, e113. [Google Scholar] [CrossRef]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the limits of transfer learning with a unified text-to-text transformer. J. Mach. Learn. Res. 2020, 21, 1–67. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Cos Range | Cos Mean ± SD | Shapiro-Wilk Test p-Value | Mann-Whitney U Test p-Value |
|---|---|---|---|---|
| ProtT5 | 0.975–1.000 | 1.000 ± 0.001 | 1.138 × 10−77 | 0.0038 |
| SeqVec | 0.966–1.000 | 0.999 ± 0.004 | 2.217 × 10−81 | 1.175 × 10−8 |
| Esm | Esm1b | ProtT5 | SeqVec | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AAM 1 | Total 2 | cos 3 | r4 | cos | r | cos | r | cos | r |
| 1 | 855 | 1.0 ± 0.0 | 0.125 | 1.0 ± 0.0 | NA | 0.999 ± 0.002 | 0.229 | 0.998 ± 0.007 | 0.197 |
| 2 | 1740 | 1.0 ± 0.0 | 0.116 | 1.0 ± 0.0 | NA | 0.999 ± 0.002 | 0.281 | 0.998 ± 0.008 | 0.222 |
| 3 | 1230 | 1.0 ± 0.0 | 0.170 | 1.0 ± 0.0 | 0.044 | 0.998 ± 0.002 | 0.293 | 0.997 ± 0.010 | 0.208 |
| 4 | 626 | 1.0 ± 0.0 | 0.158 | 1.0 ± 0.0 | 0.028 | 0.997 ± 0.003 | 0.281 | 0.996 ± 0.011 | 0.172 |
| 5 | 316 | 1.0 ± 0.0 | 0.190 | 1.0 ± 0.0 | NA | 0.997 ± 0.003 | 0.247 | 0.993 ± 0.018 | 0.158 |
| 6 | 105 | 0.999 ± 0.001 | 0.150 | 1.0 ± 0.0 | 0.057 | 0.996 ± 0.003 | 0.240 | 0.994 ± 0.011 | 0.132 |
| 7 | 42 | 0.999 ± 0.001 | 0.083 | 1.0 ± 0.0 | NA | 0.995 ± 0.005 | 0.065 | 0.994 ± 0.011 | 0.114 |
| 8 | 10 | 0.999 ± 0.001 | NA | 1.0 ± 0.0 | NA | 0.991 ± 0.008 | NA | 0.996 ± 0.003 | NA |
| 9 | 4 | 0.999 ± 0.001 | 0.816 | 1.0 ± 0.0 | 0.333 | 0.991 ± 0.007 | 0.619 | 0.995 ± 0.002 | 0.733 |
| 10 | 1 | 0.999 | NA | 1.0 | NA | 0.986 | NA | 0.890 | NA |
| 11 | 1 | 0.999 | NA | 1.0 | NA | 0.994 | NA | 0.893 | NA |
| Any | 4930 | 1.0 ± 0.0 | 0.210 | 1.0 ± 0.0 | 0.040 | 0.998 ± 0.003 | 0.348 | 0.997 ± 0.010 | 0.223 |
| MF-Alpha Precursors (PF05436) | MF a1 and a2 (PF17317) | |||||
|---|---|---|---|---|---|---|
| Sequence Identity 1 | Conservative Substitutions | Sequence Identity | Conservative Substitutions | |||
| 38.26 ± 9.54% | 57.77 ± 9.28% | 58.97 ± 11.38% | 74.06 ± 9.03% | |||
| Model | cos 2 | r 3 | r | cos | r | r |
| Esm | 0.991 ± 0.006 | 0.062 | 0.049 | 0.992 ± 0.006 | 0.449 | 0.391 |
| Esm1b | 0.993 ± 0.004 | 0.060 | 0.014 | 0.990 ± 0.007 | 0.277 | 0.292 |
| ProtT5 | 0.816 ± 0.098 | 0.108 | 0.071 | 0.882 ± 0.096 | 0.506 | 0.407 |
| SeqVec | 0.823 ± 0.077 | 0.271 | 0.205 | 0.857 ± 0.064 | 0.623 | 0.612 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, C.; Khadkikar, S.; Porollo, A. Survey of Protein Sequence Embedding Models. Int. J. Mol. Sci. 2023, 24, 3775. https://doi.org/10.3390/ijms24043775
Tran C, Khadkikar S, Porollo A. Survey of Protein Sequence Embedding Models. International Journal of Molecular Sciences. 2023; 24(4):3775. https://doi.org/10.3390/ijms24043775
Chicago/Turabian StyleTran, Chau, Siddharth Khadkikar, and Aleksey Porollo. 2023. "Survey of Protein Sequence Embedding Models" International Journal of Molecular Sciences 24, no. 4: 3775. https://doi.org/10.3390/ijms24043775