

The Roles of Epigenetic Regulation and the Tumor Microenvironment in the Mechanism of Resistance to Systemic Therapy in Hepatocellular Carcinoma

Abstract
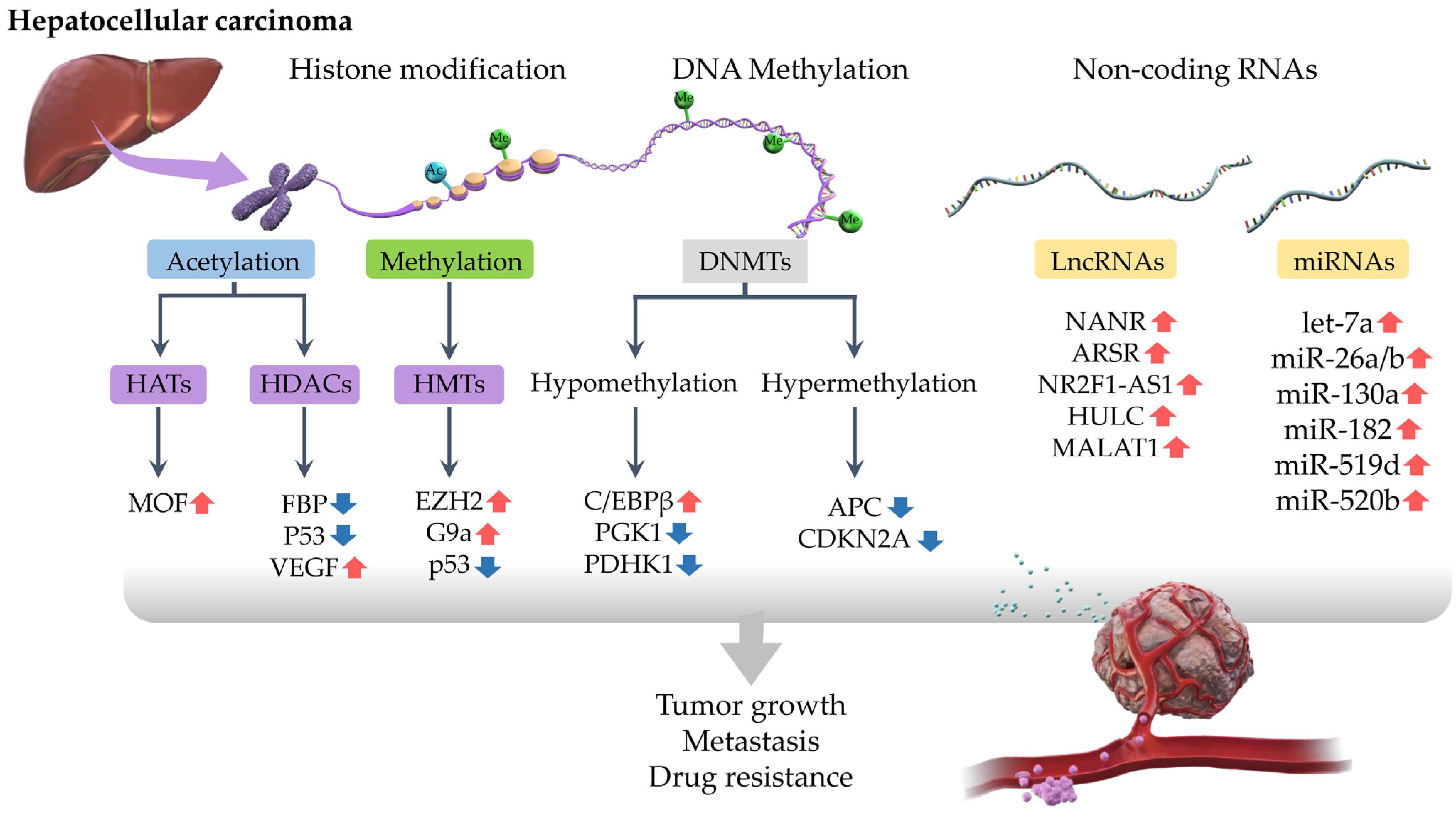
:1. Introduction
1.1. Hepatocellular Carcinoma (HCC)
1.2. Systemic Therapy for Advanced HCC
2. Epigenetic Regulation of Drug Resistance in HCC
2.1. DNA Methylation
2.2. Histone Modifications
2.3. Non-Coding RNA
3. TME and Drug Resistance in HCC
3.1. Vascular System
3.2. Transport Processes
3.3. Immune System
4. Sorafenib Drug Resistance in HCC
5. Regorafenib Drug Resistance in HCC
6. Lenvatinib Drug Resistance in HCC
7. Resistance to Other Drugs in HCC
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; Henley, S.J.; Cucinelli, J.E.; McGlynn, K.A. Changing hepatocellular carcinoma incidence and liver cancer mortality rates in the United States. Am. J. Gastroenterol. 2014, 109, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Beste, L.A.; Green, P.K.; Singal, A.G.; Tapper, E.B.; Waljee, A.K.; Sterling, R.K.; Feld, J.J.; Kaplan, D.E.; Taddei, T.H.; et al. Increased Risk for Hepatocellular Carcinoma Persists Up to 10 Years After HCV Eradication in Patients With Baseline Cirrhosis or High FIB-4 Scores. Gastroenterology 2019, 157, 1264–1278.e4. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.K.; Rich, N.E.; Ahn, C.; Turner, B.J.; Sanders, J.M.; Adamson, B.; Quirk, L.; Perryman, P.; Santini, N.O.; Singal, A.G. Evaluation of a Multifaceted Intervention to Reduce Health Disparities in Hepatitis C Screening: A Pre-Post Analysis. Hepatology 2019, 70, 40–50. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Bolondi, L.; Burroughs, A.; Dufour, J.F.; Galle, P.R.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.L.; Sangro, B. Heterogeneity of patients with intermediate (BCLC B) Hepatocellular Carcinoma: Proposal for a subclassification to facilitate treatment decisions. Semin. Liver Dis. 2012, 32, 348–359. [Google Scholar]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A.; et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 2020, 31, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kuo, H.D.; Yin, R.; Wu, R.; Liu, X.; Wang, L.; Hudlikar, R.; Peter, R.M.; Kong, A.N. Epigenetics/epigenomics of triterpenoids in cancer prevention and in health. Biochem. Pharmacol. 2020, 175, 113890. [Google Scholar] [CrossRef] [PubMed]
- Baharudin, R.; Tieng, F.Y.F.; Lee, L.H.; Ab Mutalib, N.S. Epigenetics of SFRP1: The Dual Roles in Human Cancers. Cancers 2020, 12, 445. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Dariya, B.; Kasa, P.; Peela, S.; El-Rayes, B.F. Epigenetics in hepatocellular carcinoma. Semin. Cancer Biol. 2021, 86 Pt 3, 622–632. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Mio, C.; Bulotta, S.; Russo, D.; Damante, G. Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment. Cancers 2019, 11, 61. [Google Scholar] [CrossRef]
- Taniai, M. Alcohol and hepatocarcinogenesis. Clin. Mol. Hepatol. 2020, 26, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; He, M.; Chan, A.W.H.; Song, Q.X.; Sze, S.C.; Chen, H.; Man, M.K.H.; Man, K.; Chan, S.L.; Lai, P.B.S.; et al. Genomic and Epigenomic Features of Primary and Recurrent Hepatocellular Carcinomas. Gastroenterology 2019, 157, 1630–1645.e6. [Google Scholar] [CrossRef]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef]
- Bakusic, J.; Schaufeli, W.; Claes, S.; Godderis, L. Stress, burnout and depression: A systematic review on DNA methylation mechanisms. J. Psychosom. Res. 2017, 92, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Villanueva, A.; Portela, A.; Sayols, S.; Battiston, C.; Hoshida, Y.; Mendez-Gonzalez, J.; Imbeaud, S.; Letouze, E.; Hernandez-Gea, V.; Cornella, H.; et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015, 61, 1945–1956. [Google Scholar] [CrossRef]
- Hama, N.; Totoki, Y.; Miura, F.; Tatsuno, K.; Saito-Adachi, M.; Nakamura, H.; Arai, Y.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Epigenetic landscape influences the liver cancer genome architecture. Nat. Commun. 2018, 9, 1643. [Google Scholar] [CrossRef]
- Xiong, L.; Wu, F.; Wu, Q.; Xu, L.; Cheung, O.K.; Kang, W.; Mok, M.T.; Szeto, L.L.M.; Lun, C.Y.; Lung, R.W.; et al. Aberrant enhancer hypomethylation contributes to hepatic carcinogenesis through global transcriptional reprogramming. Nat. Commun. 2019, 10, 335. [Google Scholar] [CrossRef]
- Shao, F.; Yang, X.; Wang, W.; Wang, J.; Guo, W.; Feng, X.; Shi, S.; Xue, Q.; Gao, S.; Gao, Y.; et al. Associations of PGK1 promoter hypomethylation and PGK1-mediated PDHK1 phosphorylation with cancer stage and prognosis: A TCGA pan-cancer analysis. Cancer Commun. 2019, 39, 54. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.; Hu, Y.; Wu, Y.Y.; Cheng, Z.H.; Yu, J.; Du, X.; Huang, Z.H. Quantitative methylation analysis of multiple genes using methylation-sensitive restriction enzyme-based quantitative PCR for the detection of hepatocellular carcinoma. Exp. Mol. Pathol. 2011, 91, 455–460. [Google Scholar] [CrossRef]
- Song, M.A.; Tiirikainen, M.; Kwee, S.; Okimoto, G.; Yu, H.; Wong, L.L. Elucidating the landscape of aberrant DNA methylation in hepatocellular carcinoma. PLoS ONE 2013, 8, e55761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcena-Varela, M.; Caruso, S.; Llerena, S.; Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Rebouissou, S.; Alvarez, L.; Jimenez, M.; et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology 2019, 69, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Sheng, Y.; Yang, J.; Wang, C.; Zhang, R.; Zhu, Y.; Zhang, Z.; Zhang, K.; Yan, S.; Sun, H.; et al. Osteopontin alters DNA methylation through up-regulating DNMT1 and sensitizes CD133+/CD44+ cancer stem cells to 5 azacytidine in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 179. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Puszyk, W.; Dong, H.J.; Liu, C. Epigenetic upregulation of HGF and c-Met drives metastasis in hepatocellular carcinoma. PLoS ONE 2013, 8, e63765. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.R.; Sun, H.; Wang, F.Q.; Li, Z.; Yin, Y.R.; Fang, Q.L.; Sun, Y.; Zhao, W.X.; Zhang, S.; Zhao, W.X.; et al. Integrated analysis of gene expression and DNA methylation changes induced by hepatocyte growth factor in human hepatocytes. Mol. Med. Rep. 2015, 12, 4250–4258. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Na, H.; Na, T.Y.; Shin, Y.K.; Seong, J.K.; Lee, M.O. Epigenetic control of metastasis-associated protein 1 gene expression by hepatitis B virus X protein during hepatocarcinogenesis. Oncogenesis 2012, 1, e25. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Nakamura, K.; Fujii, H.; Horichi, N.; Ohmori, T.; Hasegawa, K.; Isoe, T.; Adachi, M.; Otake, N.; Fukunaga, Y. Altered expression of topoisomerase IIalpha contributes to cross-resistant to etoposide K562/MX2 cell line by aberrant methylation. Br. J. Cancer 2005, 92, 1486–1492. [Google Scholar] [CrossRef]
- Asano, T.; Narazaki, H.; Fujita, A. Genome-wide DNA methylation profiling of CpG islands in a morpholino anthracycline derivative-resistant leukemia cell line: p38alpha as a novel candidate for resistance. Pharmacol. Res. Perspect. 2017, 5, e00285. [Google Scholar] [CrossRef]
- Galle, E.; Thienpont, B.; Cappuyns, S.; Venken, T.; Busschaert, P.; Van Haele, M.; Van Cutsem, E.; Roskams, T.; van Pelt, J.; Verslype, C.; et al. DNA methylation-driven EMT is a common mechanism of resistance to various therapeutic agents in cancer. Clin. Epigenetics 2020, 12, 27. [Google Scholar] [CrossRef]
- Ohata, Y.; Shimada, S.; Akiyama, Y.; Mogushi, K.; Nakao, K.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Acquired Resistance with Epigenetic Alterations Under Long-Term Antiangiogenic Therapy for Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1155–1165. [Google Scholar] [CrossRef]
- Nowak, S.J.; Corces, V.G. Phosphorylation of histone H3: A balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004, 20, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, M.; Narazaki, H.; Asano, T. Melatonin overcomes resistance to clofarabine in two leukemic cell lines by increased expression of deoxycytidine kinase. Exp. Hematol. 2015, 43, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Li, D.; Zeng, Z. Epigenetic regulation of histone H3 in the process of hepatocellular tumorigenesis. Biosci. Rep. 2019, 39, BSR20191815. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Cai, C.; Yao, J.; Zhou, Y.; Yu, H.; Shen, W. Histone modifications in FASN modulated by sterol regulatory element-binding protein 1c and carbohydrate responsive-element binding protein under insulin stimulation are related to NAFLD. Biochem. Biophys. Res. Commun. 2017, 483, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Suzuki, T.; Kondo, S.; Miura, H.; Fujimura, Y.; Hayashizaki, Y. Histone H3 acetylated at lysine 9 in promoter is associated with low nucleosome density in the vicinity of transcription start site in human cell. Chromosome Res. 2006, 14, 203–211. [Google Scholar] [CrossRef]
- He, C.; Xu, J.; Zhang, J.; Xie, D.; Ye, H.; Xiao, Z.; Cai, M.; Xu, K.; Zeng, Y.; Li, H.; et al. High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum. Pathol. 2012, 43, 1425–1435. [Google Scholar] [CrossRef]
- Takeda, S.; Liu, H.; Sasagawa, S.; Dong, Y.; Trainor, P.A.; Cheng, E.H.; Hsieh, J.J. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J. Clin. Investig. 2013, 123, 3154–3165. [Google Scholar] [CrossRef]
- Wang, D.Y.; Zou, L.P.; Liu, X.J.; Zhu, H.G.; Zhu, R. Hepatitis B virus X protein induces the histone H3 lysine 9 trimethylation on the promoter of p16 gene in hepatocarcinogenesis. Exp. Mol. Pathol. 2015, 99, 399–408. [Google Scholar] [CrossRef]
- Lu, M.; Zhu, W.W.; Wang, X.; Tang, J.J.; Zhang, K.L.; Yu, G.Y.; Shao, W.Q.; Lin, Z.F.; Wang, S.H.; Lu, L.; et al. ACOT12-Dependent Alteration of Acetyl-CoA Drives Hepatocellular Carcinoma Metastasis by Epigenetic Induction of Epithelial-Mesenchymal Transition. Cell Metab. 2019, 29, 886–900.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pote, N.; Cros, J.; Laouirem, S.; Raffenne, J.; Negrao, M.; Albuquerque, M.; Bedossa, P.; Godinho Ferreira, M.; Ait Si Ali, S.; Fior, R.; et al. The histone acetyltransferase hMOF promotes vascular invasion in hepatocellular carcinoma. Liver Int. 2020, 40, 956–967. [Google Scholar] [CrossRef]
- Zhao, J.; Gray, S.G.; Greene, C.M.; Lawless, M.W. Unmasking the pathological and therapeutic potential of histone deacetylases for liver cancer. Expert. Rev. Gastroenterol. Hepatol. 2019, 13, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, X.; Tao, Y.; Shan, L.; Jiang, Q.; Yu, Y.; Cai, F.; Ma, L. Prognostic and clinicopathologic significance of SIRT1 expression in hepatocellular carcinoma. Oncotarget 2017, 8, 52357–52365. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, 43864. [Google Scholar] [CrossRef]
- Salerno, D.; Chiodo, L.; Alfano, V.; Floriot, O.; Cottone, G.; Paturel, A.; Pallocca, M.; Plissonnier, M.L.; Jeddari, S.; Belloni, L.; et al. Hepatitis B protein HBx binds the DLEU2 lncRNA to sustain cccDNA and host cancer-related gene transcription. Gut 2020, 69, 2016–2024. [Google Scholar] [CrossRef]
- Cai, M.Y.; Hou, J.H.; Rao, H.L.; Luo, R.Z.; Li, M.; Pei, X.Q.; Lin, M.C.; Guan, X.Y.; Kung, H.F.; Zeng, Y.X.; et al. High expression of H3K27me3 in human hepatocellular carcinomas correlates closely with vascular invasion and predicts worse prognosis in patients. Mol. Med. 2011, 17, 12–20. [Google Scholar] [CrossRef]
- Wei, L.; Chiu, D.K.; Tsang, F.H.; Law, C.T.; Cheng, C.L.; Au, S.L.; Lee, J.M.; Wong, C.C.; Ng, I.O.; Wong, C.M. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef]
- Yang, F.; Jiang, Y.; Lv, L.Z. Long non-coding RNA XLOC_010235 correlates with poor prognosis and promotes tumorigenesis of hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4867–4874. [Google Scholar]
- Wang, J.; Yang, K.; Yuan, W.; Gao, Z. Determination of Serum Exosomal H19 as a Noninvasive Biomarker for Bladder Cancer Diagnosis and Prognosis. Med. Sci. Monit. 2018, 24, 9307–9316. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Hao, G.; Sun, Y.; Li, L.; Wang, Y. Long noncoding RNA H19 mediated the chemosensitivity of breast cancer cells via Wnt pathway and EMT process. Onco. Targets Ther. 2018, 11, 8001–8012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oura, K.; Morishita, A.; Masaki, T. Molecular and Functional Roles of MicroRNAs in the Progression of Hepatocellular Carcinoma-A Review. Int. J. Mol. Sci. 2020, 21, 8362. [Google Scholar] [CrossRef] [PubMed]
- Morishita, A.; Oura, K.; Tadokoro, T.; Fujita, K.; Tani, J.; Masaki, T. MicroRNAs in the Pathogenesis of Hepatocellular Carcinoma: A Review. Cancers 2021, 13, 514. [Google Scholar] [CrossRef]
- Tan, H.Y.; Zheng, Y.B.; Liu, J. Serum miR-199a as a potential diagnostic biomarker for detection of colorectal cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8657–8663. [Google Scholar]
- Mondal, T.; Juvvuna, P.K.; Kirkeby, A.; Mitra, S.; Kosalai, S.T.; Traxler, L.; Hertwig, F.; Wernig-Zorc, S.; Miranda, C.; Deland, L.; et al. Sense-Antisense lncRNA Pair Encoded by Locus 6p22.3 Determines Neuroblastoma Susceptibility via the USP36-CHD7-SOX9 Regulatory Axis. Cancer Cell 2018, 33, 417–434.e7. [Google Scholar] [CrossRef]
- Zhang, P.; Dong, Q.; Zhu, H.; Li, S.; Shi, L.; Chen, X. Long non-coding antisense RNA GAS6-AS1 supports gastric cancer progression via increasing GAS6 expression. Gene 2019, 696, 1–9. [Google Scholar] [CrossRef]
- Lou, Y.; Yu, Y.; Xu, X.; Zhou, S.; Shen, H.; Fan, T.; Wu, D.; Yin, J.; Li, G. Long non-coding RNA LUCAT1 promotes tumourigenesis by inhibiting ANXA2 phosphorylation in hepatocellular carcinoma. J. Cell Mol. Med. 2019, 23, 1873–1884. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Chen, T.; Liu, X.; Guo, Y.; Zhu, Q.; Tong, X.; Yang, W.; Xu, Q.; Huang, D.; et al. A novel lncRNA MCM3AP-AS1 promotes the growth of hepatocellular carcinoma by targeting miR-194-5p/FOXA1 axis. Mol. Cancer 2019, 18, 28. [Google Scholar] [CrossRef]
- Wei, L.; Wang, X.; Lv, L.; Liu, J.; Xing, H.; Song, Y.; Xie, M.; Lei, T.; Zhang, N.; Yang, M. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol. Cancer 2019, 18, 147. [Google Scholar] [CrossRef]
- Hou, Z.; Xu, X.; Zhou, L.; Fu, X.; Tao, S.; Zhou, J.; Tan, D.; Liu, S. The long non-coding RNA MALAT1 promotes the migration and invasion of hepatocellular carcinoma by sponging miR-204 and releasing SIRT1. Tumour Biol. 2017, 39, 1010428317718135. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.J.; Zheng, Y.B.; Pan, F.F.; Zhang, F.W.; Zhuang, P.; Fu, W.M. Gallic Acid Suppressed Tumorigenesis by an LncRNA MALAT1-Wnt/beta-Catenin Axis in Hepatocellular Carcinoma. Front. Pharmacol. 2021, 12, 708967. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Chen, C.Y.; Wang, S.H.; Yeh, C.T.; Su, S.C.; Ueng, S.H.; Chuang, W.Y.; Hsueh, C.; Wang, T.H. Melatonin Sensitizes Hepatocellular Carcinoma Cells to Chemotherapy Through Long Non-Coding RNA RAD51-AS1-Mediated Suppression of DNA Repair. Cancers 2018, 10, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.H.; Wu, C.H.; Yeh, C.T.; Su, S.C.; Hsia, S.M.; Liang, K.H.; Chen, C.C.; Hsueh, C.; Chen, C.Y. Melatonin suppresses hepatocellular carcinoma progression via lncRNA-CPS1-IT-mediated HIF-1alpha inactivation. Oncotarget 2017, 8, 82280–82293. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Lv, Y.; Jin, F.; Liu, Y.; Ma, Y.; Xiong, Y.; Liu, L.; Zhang, S.; Sun, Y.; Tipoe, G.L.; et al. LncRNA HANR Promotes Tumorigenesis and Increase of Chemoresistance in Hepatocellular Carcinoma. Cell Physiol. Biochem. 2017, 43, 1926–1938. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, Y.; Feng, B.; Qi, Y. Long Noncoding RNA lncARSR Promotes Doxorubicin Resistance in Hepatocellular Carcinoma via Modulating PTEN-PI3K/Akt Pathway. J. Cell Biochem. 2017, 118, 4498–4507. [Google Scholar] [CrossRef]
- Huang, H.; Chen, J.; Ding, C.M.; Jin, X.; Jia, Z.M.; Peng, J. LncRNA NR2F1-AS1 regulates hepatocellular carcinoma oxaliplatin resistance by targeting ABCC1 via miR-363. J. Cell Mol. Med. 2018, 22, 3238–3245. [Google Scholar] [CrossRef]
- Xiong, H.; Ni, Z.; He, J.; Jiang, S.; Li, X.; He, J.; Gong, W.; Zheng, L.; Chen, S.; Li, B.; et al. LncRNA HULC triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of HCC cells. Oncogene 2017, 36, 3528–3540. [Google Scholar] [CrossRef]
- Yuan, P.; Cao, W.; Zang, Q.; Li, G.; Guo, X.; Fan, J. The HIF-2alpha-MALAT1-miR-216b axis regulates multi-drug resistance of hepatocellular carcinoma cells via modulating autophagy. Biochem. Biophys. Res. Commun. 2016, 478, 1067–1073. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Ni, J.S.; Zheng, H.; Huang, Z.P.; Hong, Y.G.; Ou, Y.L.; Tao, Y.P.; Wang, M.C.; Wang, Z.G.; Yang, Y.; Zhou, W.P. MicroRNA-197-3p acts as a prognostic marker and inhibits cell invasion in hepatocellular carcinoma. Oncol. Lett. 2019, 17, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef] [PubMed]
- Oura, K.; Fujita, K.; Morishita, A.; Iwama, H.; Nakahara, M.; Tadokoro, T.; Sakamoto, T.; Nomura, T.; Yoneyama, H.; Mimura, S.; et al. Serum microRNA-125a-5p as a potential biomarker of HCV-associated hepatocellular carcinoma. Oncol. Lett. 2019, 18, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Chen, J.; Zhou, H.; Chen, Y.; Zhi, Y.; Zhang, B.; Chen, L.; Chu, X.; Wang, R.; Zhang, C. PU.1/microRNA-142–3p targets ATG5/ATG16L1 to inactivate autophagy and sensitize hepatocellular carcinoma cells to sorafenib. Cell Death Dis. 2018, 9, 312. [Google Scholar] [CrossRef]
- Tian, T.; Fu, X.; Lu, J.; Ruan, Z.; Nan, K.; Yao, Y.; Yang, Y. MicroRNA-760 Inhibits Doxorubicin Resistance in Hepatocellular Carcinoma through Regulating Notch1/Hes1-PTEN/Akt Signaling Pathway. J. Biochem. Mol. Toxicol. 2018, 32, e22167. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.M.; Yu, X.N.; Liu, T.T.; Zhu, H.R.; Shi, X.; Bilegsaikhan, E.; Guo, H.Y.; Song, G.Q.; Weng, S.Q.; Huang, X.X.; et al. microRNA-19a-3p promotes tumor metastasis and chemoresistance through the PTEN/Akt pathway in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 105, 1147–1154. [Google Scholar] [CrossRef]
- Que, K.T.; Zhou, Y.; You, Y.; Zhang, Z.; Zhao, X.P.; Gong, J.P.; Liu, Z.J. MicroRNA-31-5p regulates chemosensitivity by preventing the nuclear location of PARP1 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 268. [Google Scholar] [CrossRef]
- Xu, N.; Shen, C.; Luo, Y.; Xia, L.; Xue, F.; Xia, Q.; Zhang, J. Upregulated miR-130a increases drug resistance by regulating RUNX3 and Wnt signaling in cisplatin-treated HCC cell. Biochem. Biophys. Res. Commun. 2012, 425, 468–472. [Google Scholar] [CrossRef]
- Qin, J.; Luo, M.; Qian, H.; Chen, W. Upregulated miR-182 increases drug resistance in cisplatin-treated HCC cell by regulating TP53INP1. Gene 2014, 538, 342–347. [Google Scholar] [CrossRef]
- Tsang, W.P.; Kwok, T.T. Let-7a microRNA suppresses therapeutics-induced cancer cell death by targeting caspase-3. Apoptosis 2008, 13, 1215–1222. [Google Scholar] [CrossRef]
- Fornari, F.; Milazzo, M.; Chieco, P.; Negrini, M.; Marasco, E.; Capranico, G.; Mantovani, V.; Marinello, J.; Sabbioni, S.; Callegari, E.; et al. In hepatocellular carcinoma miR-519d is up-regulated by p53 and DNA hypomethylation and targets CDKN1A/p21, PTEN, AKT3 and TIMP2. J. Pathol. 2012, 227, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Wang, Y.; Li, M.; Zhu, Y.; Liang, H.; Wang, C.; Wang, F.; Zhang, C.Y.; Zen, K.; Li, L. MiR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017, 8, e2540. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Zhang, X.Y.; Hu, J.N.; Ke, Z.P. Apigenin sensitizes hepatocellular carcinoma cells to doxorubic through regulating miR-520b/ATG7 axis. Chem. Biol. Interact. 2018, 280, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, N.; Li, Z.; Zhu, Q.; Li, F.; Yang, C.; Han, Q.; Lv, Y.; Zhou, Z.; Liu, Z. microRNA-4717 differentially interacts with its polymorphic target in the PD1 3′ untranslated region: A mechanism for regulating PD-1 expression and function in HBV-associated liver diseases. Oncotarget 2015, 6, 18933–18944. [Google Scholar] [CrossRef]
- Naito, Y.; Yoshioka, Y.; Yamamoto, Y.; Ochiya, T. How cancer cells dictate their microenvironment: Present roles of extracellular vesicles. Cell Mol. Life Sci. 2017, 74, 697–713. [Google Scholar] [CrossRef]
- Oura, K.; Morishita, A.; Tani, J.; Masaki, T. Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review. Int. J. Mol. Sci. 2021, 22, 5801. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. The origin and function of tumor-associated macrophages. Cell Mol. Immunol. 2015, 12, 1–4. [Google Scholar] [CrossRef]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef]
- Rinaldi, L.; Vetrano, E.; Rinaldi, B.; Galiero, R.; Caturano, A.; Salvatore, T.; Sasso, F.C. HCC and Molecular Targeting Therapies: Back to the Future. Biomedicines 2021, 9, 1345. [Google Scholar] [CrossRef]
- Poon, R.T.; Fan, S.T.; Wong, J. Clinical significance of angiogenesis in gastrointestinal cancers: A target for novel prognostic and therapeutic approaches. Ann. Surg. 2003, 238, 9–28. [Google Scholar] [CrossRef]
- Motoo, Y.; Sawabu, N.; Yamaguchi, Y.; Terada, T.; Nakanuma, Y. Sinusoidal capillarization of human hepatocellular carcinoma: Possible promotion by fibroblast growth factor. Oncology 1993, 50, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; De Mol, M.; et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007, 131, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.X.; Zhu, X.D.; Zhuang, P.Y.; Zhang, J.B.; Zhang, W.; Kong, L.Q.; Wang, W.Q.; Liang, Y.; Wu, W.Z.; Wang, L.; et al. Expression and prognostic significance of placental growth factor in hepatocellular carcinoma and peritumoral liver tissue. Int. J. Cancer 2011, 128, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Belotti, D.; Pinessi, D.; Taraboletti, G. Alternative Vascularization Mechanisms in Tumor Resistance to Therapy. Cancers 2021, 13, 1912. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, J.; Zhao, W.; Peng, Z.; Liu, X.; Li, B.; Zhang, H.; Shan, B.; Zhang, C.; Duan, C. Vasculogenic mimicry in carcinogenesis and clinical applications. J. Hematol. Oncol. 2020, 13, 19. [Google Scholar] [CrossRef]
- Shi, Y.; Shang, J.; Li, Y.; Zhong, D.; Zhang, Z.; Yang, Q.; Lai, C.; Feng, T.; Yao, Y.; Huang, X. ITGA5 and ITGB1 contribute to Sorafenib resistance by promoting vasculogenic mimicry formation in hepatocellular carcinoma. Cancer Med. 2022, 1–11. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, X.; Zhu, D.; Liu, T.; Liang, X.; Liu, F.; Zhang, Y.; Dong, X.; Sun, B. HIF-1alpha promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up-regulation in hypoxic tumor microenvironment. J. Exp. Clin. Cancer Res. 2017, 36, 60. [Google Scholar] [CrossRef]
- Qiao, K.; Liu, Y.; Xu, Z.; Zhang, H.; Zhang, H.; Zhang, C.; Chang, Z.; Lu, X.; Li, Z.; Luo, C.; et al. RNA m6A methylation promotes the formation of vasculogenic mimicry in hepatocellular carcinoma via Hippo pathway. Angiogenesis 2021, 24, 83–96. [Google Scholar] [CrossRef]
- Cheng, R.; Wang, B.; Cai, X.R.; Chen, Z.S.; Du, Q.; Zhou, L.Y.; Ye, J.M.; Chen, Y.L. CD276 Promotes Vasculogenic Mimicry Formation in Hepatocellular Carcinoma via the PI3K/AKT/MMPs Pathway. Onco. Targets Ther. 2020, 13, 11485–11498. [Google Scholar] [CrossRef]
- Jue, C.; Lin, C.; Zhisheng, Z.; Yayun, Q.; Feng, J.; Min, Z.; Haibo, W.; Youyang, S.; Hisamitsu, T.; Shintaro, I.; et al. Notch1 promotes vasculogenic mimicry in hepatocellular carcinoma by inducing EMT signaling. Oncotarget 2017, 8, 2501–2513. [Google Scholar] [CrossRef]
- Meng, J.; Chen, S.; Lei, Y.Y.; Han, J.X.; Zhong, W.L.; Wang, X.R.; Liu, Y.R.; Gao, W.F.; Zhang, Q.; Tan, Q.; et al. Hsp90beta promotes aggressive vasculogenic mimicry via epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene 2019, 38, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, B.; Zhao, X.; An, J.; Zhang, Y.; Gu, Q.; Zhao, N.; Wang, Y.; Liu, F. Function of BMP4 in the Formation of Vasculogenic Mimicry in Hepatocellular Carcinoma. J. Cancer 2020, 11, 2560–2571. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Sheng, G.; Guo, L.; Yu, F.; Chen, G.; Lu, Q.; Wang, R.; Han, B.; Lu, Y. MIG7 is involved in vasculogenic mimicry formation rendering invasion and metastasis in hepatocellular carcinoma. Oncol. Rep. 2018, 39, 679–686. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Yin, M.; Bar-Zion, A.; Lee, C.R.; Butz, H.; Man, S.; Daley, F.; Vermeulen, P.B.; Yousef, G.M.; Foster, F.S.; et al. Co-option of Liver Vessels and Not Sprouting Angiogenesis Drives Acquired Sorafenib Resistance in Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2016, 108, djw030. [Google Scholar] [CrossRef]
- Rada, M.; Tsamchoe, M.; Kapelanski-Lamoureux, A.; Hassan, N.; Bloom, J.; Petrillo, S.; Kim, D.H.; Lazaris, A.; Metrakos, P. Cancer Cells Promote Phenotypic Alterations in Hepatocytes at the Edge of Cancer Cell Nests to Facilitate Vessel Co-Option Establishment in Colorectal Cancer Liver Metastases. Cancers 2022, 14, 1318. [Google Scholar] [CrossRef] [PubMed]
- Durmus, S.; Hendrikx, J.J.; Schinkel, A.H. Apical ABC transporters and cancer chemotherapeutic drug disposition. Adv. Cancer Res. 2015, 125, 1–41. [Google Scholar]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef]
- Arrighetti, N.; Corbo, C.; Evangelopoulos, M.; Pasto, A.; Zuco, V.; Tasciotti, E. Exosome-like Nanovectors for Drug Delivery in Cancer. Curr. Med. Chem. 2019, 26, 6132–6148. [Google Scholar] [CrossRef]
- Gowda, R.; Robertson, B.M.; Iyer, S.; Barry, J.; Dinavahi, S.S.; Robertson, G.P. The role of exosomes in metastasis and progression of melanoma. Cancer Treat. Rev. 2020, 85, 101975. [Google Scholar] [CrossRef]
- Takahashi, K.; Yan, I.K.; Wood, J.; Haga, H.; Patel, T. Involvement of extracellular vesicle long noncoding RNA (linc-VLDLR) in tumor cell responses to chemotherapy. Mol. Cancer Res. 2014, 12, 1377–1387. [Google Scholar] [CrossRef]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Sun, G.; Zhang, Y.; Kong, X.; Rong, D.; Song, J.; Tang, W.; Wang, X. Targeting Immune Cells in the Tumor Microenvironment of HCC: New Opportunities and Challenges. Front Cell Dev. Biol. 2021, 9, 775462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, H.; Wang, L.; Wang, W.; Yang, M.; Qin, Y. Overcoming the therapeutic resistance of hepatomas by targeting the tumor microenvironment. Front. Oncol. 2022, 12, 988956. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.K.; Qin, S.; Tai, D.W.; Lim, H.Y.; Yau, T.; et al. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.K.; Dao, T.V.; Enrico, N.; et al. Tremelimumab Plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- Bruger, A.M.; Dorhoi, A.; Esendagli, G.; Barczyk-Kahlert, K.; van der Bruggen, P.; Lipoldova, M.; Perecko, T.; Santibanez, J.; Saraiva, M.; Van Ginderachter, J.A.; et al. How to measure the immunosuppressive activity of MDSC: Assays, problems and potential solutions. Cancer Immunol. Immunother. 2019, 68, 631–644. [Google Scholar] [CrossRef]
- Li, F.; Zhao, Y.; Wei, L.; Li, S.; Liu, J. Tumor-infiltrating Treg, MDSC, and IDO expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol. Ther. 2018, 19, 695–705. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef]
- Ma, T.; Renz, B.W.; Ilmer, M.; Koch, D.; Yang, Y.; Werner, J.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Solid Tumors. Cells 2022, 11, 310. [Google Scholar] [CrossRef]
- Levring, T.B.; Kongsbak, M.; Rode, A.K.; Woetmann, A.; Odum, N.; Bonefeld, C.M.; Geisler, C. Human CD4+ T cells require exogenous cystine for glutathione and DNA synthesis. Oncotarget 2015, 6, 21853–21864. [Google Scholar] [CrossRef]
- Lu, T.; Gabrilovich, D.I. Molecular pathways: Tumor-infiltrating myeloid cells and reactive oxygen species in regulation of tumor microenvironment. Clin. Cancer Res. 2012, 18, 4877–4882. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Li, X.; Guo, X.; Lu, Y.; Xie, Y.; Huang, X.; Lin, J.; Tan, W.; Wang, C. Myeloid-derived suppressor cells promote tumor growth and sorafenib resistance by inducing FGF1 upregulation and fibrosis. Neoplasia 2022, 28, 100788. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.K.; Tse, A.P.; Xu, I.M.; Di Cui, J.; Lai, R.K.; Li, L.L.; Koh, H.Y.; Tsang, F.H.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Zhao, Z.; Song, J.; Lan, X.; Lu, S.; Chen, M.; Wang, Z.; Chen, W.; Fan, X.; Wu, F.; et al. Interactions between interleukin-6 and myeloid-derived suppressor cells drive the chemoresistant phenotype of hepatocellular cancer. Exp. Cell Res. 2017, 351, 142–149. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, M.; Sun, H.; Feng, Y.; Xu, L.; Chan, A.W.H.; Tong, J.H.; Wong, J.; Chong, C.C.N.; Lai, P.B.S.; et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 2018, 67, 931–944. [Google Scholar] [CrossRef]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Yeung, O.W.; Lo, C.M.; Ling, C.C.; Qi, X.; Geng, W.; Li, C.X.; Ng, K.T.; Forbes, S.J.; Guan, X.Y.; Poon, R.T.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef]
- Sumitomo, R.; Hirai, T.; Fujita, M.; Murakami, H.; Otake, Y.; Huang, C.L. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp. Ther. Med. 2019, 18, 4490–4498. [Google Scholar] [CrossRef]
- Kakoschky, B.; Pleli, T.; Schmithals, C.; Zeuzem, S.; Brune, B.; Vogl, T.J.; Korf, H.W.; Weigert, A.; Piiper, A. Selective targeting of tumor associated macrophages in different tumor models. PLoS ONE 2018, 13, e0193015. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Shi, X.; Wang, S.; Gao, Y.; Kuang, Z.; Xie, Q.; Li, Y.; Deng, H.; Wu, Y.; Li, M.; et al. M2 macrophages mediate sorafenib resistance by secreting HGF in a feed-forward manner in hepatocellular carcinoma. Br. J. Cancer 2019, 121, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.T.; Song, K.; Zhou, J.; Shi, Y.H.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Ding, Z.B.; Fan, J. Tumor-associated macrophages modulate resistance to oxaliplatin via inducing autophagy in hepatocellular carcinoma. Cancer Cell Int. 2019, 19, 71. [Google Scholar] [CrossRef]
- Yang, X.H.; Yamagiwa, S.; Ichida, T.; Matsuda, Y.; Sugahara, S.; Watanabe, H.; Sato, Y.; Abo, T.; Horwitz, D.A.; Aoyagi, Y. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J. Hepatol. 2006, 45, 254–262. [Google Scholar] [CrossRef]
- Chiu, D.K.; Yuen, V.W.; Cheu, J.W.; Wei, L.L.; Ting, V.; Fehlings, M.; Sumatoh, H.; Nardin, A.; Newell, E.W.; Ng, I.O. Hepatocellular Carcinoma Cells Up-regulate PVRL1, Stabilizing PVR and Inhibiting the Cytotoxic T-Cell Response via TIGIT to Mediate Tumor Resistance to PD1 Inhibitors in Mice. Gastroenterology 2020, 159, 609–623. [Google Scholar] [CrossRef]
- Shrestha, R.; Prithviraj, P.; Bridle, K.R.; Crawford, D.H.G.; Jayachandran, A. Combined Inhibition of TGF-beta1-Induced EMT and PD-L1 Silencing Re-Sensitizes Hepatocellular Carcinoma to Sorafenib Treatment. J. Clin. Med. 2021, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; You, M.; Fu, J.; Tian, M.; Zhong, X.; Du, C.; Hong, Z.; Zhu, Z.; Liu, J.; Markowitz, G.J.; et al. Intratumoral stem-like CCR4+ regulatory T cells orchestrate the immunosuppressive microenvironment in HCC associated with hepatitis B. J. Hepatol. 2022, 76, 148–159. [Google Scholar] [CrossRef]
- Karabicici, M.; Azbazdar, Y.; Ozhan, G.; Senturk, S.; Firtina Karagonlar, Z.; Erdal, E. Changes in Wnt and TGF-beta Signaling Mediate the Development of Regorafenib Resistance in Hepatocellular Carcinoma Cell Line HuH7. Front Cell Dev. Biol. 2021, 9, 639779. [Google Scholar] [CrossRef]
- Sun, C.; Sun, H.; Zhang, C.; Tian, Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol. Immunol. 2015, 12, 292–302. [Google Scholar] [CrossRef]
- Peng, H.; Wisse, E.; Tian, Z. Liver natural killer cells: Subsets and roles in liver immunity. Cell Mol. Immunol. 2016, 13, 328–336. [Google Scholar] [CrossRef]
- Konjevic, G.M.; Vuletic, A.M.; Mirjacic Martinovic, K.M.; Larsen, A.K.; Jurisic, V.B. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine 2019, 117, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Ren, X.; Cao, D.; Zhao, H.; Zhai, Z.; Li, H.; Li, Y.; Fu, X.; He, J.; Zhao, H. CNOT7 depletion reverses natural killer cell resistance by modulating the tumor immune microenvironment of hepatocellular carcinoma. FEBS Open Bio. 2020, 10, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Bugide, S.; Green, M.R.; Wajapeyee, N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl. Acad. Sci. USA 2018, 115, E3509–E3518. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.F.; Yip, C.W.; Ng, L.W.; Wong, C.K.; Cheung, T.T.; Lo, C.M.; Fan, S.T.; Cheung, S.T. Restoration of natural killer activity in hepatocellular carcinoma by treatment with antibody against granulin-epithelin precursor. Oncoimmunology 2015, 4, e1016706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Zhang, Q.; Zhou, H.; Zhou, J.; Zhang, J.; Jiang, Y.; Wang, J.; Meng, X.; Zeng, L.; Jiang, X. microRNA-889 is downregulated by histone deacetylase inhibitors and confers resistance to natural killer cytotoxicity in hepatocellular carcinoma cells. Cytotechnology 2018, 70, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Immune Checkpoint Inhibition in Hepatocellular Carcinoma: Basics and Ongoing Clinical Trials. Oncology 2017, 92 (Suppl. 1), 50–62. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yang, Y.; Chen, Z.; Jiang, Z.; Gu, Y.; Liu, Y.; Xu, S.; Lin, C.; Pan, Z.; Zhou, W.; et al. Human hepatocellular carcinoma-infiltrating CD4+CD69+Foxp3− regulatory T cell suppresses T cell response via membrane-bound TGF-beta1. J. Mol. Med. 2014, 92, 539–550. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.Z.; Liu, Z.W.; Zhang, J.Y.; Yang, Y.P.; Tien, P.; Wang, F.S. PD-1 and PD-L1 upregulation promotes CD8+ T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011, 128, 887–896. [Google Scholar] [CrossRef]
- Mengshol, J.A.; Golden-Mason, L.; Arikawa, T.; Smith, M.; Niki, T.; McWilliams, R.; Randall, J.A.; McMahan, R.; Zimmerman, M.A.; Rangachari, M.; et al. A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS ONE 2010, 5, e9504. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Abeni, E.; Salvi, A.; Marchina, E.; Traversa, M.; Arici, B.; De Petro, G. Sorafenib induces variations of the DNA methylome in HA22T/VGH human hepatocellular carcinoma-derived cells. Int. J. Oncol. 2017, 51, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Qin, Z.Y.; Wen, L.Z.; Guo, Y.; Liu, Q.; Lei, Z.J.; Pan, W.; Liu, K.J.; Wang, X.W.; Lai, S.J.; et al. Epigenetic restriction of Hippo signaling by MORC2 underlies stemness of hepatocellular carcinoma cells. Cell Death Differ. 2018, 25, 2086–2100. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, C.S.; Laggai, S.; Czepukojc, B.; Hussein, U.K.; List, M.; Barghash, A.; Tierling, S.; Hosseini, K.; Golob-Schwarzl, N.; Pokorny, J.; et al. The long non-coding RNA H19 suppresses carcinogenesis and chemoresistance in hepatocellular carcinoma. Cell Stress 2017, 1, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.L.; Ng, K.Y.; Tan, K.V.; Chan, L.H.; Zhou, L.; Che, N.; Hoo, R.L.C.; Lee, T.K.; Richard, S.; Lo, C.M.; et al. CRAF Methylation by PRMT6 Regulates Aerobic Glycolysis-Driven Hepatocarcinogenesis via ERK-Dependent PKM2 Nuclear Relocalization and Activation. Hepatology 2020, 71, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Blanco, C.; Fondevila, F.; Fernandez-Palanca, P.; Garcia-Palomo, A.; Pelt, J.V.; Verslype, C.; Gonzalez-Gallego, J.; Mauriz, J.L. Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells. Cancers 2019, 11, 1984. [Google Scholar] [CrossRef]
- Li, W.; Dong, X.; He, C.; Tan, G.; Li, Z.; Zhai, B.; Feng, J.; Jiang, X.; Liu, C.; Jiang, H.; et al. LncRNA SNHG1 contributes to sorafenib resistance by activating the Akt pathway and is positively regulated by miR-21 in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 183. [Google Scholar] [CrossRef]
- Zhang, T.; Cao, C.; Wu, D.; Liu, L. SNHG3 correlates with malignant status and poor prognosis in hepatocellular carcinoma. Tumour Biol. 2016, 37, 2379–2385. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, R.; Du, X.; Chai, W.; Zhou, Q. Long noncoding RNA SNHG16 induces sorafenib resistance in hepatocellular carcinoma cells through sponging miR-140-5p. Onco. Targets Ther. 2019, 12, 415–422. [Google Scholar] [CrossRef]
- Chen, S.; Xia, X. Long noncoding RNA NEAT1 suppresses sorafenib sensitivity of hepatocellular carcinoma cells via regulating miR-335-c-Met. J. Cell Physiol. 2019, 234, 14999–15009. [Google Scholar] [CrossRef] [PubMed]
- Sui, C.; Dong, Z.; Yang, C.; Zhang, M.; Dai, B.; Geng, L.; Lu, J.; Yang, J.; Xu, M. LncRNA FOXD2-AS1 as a competitive endogenous RNA against miR-150-5p reverses resistance to sorafenib in hepatocellular carcinoma. J. Cell Mol. Med. 2019, 23, 6024–6033. [Google Scholar] [CrossRef]
- Azumi, J.; Tsubota, T.; Sakabe, T.; Shiota, G. miR-181a induces sorafenib resistance of hepatocellular carcinoma cells through downregulation of RASSF1 expression. Cancer Sci. 2016, 107, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Pollutri, D.; Patrizi, C.; La Bella, T.; Marinelli, S.; Casadei Gardini, A.; Marisi, G.; Baron Toaldo, M.; Baglioni, M.; Salvatore, V.; et al. In Hepatocellular Carcinoma miR-221 Modulates Sorafenib Resistance through Inhibition of Caspase-3-Mediated Apoptosis. Clin. Cancer Res. 2017, 23, 3953–3965. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, H.; Hong, H.; Zhang, Z. MiR-374b re-sensitizes hepatocellular carcinoma cells to sorafenib therapy by antagonizing PKM2-mediated glycolysis pathway. Am. J. Cancer. Res. 2019, 9, 765–778. [Google Scholar] [PubMed]
- Pollutri, D.; Patrizi, C.; Marinelli, S.; Giovannini, C.; Trombetta, E.; Giannone, F.A.; Baldassarre, M.; Quarta, S.; Vandewynckel, Y.P.; Vandierendonck, A.; et al. The epigenetically regulated miR-494 associates with stem-cell phenotype and induces sorafenib resistance in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 4. [Google Scholar] [CrossRef]
- Dietrich, P.; Koch, A.; Fritz, V.; Hartmann, A.; Bosserhoff, A.K.; Hellerbrand, C. Wild type Kirsten rat sarcoma is a novel microRNA-622-regulated therapeutic target for hepatocellular carcinoma and contributes to sorafenib resistance. Gut 2018, 67, 1328–1341. [Google Scholar] [CrossRef]
- Liu, L.P.; Ho, R.L.; Chen, G.G.; Lai, P.B. Sorafenib inhibits hypoxia-inducible factor-1alpha synthesis: Implications for antiangiogenic activity in hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 5662–5671. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar]
- Mendez-Blanco, C.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Zhao, D.; Zhai, B.; He, C.; Tan, G.; Jiang, X.; Pan, S.; Dong, X.; Wei, Z.; Ma, L.; Qiao, H.; et al. Upregulation of HIF-2alpha induced by sorafenib contributes to the resistance by activating the TGF-alpha/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014, 26, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zheng, L.; Chen, J.; Sun, Y.; Lin, H.; Jin, R.A.; Tang, M.; Liang, X.; Cai, X. Increasing AR by HIF-2alpha inhibitor (PT-2385) overcomes the side-effects of sorafenib by suppressing hepatocellular carcinoma invasion via alteration of pSTAT3, pAKT and pERK signals. Cell Death Dis. 2017, 8, e3095. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Briz, O.; Monte, M.J.; Sanchez-Vicente, L.; Abete, L.; Lozano, E.; Mazzanti, G.; Di Sotto, A.; Marin, J.J.G. Chemosensitization of hepatocellular carcinoma cells to sorafenib by beta-caryophyllene oxide-induced inhibition of ABC export pumps. Arch. Toxicol. 2019, 93, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qian, Z.; Zhao, H.; Zhang, X.; Che, S.; Zhang, H.; Shang, H.; Bao, J.; Hao, C.; Liu, J.; et al. CSN5 silencing reverses sorafenib resistance of human hepatocellular carcinoma HepG2 cells. Mol. Med. Rep. 2015, 12, 3902–3908. [Google Scholar] [CrossRef] [PubMed]
- Tomonari, T.; Takeishi, S.; Taniguchi, T.; Tanaka, T.; Tanaka, H.; Fujimoto, S.; Kimura, T.; Okamoto, K.; Miyamoto, H.; Muguruma, N.; et al. MRP3 as a novel resistance factor for sorafenib in hepatocellular carcinoma. Oncotarget 2016, 7, 7207–7215. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, C.; Shi, Y.; Zhao, L. Exosomes derived from siRNA against GRP78 modified bone-marrow-derived mesenchymal stem cells suppress Sorafenib resistance in hepatocellular carcinoma. J. Nanobiotechnology 2018, 16, 103. [Google Scholar] [CrossRef]
- Yao, W.; Ba, Q.; Li, X.; Li, H.; Zhang, S.; Yuan, Y.; Wang, F.; Duan, X.; Li, J.; Zhang, W.; et al. A Natural CCR2 Antagonist Relieves Tumor-associated Macrophage-mediated Immunosuppression to Produce a Therapeutic Effect for Liver Cancer. EBioMedicine 2017, 22, 58–67. [Google Scholar] [CrossRef]
- Chen, Y.; Ramjiawan, R.R.; Reiberger, T.; Ng, M.R.; Hato, T.; Huang, Y.; Ochiai, H.; Kitahara, S.; Unan, E.C.; Reddy, T.P.; et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 2015, 61, 1591–1602. [Google Scholar] [CrossRef]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef]
- Chen, J.; Ji, T.; Zhao, J.; Li, G.; Zhang, J.; Jin, R.; Liu, J.; Liu, X.; Liang, X.; Huang, D.; et al. Sorafenib-resistant hepatocellular carcinoma stratified by phosphorylated ERK activates PD-1 immune checkpoint. Oncotarget 2016, 7, 41274–41284. [Google Scholar] [CrossRef]
- Juengpanich, S.; Topatana, W.; Lu, C.; Staiculescu, D.; Li, S.; Cao, J.; Lin, J.; Hu, J.; Chen, M.; Chen, J.; et al. Role of cellular, molecular and tumor microenvironment in hepatocellular carcinoma: Possible targets and future directions in the regorafenib era. Int. J. Cancer 2020, 147, 1778–1792. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, J.; Zhang, Y.; Cai, H.; Chen, X.; Sun, D. Regorafenib reverses HGF-induced sorafenib resistance by inhibiting epithelial-mesenchymal transition in hepatocellular carcinoma. FEBS Open Bio. 2019, 9, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Fondevila, F.; Mendez-Blanco, C.; Fernandez-Palanca, P.; Payo-Serafin, T.; van Pelt, J.; Verslype, C.; Gonzalez-Gallego, J.; Mauriz, J.L. Autophagy-Related Chemoprotection against Sorafenib in Human Hepatocarcinoma: Role of FOXO3 Upregulation and Modulation by Regorafenib. Int. J. Mol. Sci. 2021, 22, 11770. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, K.; Matsui, A.; Kikuchi, H.; Klein, S.; Mamessier, E.; Chen, I.X.; Aoki, S.; Kitahara, S.; Inoue, K.; Shigeta, A.; et al. Regorafenib combined with PD1 blockade increases CD8 T-cell infiltration by inducing CXCL10 expression in hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e001435. [Google Scholar] [CrossRef] [PubMed]
- Teufel, M.; Seidel, H.; Kochert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers Associated With Response to Regorafenib in Patients With Hepatocellular Carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Che, N.; Zhou, L.; Luk, S.T.; Kau, P.W.; Chai, S.; Ngan, E.S.; Lo, C.M.; Man, K.; Ding, J.; et al. Efficacy of annexin A3 blockade in sensitizing hepatocellular carcinoma to sorafenib and regorafenib. J. Hepatol. 2018, 69, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, N.; Han, Q.; Lu, W.; Wang, L.; Yang, D.; Zheng, M.; Zhang, Z.; Liu, H.; Lee, T.H.; et al. Pin1 inhibition reverses the acquired resistance of human hepatocellular carcinoma cells to Regorafenib via the Gli1/Snail/E-cadherin pathway. Cancer Lett. 2019, 444, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-alpha is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef]
- Wuputra, K.; Hsiao, P.J.; Chang, W.T.; Wu, P.H.; Chen, L.A.; Huang, J.W.; Su, W.L.; Yang, Y.H.; Wu, D.C.; Yokoyama, K.K.; et al. FOXM1-CD44 Signaling Is Critical for the Acquisition of Regorafenib Resistance in Human Liver Cancer Cells. Int. J. Mol. Sci. 2022, 23, 7782. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, Q.; Li, X.; Ren, X.; Li, J.; Zhang, Y.; Zeng, S.; Xu, L.; Dong, X.; Zhai, B. TOP2A inhibition reverses drug resistance of hepatocellular carcinoma to regorafenib. Am. J. Cancer Res. 2022, 12, 4343–4360. [Google Scholar]
- Sofer, S.; Lamkiewicz, K.; Armoza Eilat, S.; Partouche, S.; Marz, M.; Moskovits, N.; Stemmer, S.M.; Shlomai, A.; Sklan, E.H. A genome-wide CRISPR activation screen reveals Hexokinase 1 as a critical factor in promoting resistance to multi-kinase inhibitors in hepatocellular carcinoma cells. FASEB J. 2022, 36, e22191. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wang, X.; Peng, R.; Zhang, B.; Han, Q.; Lin, J.; Wang, J.; Lin, J.; Jiang, M.; Liu, H.; et al. Induction of IL-6Ralpha by ATF3 enhances IL-6 mediated sorafenib and regorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2022, 524, 161–171. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, S.; Ma, D.; Yan, D.; Zhang, G.; Cao, Y.; Wang, Z.; Wu, J.; Jiang, C. Targeting SphK2 Reverses Acquired Resistance of Regorafenib in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 694. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Y.; Tan, C.T.; Chen, C.W.; Ou, D.L.; Cheng, A.L.; Hsu, C. Immunomodulatory Effects of Current Targeted Therapies on Hepatocellular Carcinoma: Implication for the Future of Immunotherapy. Semin. Liver Dis. 2018, 38, 379–388. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, H.; Yuan, X.; Fan, X.; Zhang, C. Advances in Immune Checkpoint Inhibitors for Advanced Hepatocellular Carcinoma. Front. Immunol. 2022, 13, 896752. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K.; Minoshima, Y.; Iwata, M.; Funahashi, Y. Antitumor activity of lenvatinib (e7080): An angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef]
- Chen, X.; Ye, Q.; Chen, Z.; Lin, Q.; Chen, W.; Xie, C.; Wang, X. Long non-coding RNA muskelin 1 antisense RNA as a potential therapeutic target in hepatocellular carcinoma treatment. Bioengineered 2022, 13, 12237–12247. [Google Scholar] [CrossRef]
- Wei, Y.; Wei, L.; Han, T.; Ding, S. miR-3154 promotes hepatocellular carcinoma progression via suppressing HNF4alpha. Carcinogenesis 2022, 43, 1002–1014. [Google Scholar] [CrossRef]
- Iseda, N.; Itoh, S.; Toshida, K.; Tomiyama, T.; Morinaga, A.; Shimokawa, M.; Shimagaki, T.; Wang, H.; Kurihara, T.; Toshima, T.; et al. Ferroptosis is induced by lenvatinib through fibroblast growth factor receptor-4 inhibition in hepatocellular carcinoma. Cancer Sci. 2022, 113, 2272–2287. [Google Scholar] [CrossRef]
- Ma, X.; Qiu, Y.; Sun, Y.; Zhu, L.; Zhao, Y.; Li, T.; Lin, Y.; Ma, D.; Qin, Z.; Sun, C.; et al. NOD2 inhibits tumorigenesis and increases chemosensitivity of hepatocellular carcinoma by targeting AMPK pathway. Cell Death Dis. 2020, 11, 174. [Google Scholar] [CrossRef]
- Duan, A.; Li, H.; Yu, W.; Zhang, Y.; Yin, L. Long Noncoding RNA XIST Promotes Resistance to Lenvatinib in Hepatocellular Carcinoma Cells via Epigenetic Inhibition of NOD2. J. Oncol. 2022, 2022, 4537343. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tan, K.; Hu, W.; Hou, Y.; Yang, G. LncRNA AC026401.3 interacts with OCT1 to intensify sorafenib and lenvatinib resistance by activating E2F2 signaling in hepatocellular carcinoma. Exp. Cell Res. 2022, 420, 113335. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jiang, W.; Han, P.; Zhang, J.; Tong, L.; Sun, X. MicroRNA-128-3p Mediates Lenvatinib Resistance of Hepatocellular Carcinoma Cells by Downregulating c-Met. J. Hepatocell Carcinoma 2022, 9, 113–126. [Google Scholar] [CrossRef]
- Hao, X.; Zhang, Y.; Shi, X.; Liu, H.; Zheng, Z.; Han, G.; Rong, D.; Zhang, C.; Tang, W.; Wang, X. CircPAK1 promotes the progression of hepatocellular carcinoma via modulation of YAP nucleus localization by interacting with 14-3-3zeta. J. Exp. Clin. Cancer Res. 2022, 41, 281. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, H.; Wen, P.; Wang, Y.; Cui, Y.; Wu, J. circRNA circMED27 acts as a prognostic factor and mediator to promote lenvatinib resistance of hepatocellular carcinoma. Mol. Ther. Nucleic Acids 2022, 27, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Liu, J.; Wang, Y.; Dong, J. Co-administration of MDR1 and BCRP or EGFR/PI3K inhibitors overcomes lenvatinib resistance in hepatocellular carcinoma. Front. Oncol. 2022, 12, 944537. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zou, T.; Qin, W.; Shen, X.; Su, Y.; Li, J.; Chen, Y.; Zhang, Z.; Sun, H.; Zheng, Y.; et al. Inhibition of EGFR Overcomes Acquired Lenvatinib Resistance Driven by STAT3-ABCB1 Signaling in Hepatocellular Carcinoma. Cancer Res. 2022, 82, 3845–3857. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Palanca, P.; Payo-Serafin, T.; San-Miguel, B.; Mendez-Blanco, C.; Tunon, M.J.; Gonzalez-Gallego, J.; Mauriz, J.L. Hepatocellular carcinoma cells loss lenvatinib efficacy in vitro through autophagy and hypoxia response-derived neuropilin-1 degradation. Acta Pharmacol. Sin. 2022, 1–17. [Google Scholar] [CrossRef]
- Takahashi, M.; Okada, K.; Ouch, R.; Konno, T.; Usui, K.; Suzuki, H.; Satoh, M.; Kogure, T.; Satoh, K.; Watanabe, Y.; et al. Fibronectin plays a major role in hypoxia-induced lenvatinib resistance in hepatocellular carcinoma PLC/PRF/5 cells. Pharmazie 2021, 76, 594–601. [Google Scholar]
- Hamaya, S.; Fujihara, S.; Iwama, H.; Fujita, K.; Shi, T.; Nakabayashi, R.; Mizuo, T.; Takuma, K.; Nakahara, M.; Oura, K.; et al. Characterization of Cisplatin Effects in Lenvatinib-resistant Hepatocellular Carcinoma Cells. Anticancer Res. 2022, 42, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Ao, J.; Chiba, T.; Shibata, S.; Kurosugi, A.; Qiang, N.; Ma, Y.; Kan, M.; Iwanaga, T.; Sakuma, T.; Kanzaki, H.; et al. Acquisition of mesenchymal-like phenotypes and overproduction of angiogenic factors in lenvatinib-resistant hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2021, 549, 171–178. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, D.; Wu, F.; Tu, J.; Song, J.; Xu, M.; Ji, J. Sophoridine suppresses lenvatinib-resistant hepatocellular carcinoma growth by inhibiting RAS/MEK/ERK axis via decreasing VEGFR2 expression. J. Cell Mol. Med. 2021, 25, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ma, Z.; Zhou, Q.; Wang, A.; Gong, Y.; Li, Z.; Wang, S.; Yan, Q.; Wang, D.; Hou, B.; et al. Genome-Wide CRISPR/Cas9 Library Screening Identified that DUSP4 Deficiency Induces Lenvatinib Resistance in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2022, 18, 4357–4371. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.L.; Yau, T.; Furuse, J.; Park, J.W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.H.; He, A.R.; Ryoo, B.Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Cochin, V.; Gross-Goupil, M.; Ravaud, A.; Godbert, Y.; Le Moulec, S. Cabozantinib: Mechanism of action, efficacy and indications. Bull. Cancer 2017, 104, 393–401. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Hessel, C.; Halabi, S.; Sanford, B.; Michaelson, M.D.; Hahn, O.; Walsh, M.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur. J. Cancer 2018, 94, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chen, Z.; Fang, W.; Ren, Z.; Xu, R.; Ryoo, B.Y.; Meng, Z.; Bai, Y.; Chen, X.; Liu, X.; et al. Pembrolizumab Versus Placebo as Second-Line Therapy in Patients From Asia With Advanced Hepatocellular Carcinoma: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2022, 1–13. [Google Scholar] [CrossRef]


{kind=link}
| Molecules | Expression | Major Effects | References |
|---|---|---|---|
| DNA methylation | |||
| MORC | Upregulated | MORC forms a complex with DNMT3 on the promoters of NF2 and KIBRA to cause DNA hypermethylation. | [164] |
| PRMT6 | - | PRMT6 methylates CRAF and inhibits FRAS/RAF binding ability, thereby altering ERK-mediated nuclear transport of PKM2 and reducing sorafenib resistance. | [166] |
| BNIP3 | Downregulated | Epigenetic silencing of BNIP3 is associated with sorafenib resistance. Promoter demethylation and restoration of BNIP3 can overcome sorafenib resistance. | [167] |
| Long non-coding RNAs | |||
| H19 | Downregulated | The promoter methylation of the H19 gene is associated with sorafenib resistance. Overexpression of H19 sensitizes HCC cells to sorafenib. | [165] |
| SNHG1 | Upregulated | Sorafenib induces miR-21 to enter the nucleus and promote SNHG1 expression, leading to activation of AKT pathway and contributing to sorafenib resistance in HCC cells. | [168] |
| SNHG3 | Upregulated | SNHG3 causes EMT of HCC cells through miR-128/CD151 cascade activation and is involved in sorafenib resistance. | [169] |
| SNHG16 | Upregulated | Knockdown of SNHG16, which is upregulated in sorafenib-resistant HCC, improves sensitivity to sorafenib and functions as endogenous sponge for miR-140-5p. | [170] |
| NEAT1 | Upregulated | NEAT1 negatively regulates miR-335 expression and inhibits the cMet-AKT pathway, which is associated with sorafenib resistance in HCC. | [171] |
| FOXD2-AS1 | Downregulated | FOXD2-AS1 functions as a sponge for miR-150-5p and contributes to sorafenib resistance in HCC by suppressing TMEM9. | [172] |
| MicroRNAs | |||
| miR-19a-3p | Upregulated | Aberrant expression of miR-19a-3p induces sorafenib resistance in HCC cells by regulating the PTEN/AKT pathway. | [86] |
| miR-181a | Upregulated | miR-181a induces sorafenib resistance in HCC via the suppression of RASSF1. | [173] |
| miR-221 | Upregulated | mir-221 exerts anti-apoptotic activity by targeting caspase-3 and is involved in sorafenib resistance in HCC. | [174] |
| miR-374b | Downregulated | In sorafenib-resistant HCC cells and xenograft mice, miR-375b overcomes drug resistance by suppressing the hnRNPA1/PKM2 axis. | [175] |
| miR-494 | Upregulated | Overexpression of miR-494 enhances sorafenib resistance in HCC cells by activating mTOR. | [176] |
| miR-622 | Downregulated | MiR-622 contributes to the abrogation of drug resistance in sorafenib-resistant HCC cells by targeting KRAS and inhibiting RAF/ERK and PI3K/AKT signaling. | [177] |
| Molecules | Expression | Major Effects | Reference |
|---|---|---|---|
| Epithelial–mesenchymal transition | |||
| Pin1 | Upregulated | Pin1 regulates the expression of EMT-related molecules such as E-cadherin and promotes HCC progression, invasion, and metastasis in regorafenib resistance of HCC. | [197] |
| Cell cycle | |||
| FOXM1 | Upregulated | FOXM1 is overexpressed in regorafenib-resistant HCC cells and elevated FOXM1 expression, correlating with drug resistance and decreased survival. | [199] |
| Apoptosis | |||
| TOP2A | Upregulated | Elevated TOP2A expression correlates with regorafenib resistance and poor prognosis in patients with HCC. | [200] |
| Others | |||
| TGF-β | Upregulated | Regorafenib-resistant HCC cells deactivate Wnt/β-catenin signaling and activate TGF-β signaling. Regorafenib resistance is restored by TGF-β type 1 receptor inhibition. | [148] |
| HK1 | Upregulated | Regorafenib-resistant HCC cells increase the expression of HK1, which catalyzes glucose metabolism, and HK1 expression correlates with drug resistance. | [201] |
| ATF3 | Upregulated | ATF3-mediated upregulation of IL-6α induces multifunctional cytokines and promotes regorafenib resistance against HCC cells. | [202] |
| SphK2 | Upregulated | Regorafenib-resistant HCC cells show high expression of SphK2. SphK2/S1P causes regorafenib resistance via the activation of NF-κB and STAT3. | [203] |
| Molecules | Expression | Major Effects | Reference |
|---|---|---|---|
| Non-coding RNAs | |||
| lncXIST | Upregulated | XIST promotes lenvatinib resistance in HCC cells via the activation of EZH2/NOD2/ERK axis. | [211] |
| lncAC026401.3 | Upregulated | AC026401.3 is upregulated in HCC tissues and correlates with poor prognosis in HCC patients. AC026401.3 interacts with OCT1 to activate E2F2 and enhances lenvatinib resistance in HCC. | [212] |
| miR-128-3p | Downregulated | Downregulation of miR-128-3p is involved in lenvatinib resistance via AKT and ERK. | [213] |
| circPAK1 | Upregulated | CircRAK1 is highly expressed in HCC cells and tissues and correlates with poor prognosis in HCC patients. CircPAK1 is transported by exosomes to induce lenvatinib resistance. | [214] |
| circMED27 | Upregulated | Serum circMED27 is significantly elevated in HCC patients, correlating with poor prognosis. Competing with miR655-3p, cirMED27 upregulates USP28 to promote lenvatinib resistance. | [215] |
| Transport processes | |||
| BCRP | Upregulated | BCRP transporter expression is elevated in lenvatinib-resistant HCC cells. Activation of EGFR-, MEK/ERK-, and PI3K/Akt-signaling pathways are associated with lenvatinib resistance. | [216] |
| ABCB1 | Upregulated | Activation of EGFR- and STAT3/ABCB1-signaling pathways and enhancement of exocytosis cause lenvatinib resistance in HCC cells. | [217] |
| Hypoxia | |||
| NRP1 | - | NRP1 gene silencing significantly enhances the anti-tumor effect of lenvatinib resistance. HIF1α directly regulates NRP1 expression in hypoxic microenvironment. | [218] |
| HIF-1α | Upregulated | Under hypoxic conditions, transcription factors, including HIF-1α, are induced, increasing fibronectin expression, leading to lenvatinib resistance in HCC. | [219] |
| Others | |||
| ERK1 | Upregulated | Activation of ERK1 signaling is observed in lenvatinib-resistant HCC cells. Cisplatin shows an effective anti-tumor effect on lenvatinib-resistant HCC cells via ATM/ATR-Chk1/Chk2 signaling. | [220] |
| ERK | Upregulated | Activation of MAPK/MEK/ERK signaling and increased expression of EMT markers are observed in lenvatinib-resistant HCC cells. | [221] |
| ERK | Upregulated | VEGFR2 expression and its downstream RAS/MEK/ERK signaling are elevated in renvatinib-resistant HCC cells. | [222] |
| ERK | Upregulated | Knockout of DUSP4, a gene associated with lenvatinib resistance, activates ERK/MEK signaling at the phosphorylation level and induces lenvatinib resistance in HCC cells. | [223] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oura, K.; Morishita, A.; Hamaya, S.; Fujita, K.; Masaki, T. The Roles of Epigenetic Regulation and the Tumor Microenvironment in the Mechanism of Resistance to Systemic Therapy in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 2805. https://doi.org/10.3390/ijms24032805
Oura K, Morishita A, Hamaya S, Fujita K, Masaki T. The Roles of Epigenetic Regulation and the Tumor Microenvironment in the Mechanism of Resistance to Systemic Therapy in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2023; 24(3):2805. https://doi.org/10.3390/ijms24032805
Chicago/Turabian StyleOura, Kyoko, Asahiro Morishita, Sae Hamaya, Koji Fujita, and Tsutomu Masaki. 2023. "The Roles of Epigenetic Regulation and the Tumor Microenvironment in the Mechanism of Resistance to Systemic Therapy in Hepatocellular Carcinoma" International Journal of Molecular Sciences 24, no. 3: 2805. https://doi.org/10.3390/ijms24032805