
Computer-Assisted Design of Peptide-Based Radiotracers
 ,
,  ,
,  and
and 
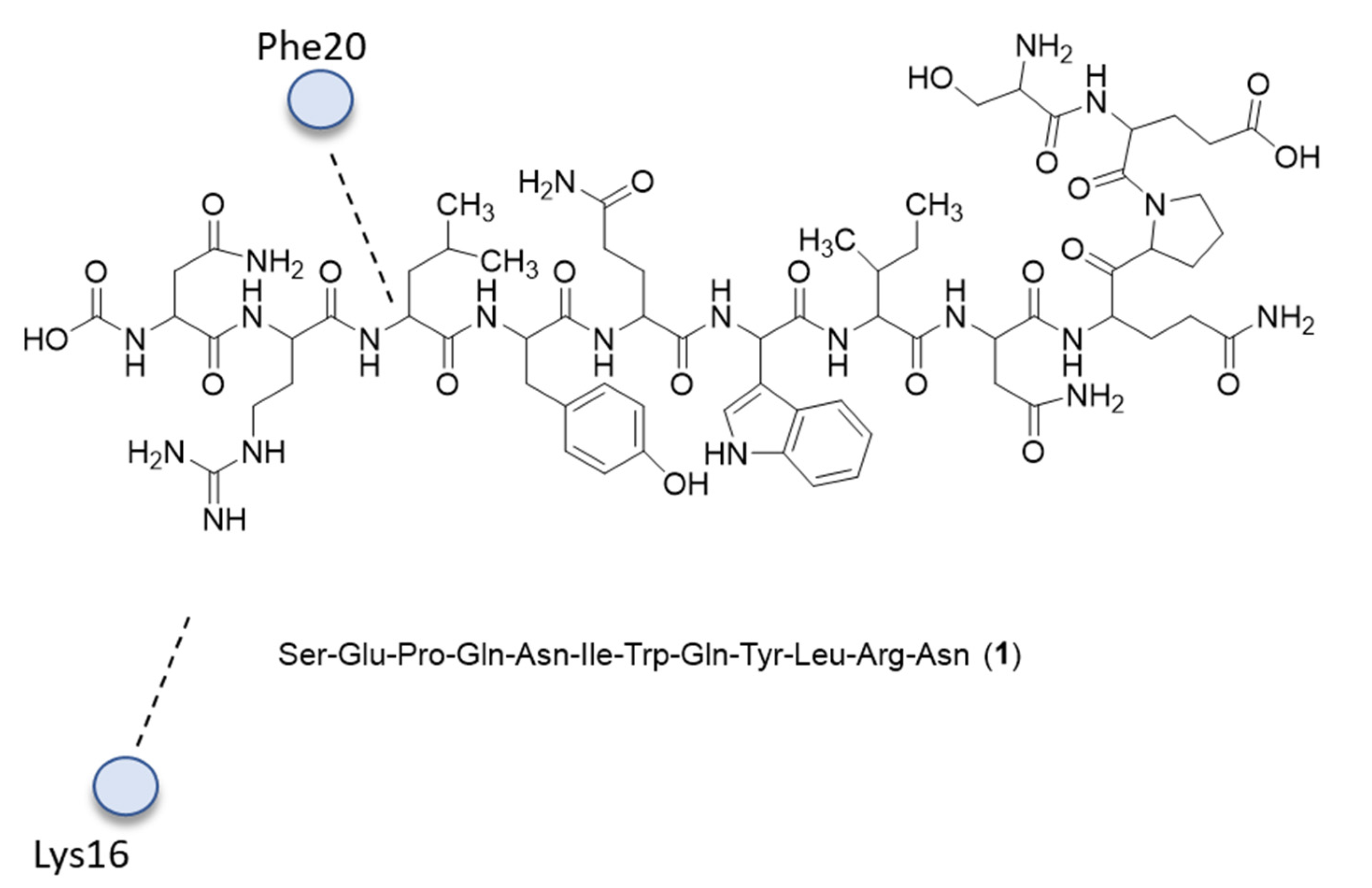{kind=link}
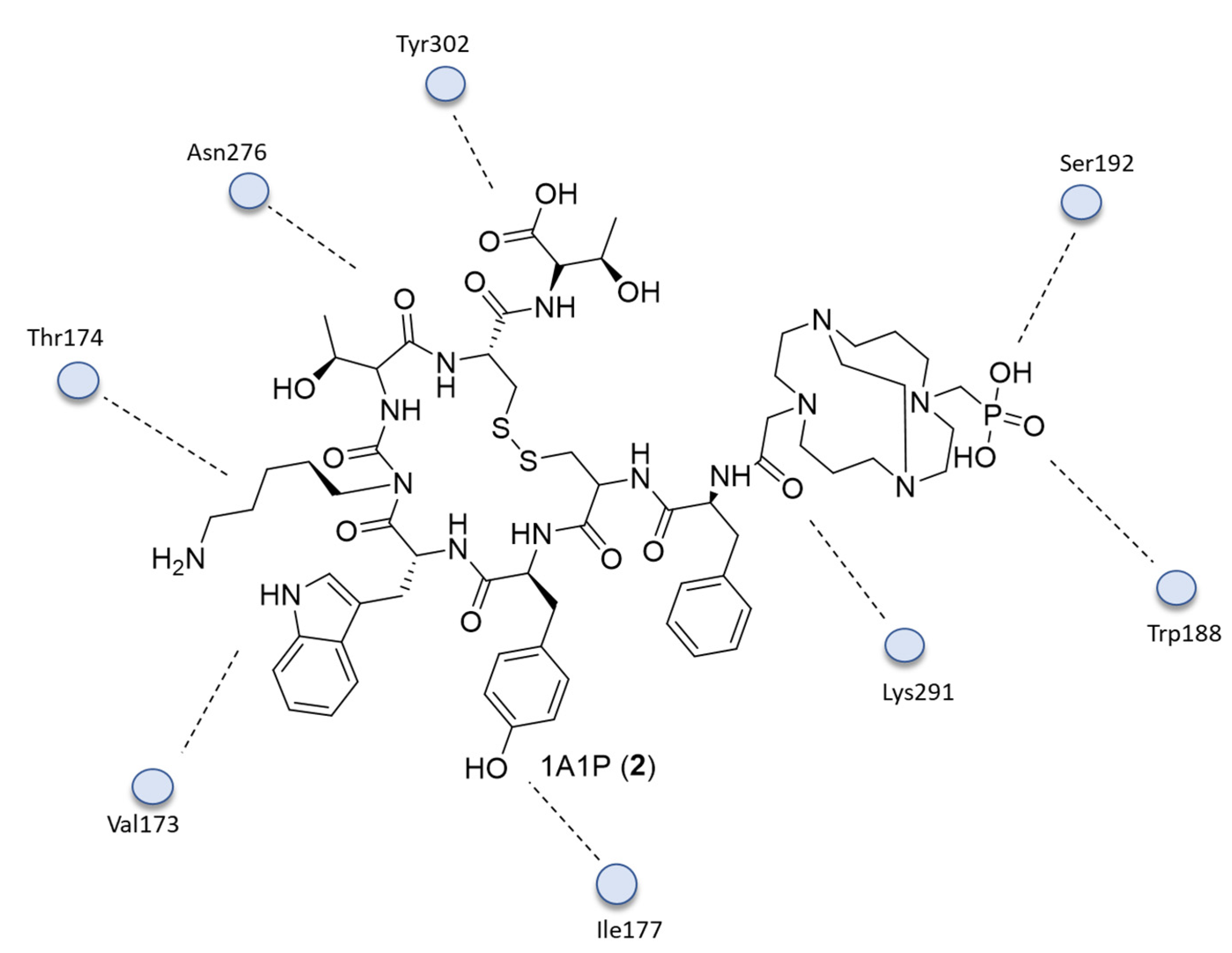{kind=link}
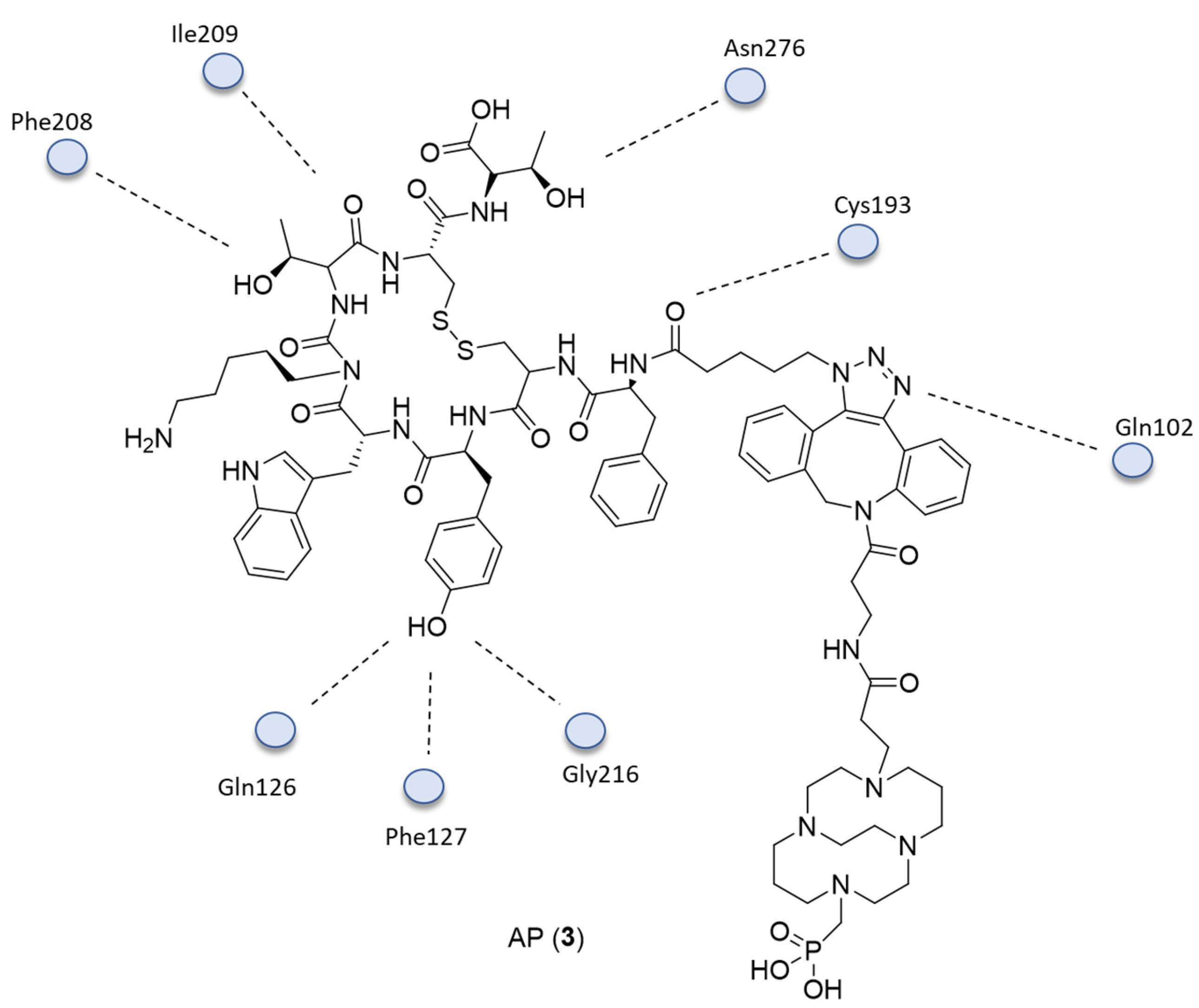{kind=link}
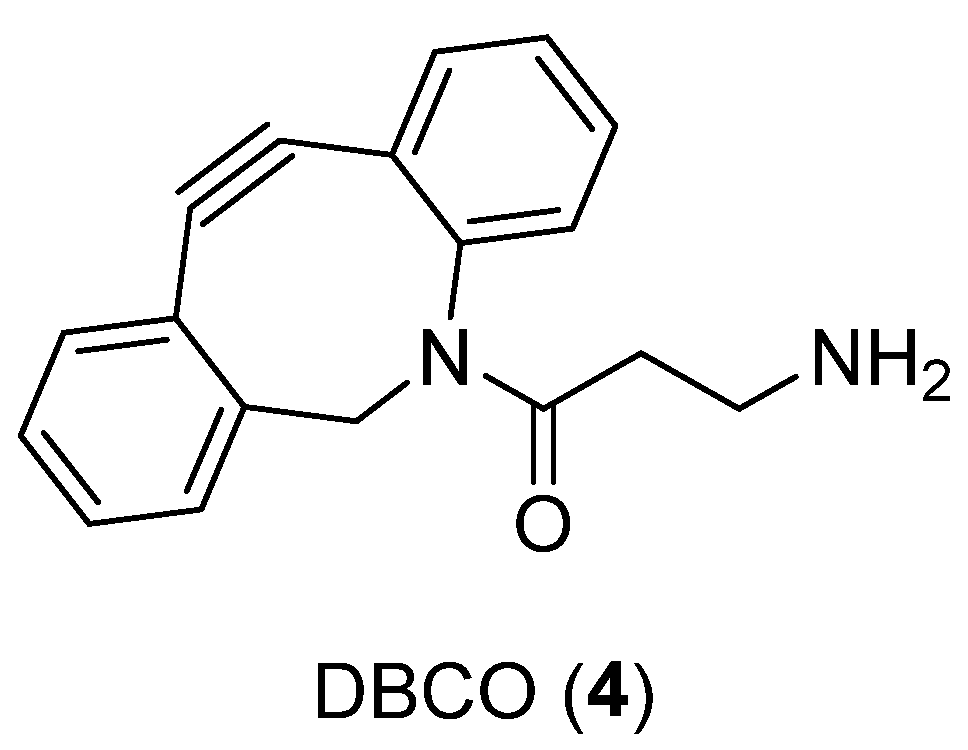{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
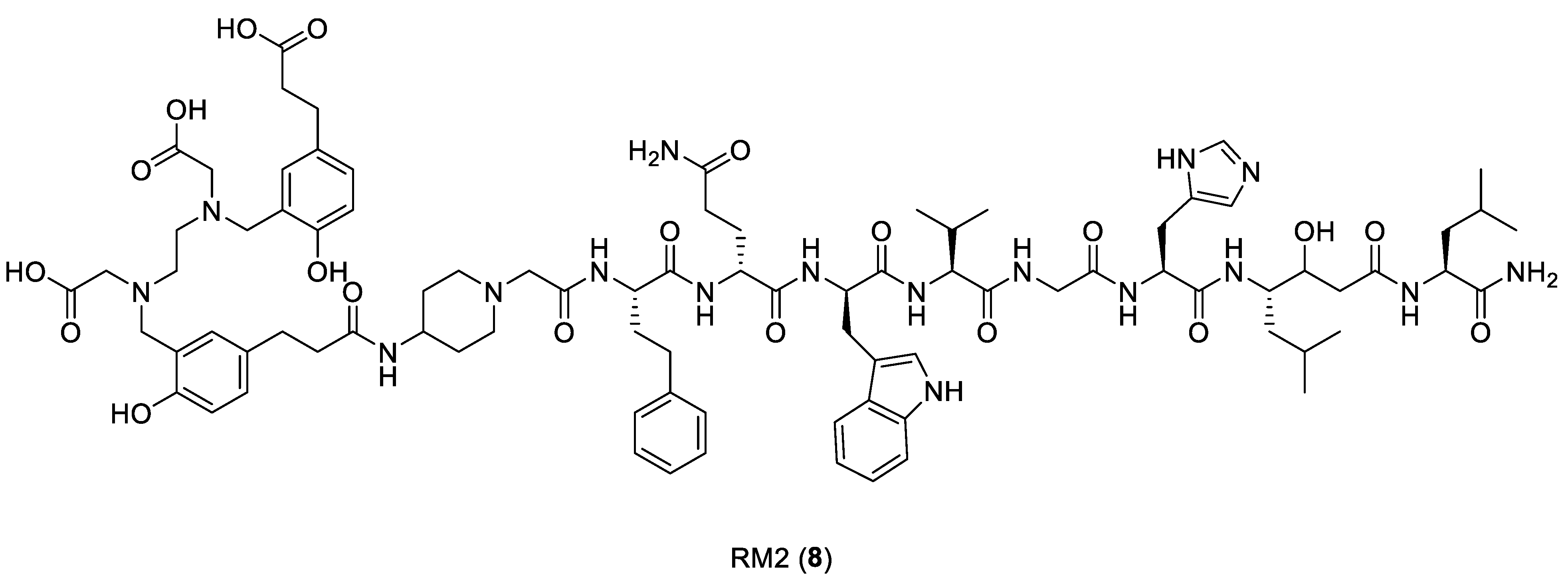{kind=link}
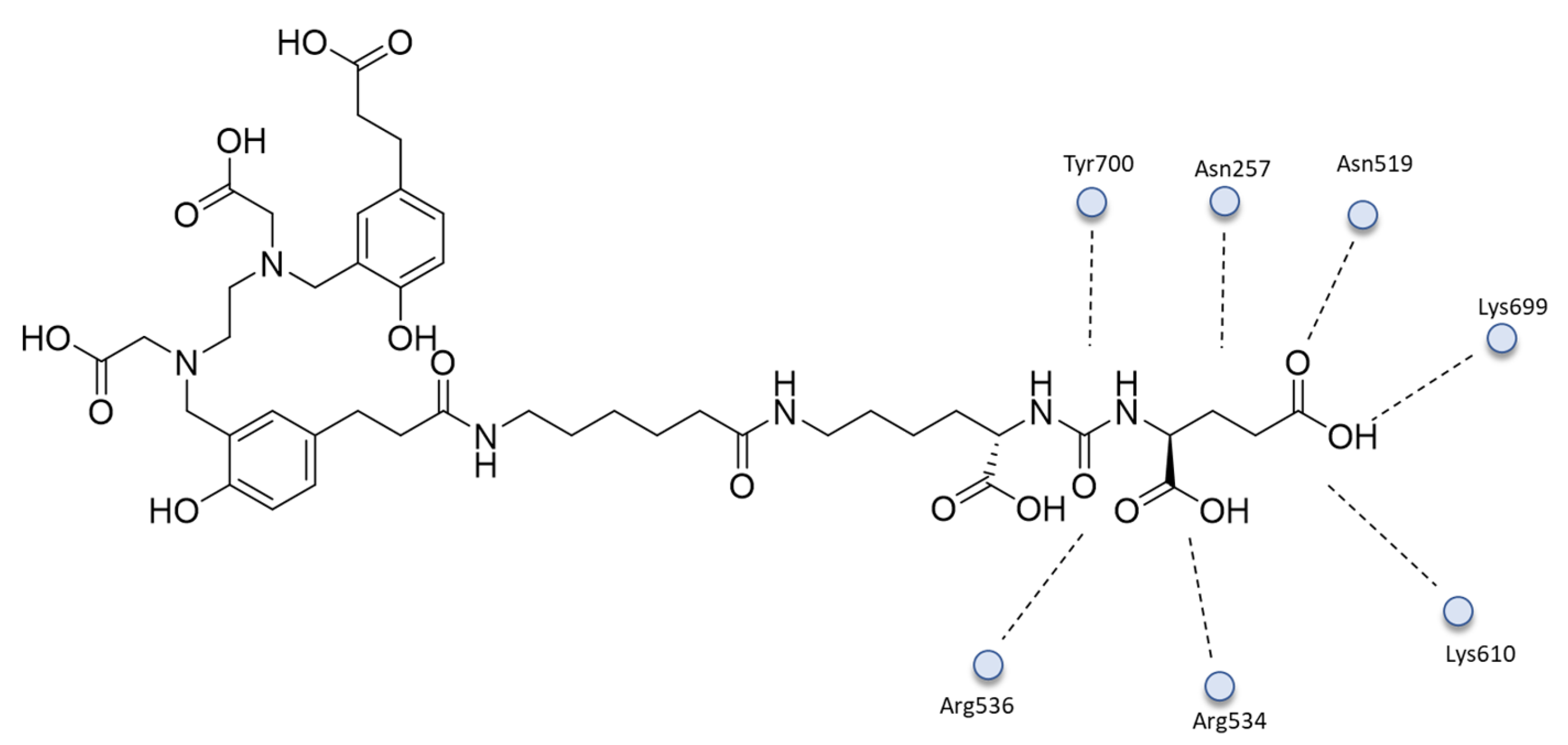{kind=link}
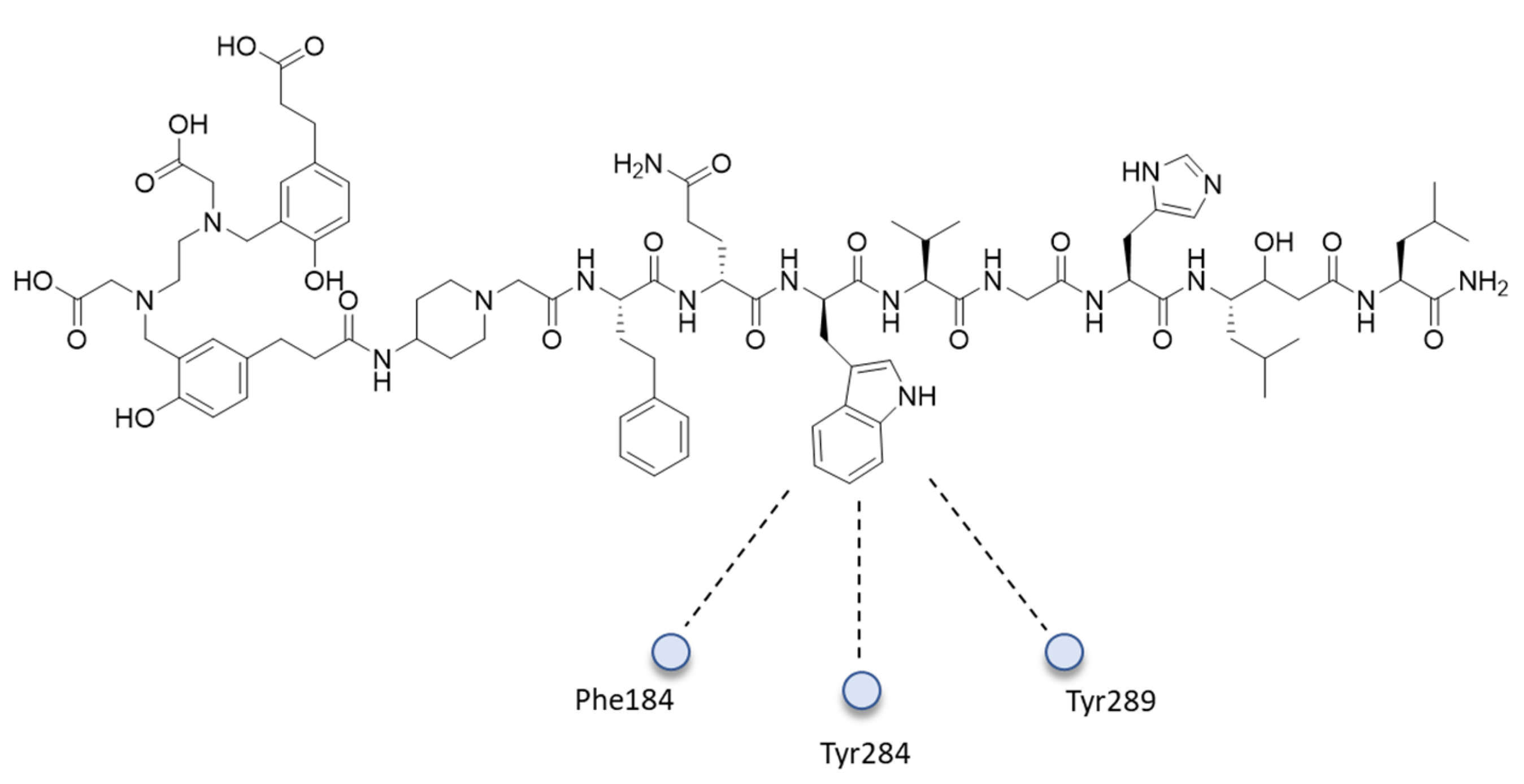{kind=link}
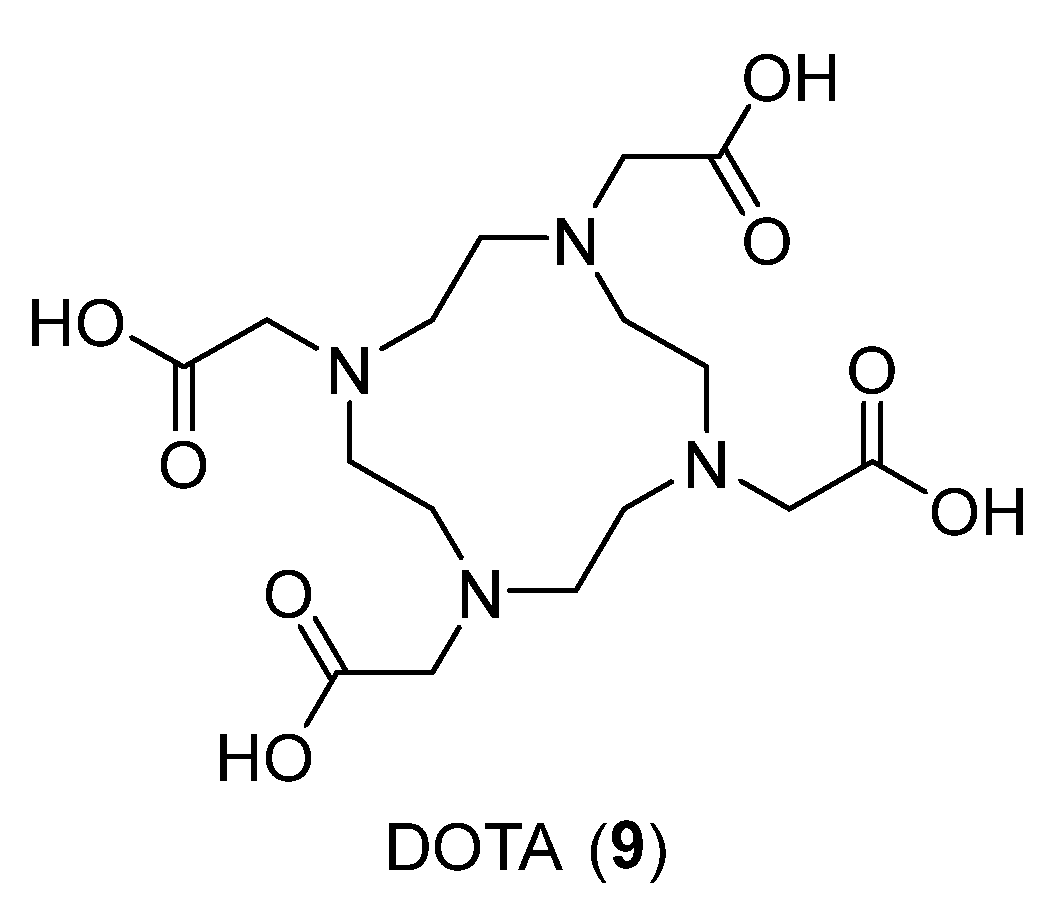{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discussions
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABPP | Aβ binding peptide probe |
| AD | Alzheimer’s disease |
| Ahx | 6-aminohexanoic acid |
| Aβ42 | amyloid-beta 42 |
| BB2R | bombesin receptor 2 |
| BB3 | bombesin receptor 3 |
| BCa | breast cancer |
| BMG-1 | cerebral glioma cell line |
| CADD | computer-aided drug design |
| CCK | cholecystokinin receptors |
| CCK-2R | cholecystokinin receptor subtype 2 |
| CCP | positively charged octamer |
| cDNA | complementary DNA |
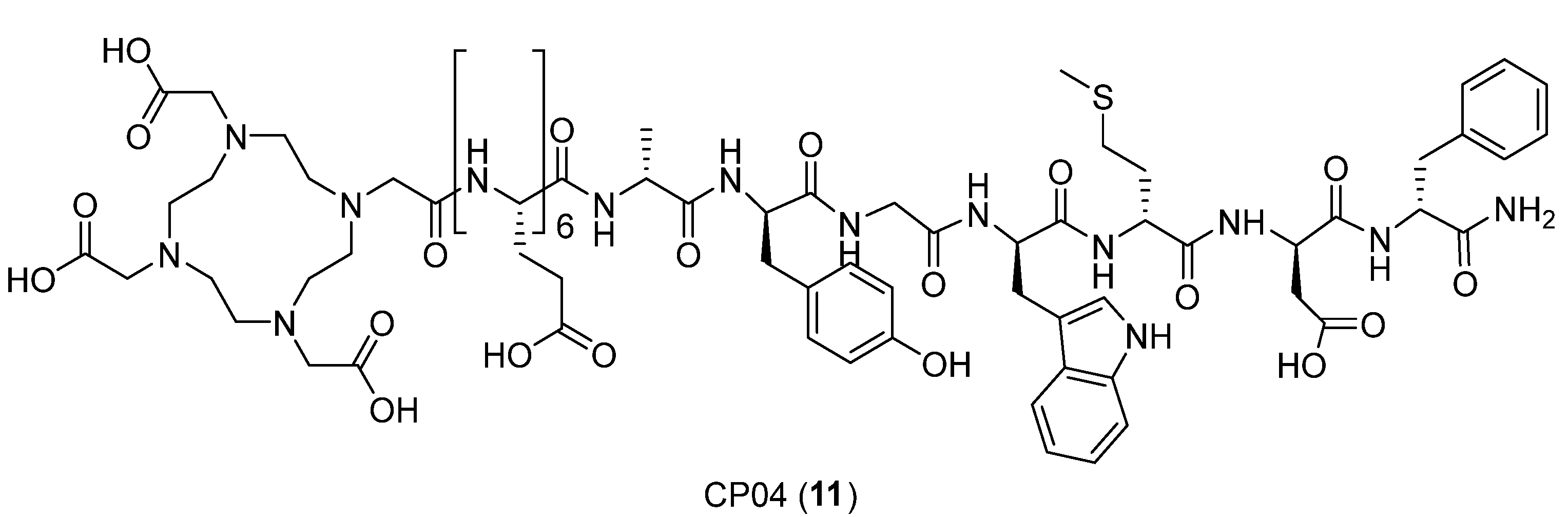
| CP04 | minigastrin analog DOTA-(dGlu)-6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 |
| CSF | cerebral-cerebrospinal fluid |
| C-X-C | chemokine receptors |
| CXCR4 | chemokine receptor 4 |
| DBCO | dibenzocyclooctyne |
| DCIBzL | ((1-carboxy-5-(4-iodobenzamido)pentyl)carbamoyl)glutamic acid |
| DOTA | 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid |
| DOTATOC | 1,4,7,10-tetraazacyclododecan-4,7,10-tricarboxy-meth-1-yl-acetyl-d-Phe-Tyr3-octreotide |
| DTPA | pentetic acid, diethylenetriaminepentaacetic acid |
| EAT | Xenografted Ehrlich Ascites |
| ELISA | enzyme-linked immunosorbent assay |
| ESCC | esophageal squamous cell carcinoma |
| Fmoc | fluorenylmethoxycarbonyl protecting group |
| GCP II | glutamate carboxypeptidase II |
| GGT | gamma glutamyl transferase |
| GRPR | gastrin releasing peptide receptor |
| GSH | glutathione |
| HBED-CC | N,N′-bis-[2-hydroxy-5-(carboxyethyl)benzyl]ethylenediamine-N,N-diacetic acid |
| HIV-1 | human immunodeficiency virus |
| MD | molecular dynamics |
| MMPs | matrix metalloproteases |
| MOLCAD | Computer-Aided Visualization and Manipulation of Models |
| MTC | medullary thyroid carcinoma cells |
| NOTA | 2,2′,2″-(1,4,7-triazacyclononane-1,4,7-triyl)triacetic acid |
| NOTA-CP01 | cyclo[Phe-Tyr-Lys-(iPr)-d-Arg-2-Nal-Gly-d-Glu]-Lys-(iPr)-butane-1,4-diamine-bis-t-butyl NOTA-NH2 |
| PCa | prostate cancer |
| PEG | polyethylene glycol |
| PET | positron emission tomography |
| PSMA | prostate-specific membrane antigen |
| RM2 | gastrin-releasing peptide receptor antagonist |
| S1 | non-prime section |
| S1′ | prime section |
| SAAC | single amino acid chelated |
| SAR | structure–activity relationship |
| SDF-1/CXCL12 | stromal cell-derived factor 1 |
| Siglec-9 | sialic acid-binding Ig-like lectin 9 |
| SPECT | single-photon emission computed tomography |
| SSTR2 | somatostatin receptor subtype 2 |
| TE1K1P−PEG4−DBCO−Y3-TATE | |
| TRNT | targeted radionuclide therapy |
| VAP-1 | vascular adhesion protein-1 |
| Y3-TATE | tyrosine 3-octreotate |
References
- Blower, P.J. A nuclear chocolate box: The periodic table of nuclear medicine. Dalton Trans. 2015, 44, 4819–4844. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Schär, J.C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Mäcke, H.R. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [Google Scholar] [CrossRef]
- Graham, M.M.; Gu, X.; Ginader, T.; Breheny, P.; Sunderland, J.J. 68Ga-DOTATOC imaging of neuroendocrine tumors: A systematic review and metaanalysis. J. Nucl. Med. 2017, 58, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleynhans, J.; Kruger, H.G.; Cloete, T.; Zeevaart, J.R.; Ebenhan, T. In silico modelling in the development of novel radiolabelled peptide probes. Curr. Med. Chem. 2020, 27, 7048–7063. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Cilibrizzi, A.; Abbate, V.; Spampinato, A.; Zagni, C.; Rescifina, A. FABP4 inhibitors 3D-QSAR model and isosteric replacement of BMS309403 datasets. Data Brief 2019, 22, 471–483. [Google Scholar] [CrossRef]
- Floresta, G.; Zagni, C.; Gentile, D.; Patamia, V.; Rescifina, A. Artificial Intelligence Technologies for COVID-19 De Novo Drug Design. Int. J. Mol. Sci. 2022, 23, 3261. [Google Scholar] [CrossRef]
- Hsieh, C.-J.; Giannakoulias, S.; Petersson, E.J.; Mach, R.H. Computational Chemistry for the Identification of Lead Compounds for Radiotracer Development. Pharmaceuticals 2023, 16, 317. [Google Scholar] [CrossRef]
- Failla, M.; Floresta, G.; Abbate, V. Peptide-based positron emission tomography probes: Current strategies for synthesis and radiolabelling. RSC Med. Chem. 2023. [Google Scholar] [CrossRef]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Diao, L.; Meibohm, B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin. Pharm. 2013, 52, 855–868. [Google Scholar] [CrossRef]
- Onda, M. Reducing the immunogenicity of protein therapeutics. Curr. Drug Targets 2009, 10, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Van Beers, M.M.C.; Bardor, M. Minimizing immunogenicity of biopharmaceuticals by controlling critical quality attributes of proteins. Biotechnol. J. 2012, 7, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.; Bustos, R.H.; Zapata, C.; Garcia, J.; Jauregui, E.; Ashraf, G.M. Immunogenicity in Protein and Peptide Based-Therapeutics: An Overview. Curr. Protein Pept. Sci. 2018, 19, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Abbate, V. Recent progress in the imaging of c-Met aberrant cancers with positron emission tomography. Med. Res. Rev. 2022, 42, 1588–1606. [Google Scholar] [CrossRef]
- Cheetham, A.G.; Keith, D.; Zhang, P.; Lin, R.; Su, H.; Cui, H. Targeting tumors with small molecule peptides. Curr. Cancer Drug Targets 2016, 16, 489–508. [Google Scholar] [CrossRef]
- Floresta, G.; Memdouh, S.; Pham, T.; Ma, M.T.; Blower, P.J.; Hider, R.C.; Abbate, V.; Cilibrizzi, A. Targeting integrin αvβ6 with gallium-68 tris (hydroxypyridinone) based PET probes. Dalton Trans. 2022, 51, 12796–12803. [Google Scholar] [CrossRef]
- Floresta, G.; Keeling, G.P.; Memdouh, S.; Meszaros, L.K.; de Rosales, R.T.; Abbate, V. NHS-Functionalized THP Derivative for Efficient Synthesis of Kit-Based Precursors for 68Ga Labeled PET Probes. Biomedicines 2021, 9, 367. [Google Scholar] [CrossRef]
- Rescifina, A.; Zagni, C.; Mineo, P.G.; Giofrè, S.V.; Chiacchio, U.; Tommasone, S.; Talotta, C.; Gaeta, C.; Neri, P. DNA Recognition with Polycyclic-Aromatic-Hydrocarbon-Presenting Calixarene Conjugates. Eur. J. Org. Chem. 2014, 2014, 7605–7613. [Google Scholar] [CrossRef]
- Szczepańska, K.; Podlewska, S.; Dichiara, M.; Gentile, D.; Patamia, V.; Rosier, N.; Mönnich, D.; Ruiz Cantero, M.C.; Karcz, T.; Łażewska, D.; et al. Structural and molecular insight into piperazine and piperidine derivatives as histamine H3 and sigma-1 receptor antagonists with promising antinociceptive properties. ACS Chem. Neurosci. 2021, 13, 1–15. [Google Scholar] [CrossRef]
- Kim, S.-H.; Lee, E.-H.; Kim, H.-J.; Kim, A.-R.; Kim, Y.-E.; Lee, J.-H.; Yoon, M.-Y.; Koh, S.-H. Development of a Low-Molecular-Weight Aβ42 Detection System Using a Enzyme-Linked Peptide Assay. Biomolecules 2021, 11, 1818. [Google Scholar] [CrossRef]
- Janssen, L.; Sobott, F.; De Deyn, P.P.; Van Dam, D. Signal loss due to oligomerization in ELISA analysis of amyloid-beta can be recovered by a novel sample pre-treatment method. MethodsX 2015, 2, 112–123. [Google Scholar] [CrossRef]
- Giordano, R.J.; Cardó-Vila, M.; Lahdenranta, J.; Pasqualini, R.; Arap, W. Biopanning and rapid analysis of selective interactive ligands. Nat. Med. 2001, 7, 1249–1253. [Google Scholar] [CrossRef]
- McGuire, M.J.; Li, S.; Brown, K.C. Biopanning of phage displayed peptide libraries for the isolation of cell-specific ligands. Methods Mol. Biol. 2009, 504, 291–321. [Google Scholar] [CrossRef] [Green Version]
- Panagides, N.; Zacchi, L.F.; De Souza, M.J.; Morales, R.A.V.; Karnowski, A.; Liddament, M.T.; Owczarek, C.M.; Mahler, S.M.; Panousis, C.; Jones, M.L.; et al. Evaluation of Phage Display Biopanning Strategies for the Selection of Anti-Cell Surface Receptor Antibodies. Int. J. Mol. Sci. 2022, 23, 8470. [Google Scholar] [CrossRef]
- Spitzer, R.; Jain, A.N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput.-Aided Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef]
- Cai, Z.; Ouyang, Q.; Zeng, D.; Nguyen, K.N.; Modi, J.; Wang, L.; White, A.G.; Rogers, B.E.; Xie, X.-Q.; Anderson, C.J. 64Cu-labeled somatostatin analogues conjugated with cross-bridged phosphonate-based chelators via strain-promoted click chemistry for PET imaging: In silico through in vivo studies. J. Med. Chem. 2014, 57, 6019–6029. [Google Scholar] [CrossRef]
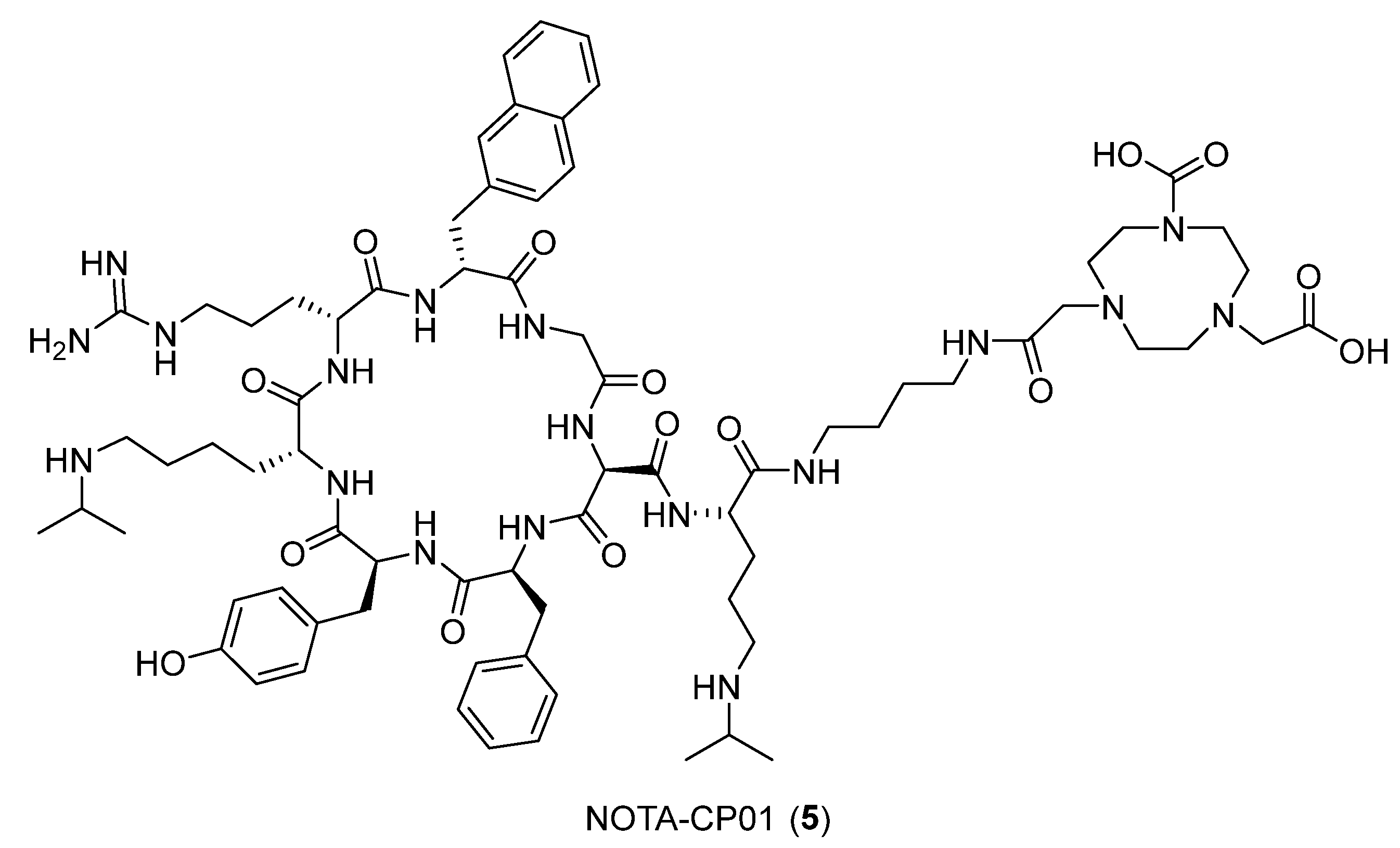
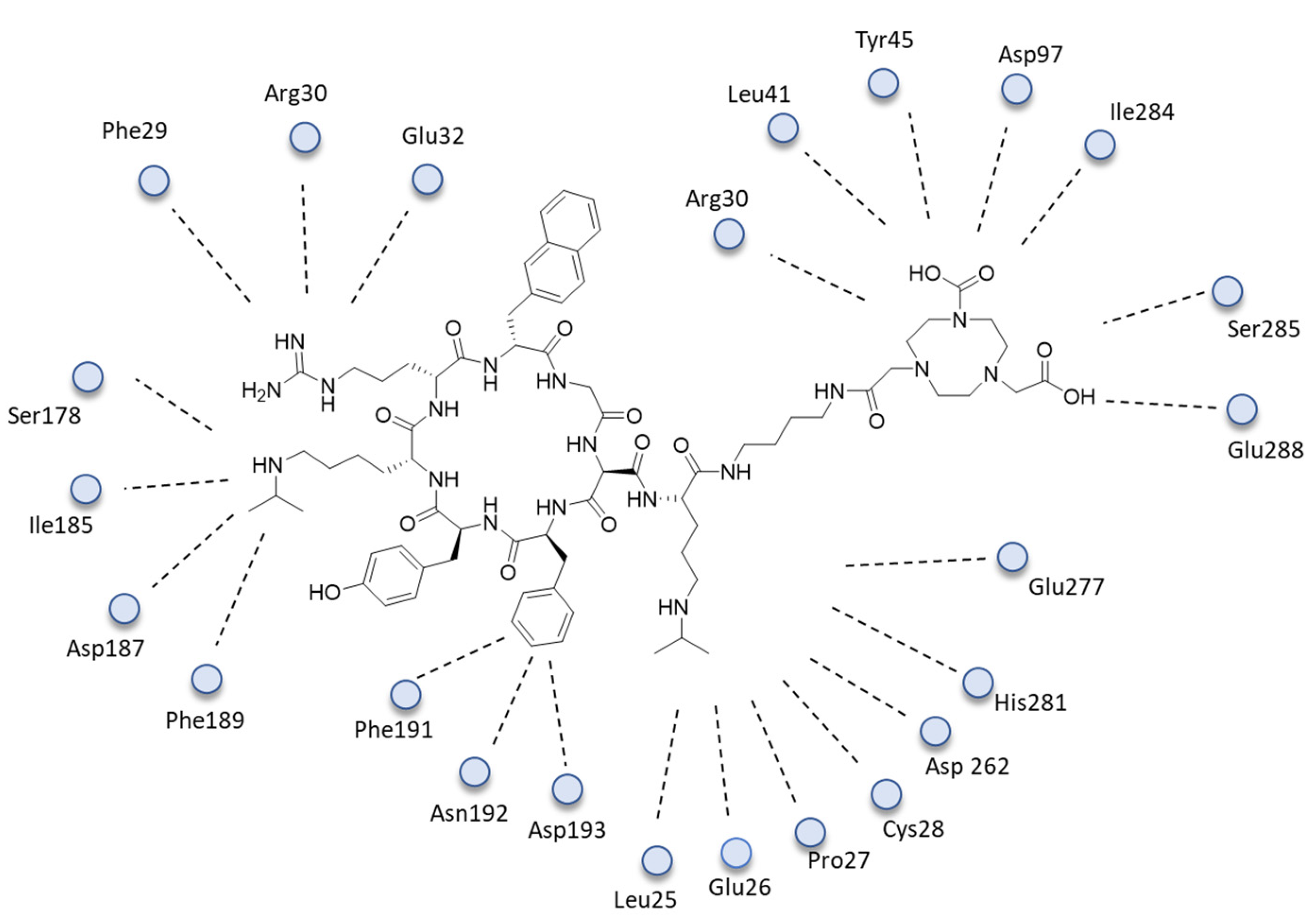
- Peng, T.; Wang, X.; Li, Z.; Bi, L.; Gao, J.; Yang, M.; Wang, Y.; Yao, X.; Shan, H.; Jin, H. Preclinical evaluation of [64Cu] NOTA-CP01 as a PET imaging agent for metastatic esophageal squamous cell carcinoma. Mol. Pharm. 2021, 18, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Liolios, C.; Patsis, C.; Lambrinidis, G.; Tzortzini, E.; Roscher, M.; Bauder-Wüst, U.; Kolocouris, A.; Kopka, K. Investigation of tumor cells and receptor-ligand simulation models for the development of PET imaging probes targeting PSMA and GRPR and a possible crosstalk between the two receptors. Mol. Pharm. 2022, 19, 2231–2247. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aalto, K.; Autio, A.; Kiss, E.A.; Elima, K.; Nymalm, Y.; Veres, T.Z.; Marttila-Ichihara, F.; Elovaara, H.; Saanijoki, T.; Crocker, P.R. Siglec-9 is a novel leukocyte ligand for vascular adhesion protein-1 and can be used in PET imaging of inflammation and cancer. Blood J. Am. Soc. Hematol. 2011, 118, 3725–3733. [Google Scholar] [CrossRef]
- Demmer, O.; Dijkgraaf, I.; Schumacher, U.; Marinelli, L.; Cosconati, S.; Gourni, E.; Wester, H.-J.; Kessler, H. Design, synthesis, and functionalization of dimeric peptides targeting chemokine receptor CXCR4. J. Med. Chem. 2011, 54, 7648–7662. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
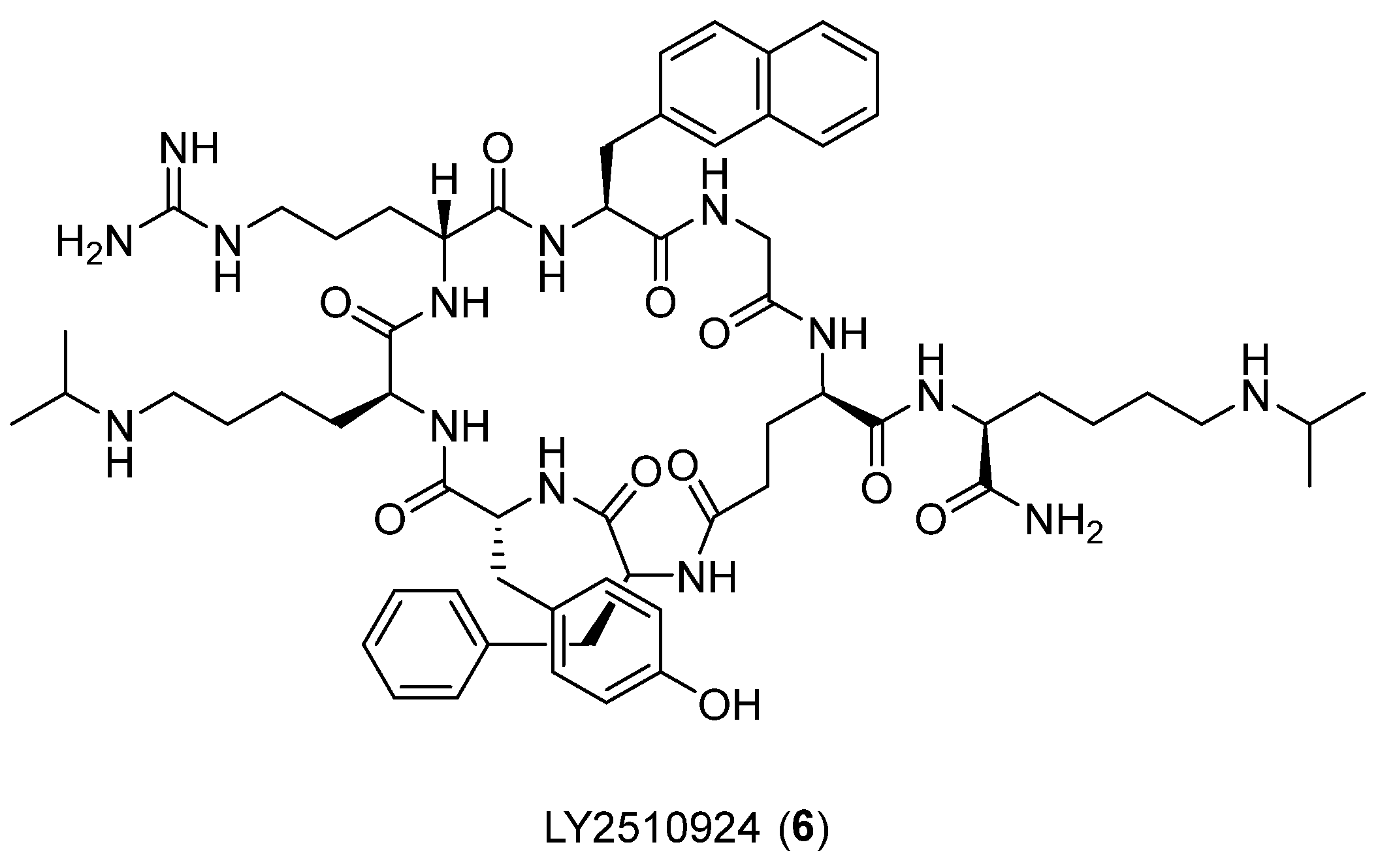
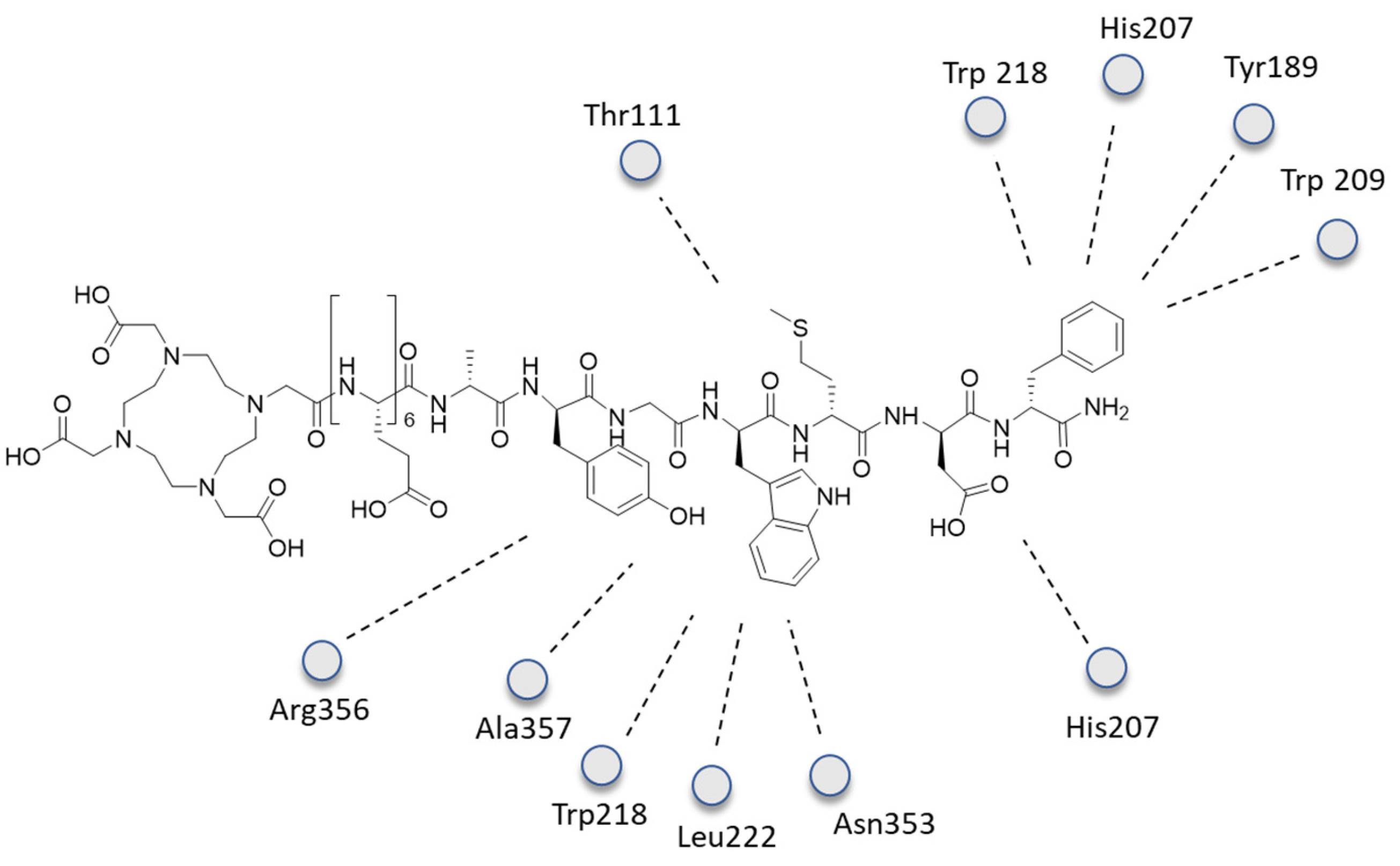
- Lipiński, P.F.; Garnuszek, P.; Maurin, M.; Stoll, R.; Metzler-Nolte, N.; Wodyński, A.; Dobrowolski, J.C.; Dudek, M.K.; Orzełowska, M.; Mikołajczak, R. Structural studies on radiopharmaceutical DOTA-minigastrin analogue (CP04) complexes and their interaction with CCK2 receptor. EJNMMI Res. 2018, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput.-Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
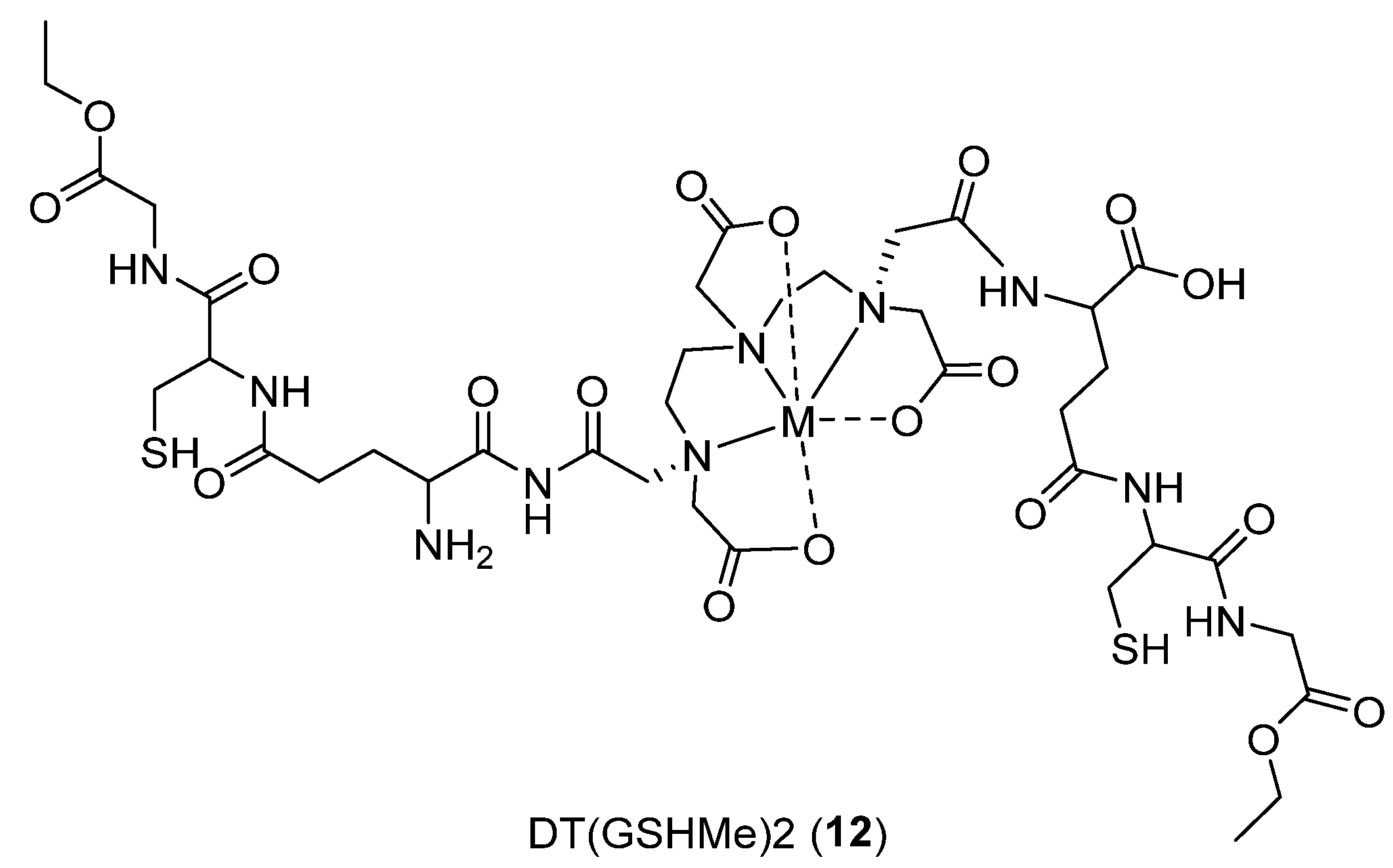
- Khurana, H.; Meena, V.K.; Prakash, S.; Chuttani, K.; Chadha, N.; Jaswal, A.; Dhawan, D.K.; Mishra, A.K.; Hazari, P.P. Preclinical evaluation of a potential GSH ester based PET/SPECT imaging probe DT (GSHMe) 2 to detect Gamma Glutamyl Transferase over expressing tumors. PLoS ONE 2015, 10, e0134281. [Google Scholar] [CrossRef]
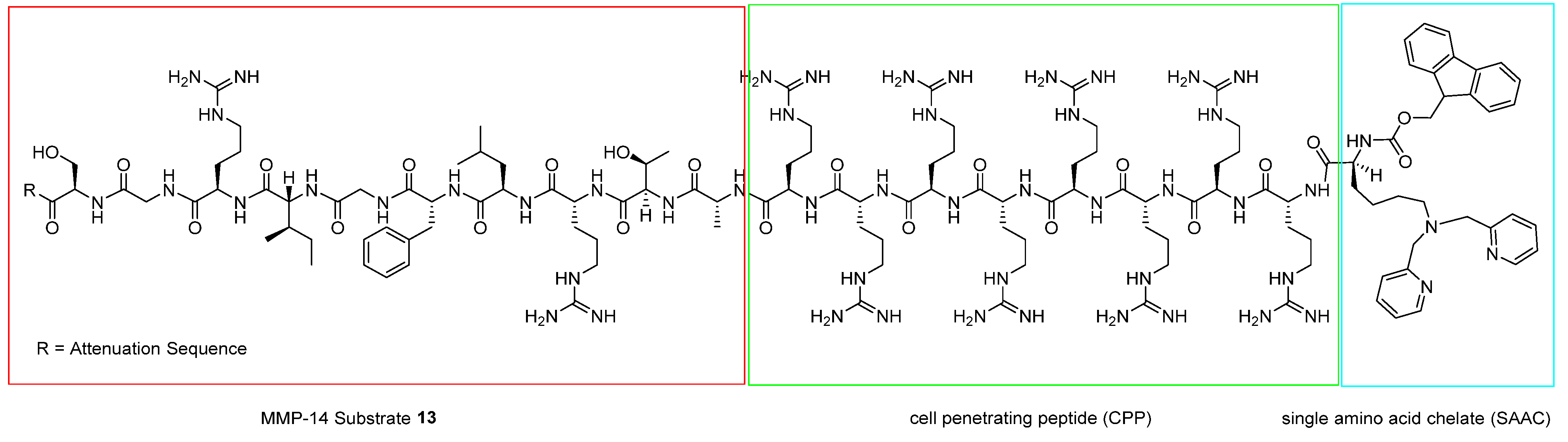
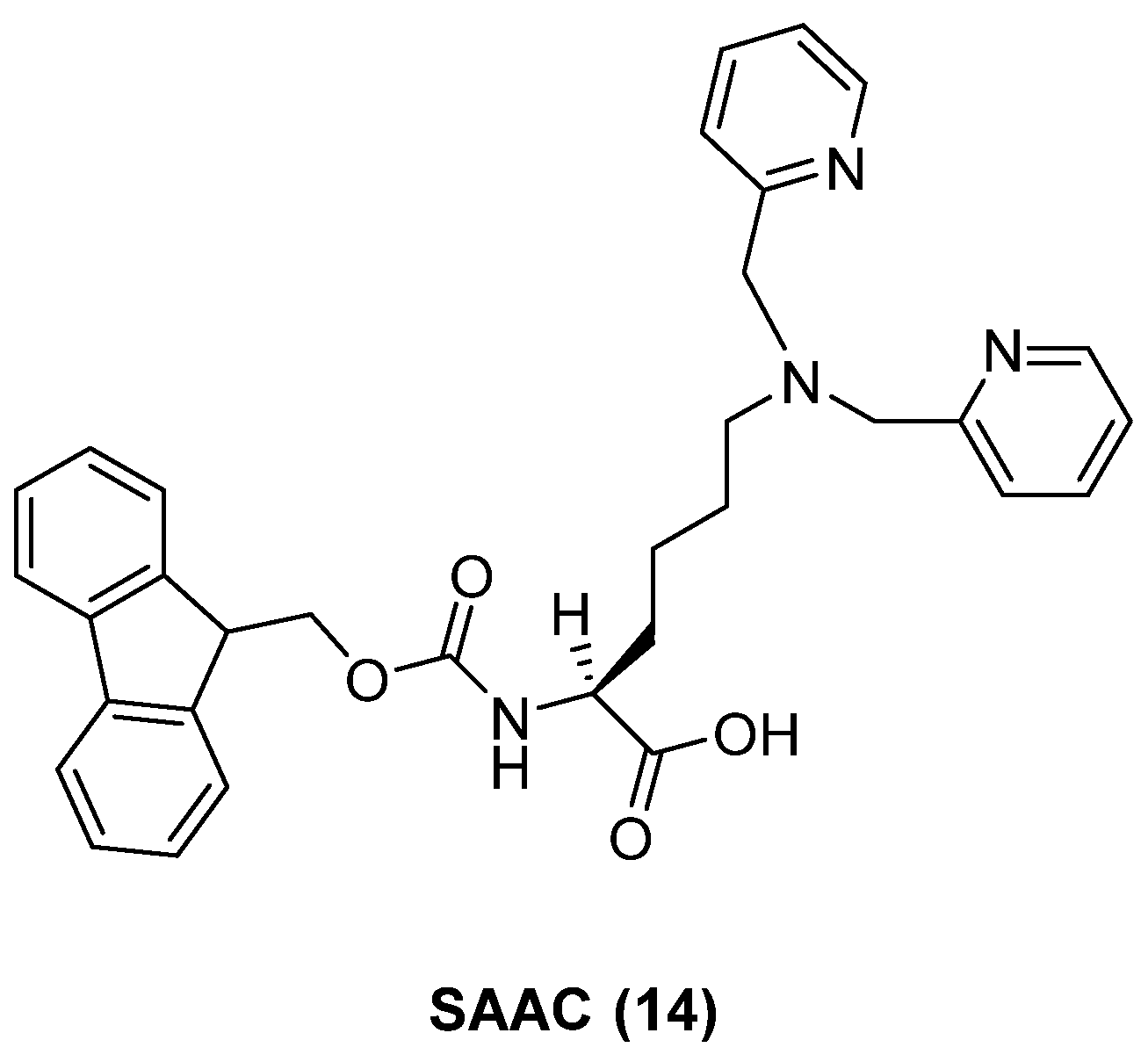
- Watkins, G.A.; Jones, E.F.; Shell, M.S.; VanBrocklin, H.F.; Pan, M.-H.; Hanrahan, S.M.; Feng, J.J.; He, J.; Sounni, N.E.; Dill, K.A. Development of an optimized activatable MMP-14 targeted SPECT imaging probe. Bioorg. Med. Chem. 2009, 17, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Kaul, A.; Tiwari, A.K.; Varshney, R.; Mishra, A.K. Synthesis, in silico screening and preclinical evaluation studies of a hexapeptide analogue for its antimicrobial efficacy. RSC Adv. 2015, 5, 97180–97186. [Google Scholar] [CrossRef]
- Mukai, H.; Watanabe, Y. Review: PET imaging with macro- and middle-sized molecular probes. Nucl. Med. Biol. 2021, 92, 156–170. [Google Scholar] [CrossRef]
- Chen, K.; Conti, P.S. Target-specific delivery of peptide-based probes for PET imaging. Adv. Drug Deliv. Rev. 2010, 62, 1005–1022. [Google Scholar] [CrossRef] [PubMed]
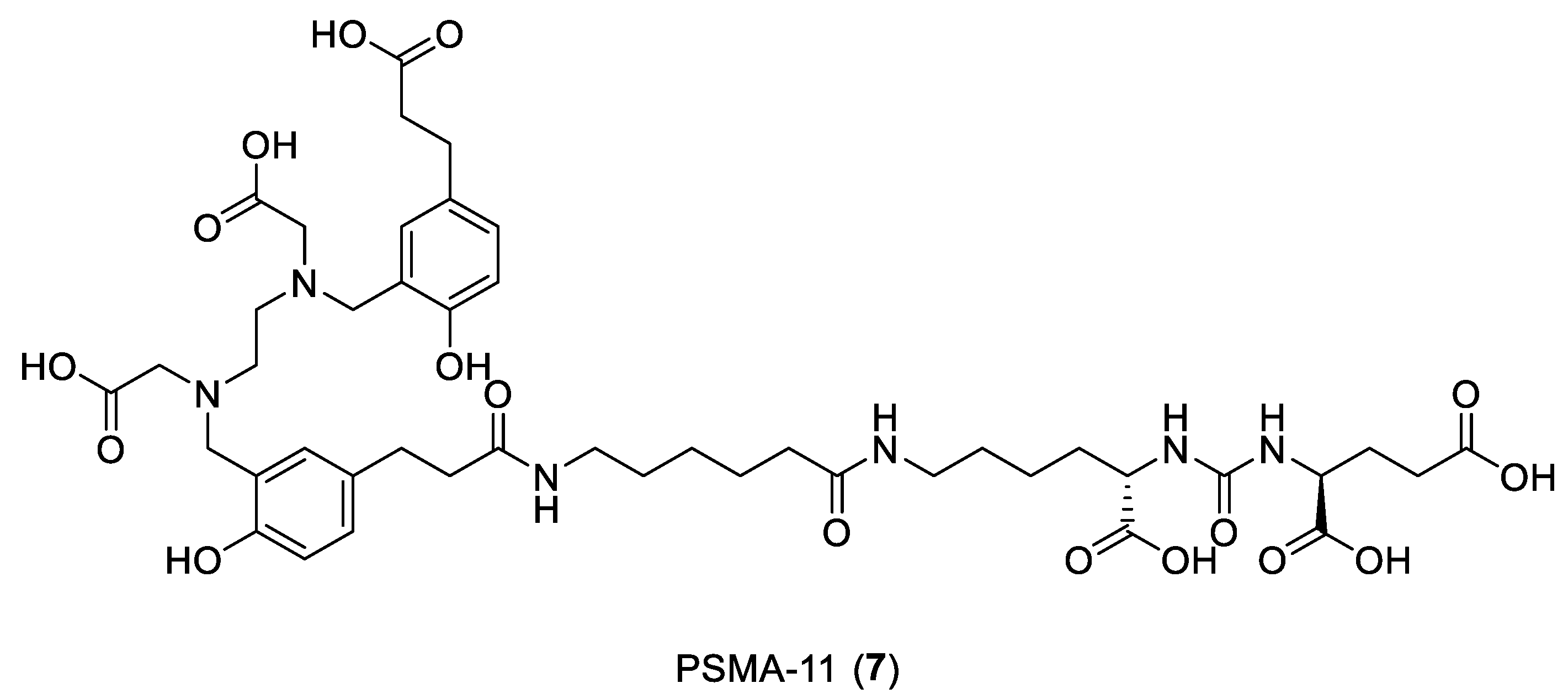
- Young, J.D.; Abbate, V.; Imberti, C.; Meszaros, L.K.; Ma, M.T.; Terry, S.Y.A.; Hider, R.C.; Mullen, G.E.; Blower, P.J. 68Ga-THP-PSMA: A PET Imaging Agent for Prostate Cancer Offering Rapid, Room-Temperature, 1-Step Kit-Based Radiolabeling. J. Nucl. Med. 2017, 58, 1270–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.D.; Ma, M.T.; Eykyn, T.R.; Atkinson, R.A.; Abbate, V.; Cilibrizzi, A.; Hider, R.C.; Blower, P.J. Dipeptide inhibitors of the prostate specific membrane antigen (PSMA): A comparison of urea and thiourea derivatives. Bioorg. Med. Chem. Lett. 2021, 42, 128044. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L. Grand Challenges of Computer-Aided Drug Design: The Road Ahead. Front. Drug Discov. 2021, 1, 728551. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]





















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patamia, V.; Zagni, C.; Brullo, I.; Saccullo, E.; Coco, A.; Floresta, G.; Rescifina, A. Computer-Assisted Design of Peptide-Based Radiotracers. Int. J. Mol. Sci. 2023, 24, 6856. https://doi.org/10.3390/ijms24076856
Patamia V, Zagni C, Brullo I, Saccullo E, Coco A, Floresta G, Rescifina A. Computer-Assisted Design of Peptide-Based Radiotracers. International Journal of Molecular Sciences. 2023; 24(7):6856. https://doi.org/10.3390/ijms24076856
Chicago/Turabian StylePatamia, Vincenzo, Chiara Zagni, Ilaria Brullo, Erika Saccullo, Alessandro Coco, Giuseppe Floresta, and Antonio Rescifina. 2023. "Computer-Assisted Design of Peptide-Based Radiotracers" International Journal of Molecular Sciences 24, no. 7: 6856. https://doi.org/10.3390/ijms24076856