
New Piperazine Derivatives of 6-Acetyl-7-hydroxy-4-methylcoumarin as 5-HT1A Receptor Agents
 , ,
, ,
Abstract
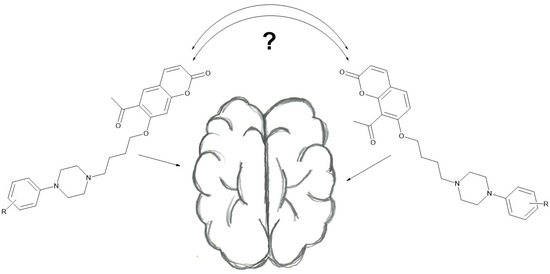
:
1. Introduction
2. Results and Discussion
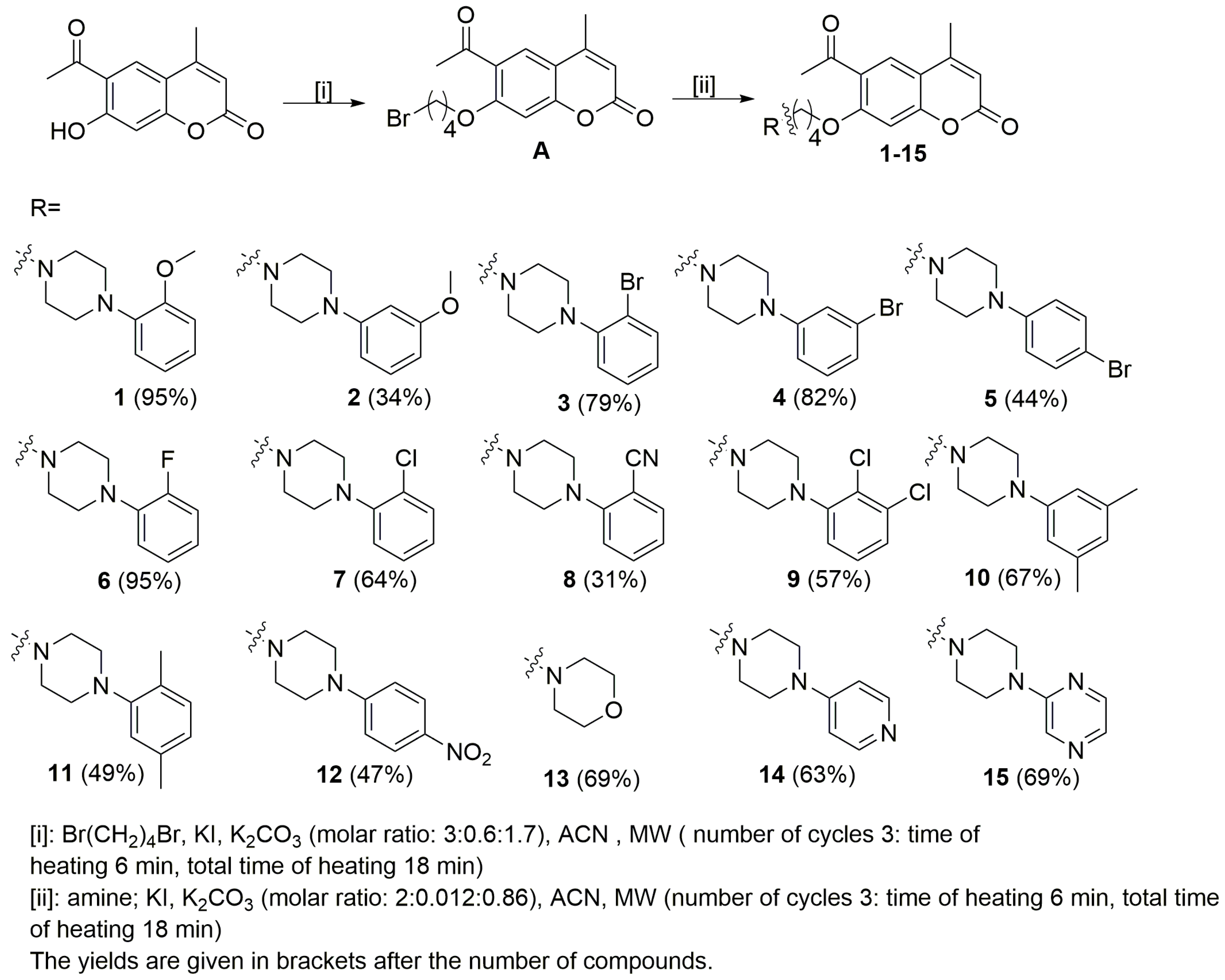
2.1. Chemistry
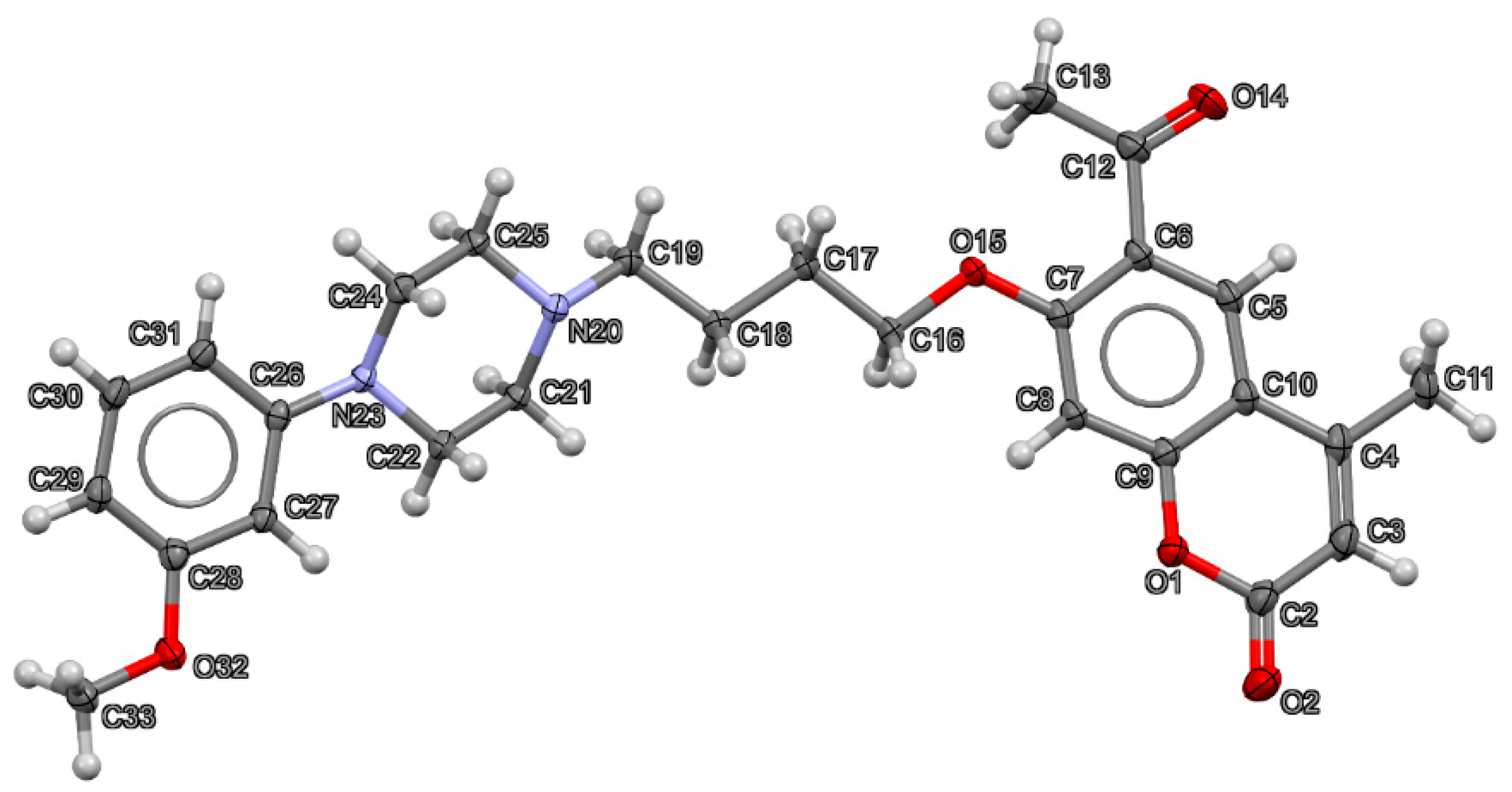
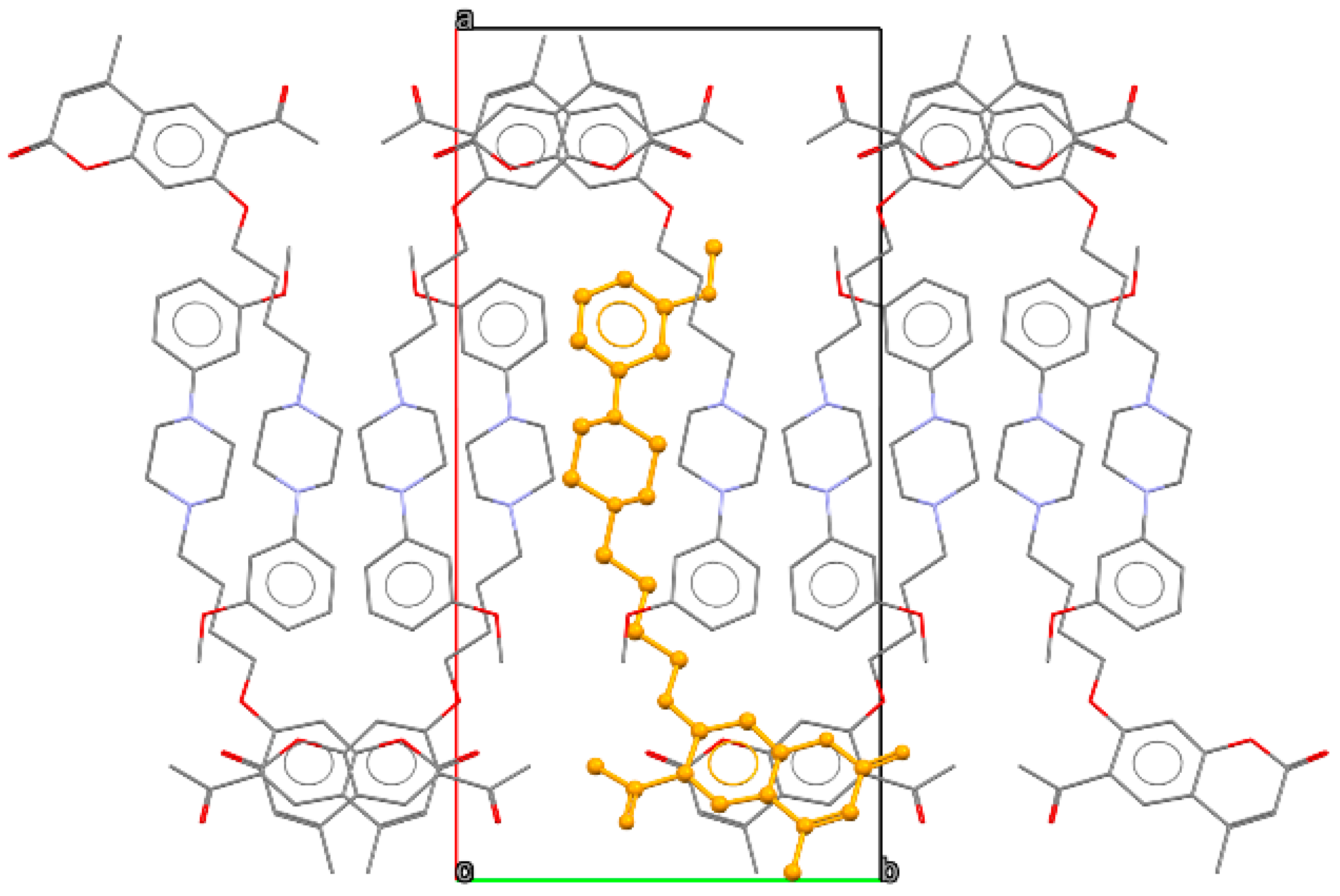
2.2. X-ray Crystallography
2.3. Computational Studies
2.3.1. ADME Properties
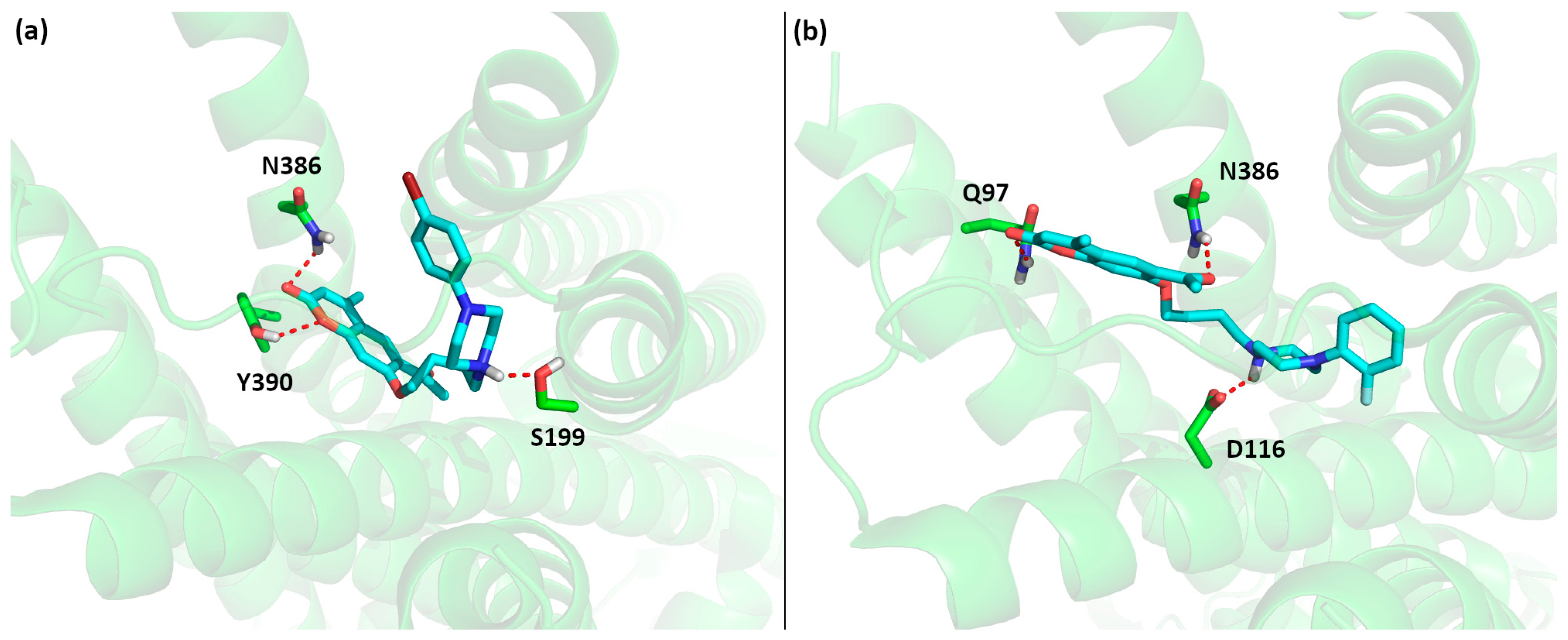
2.3.2. Molecular Docking
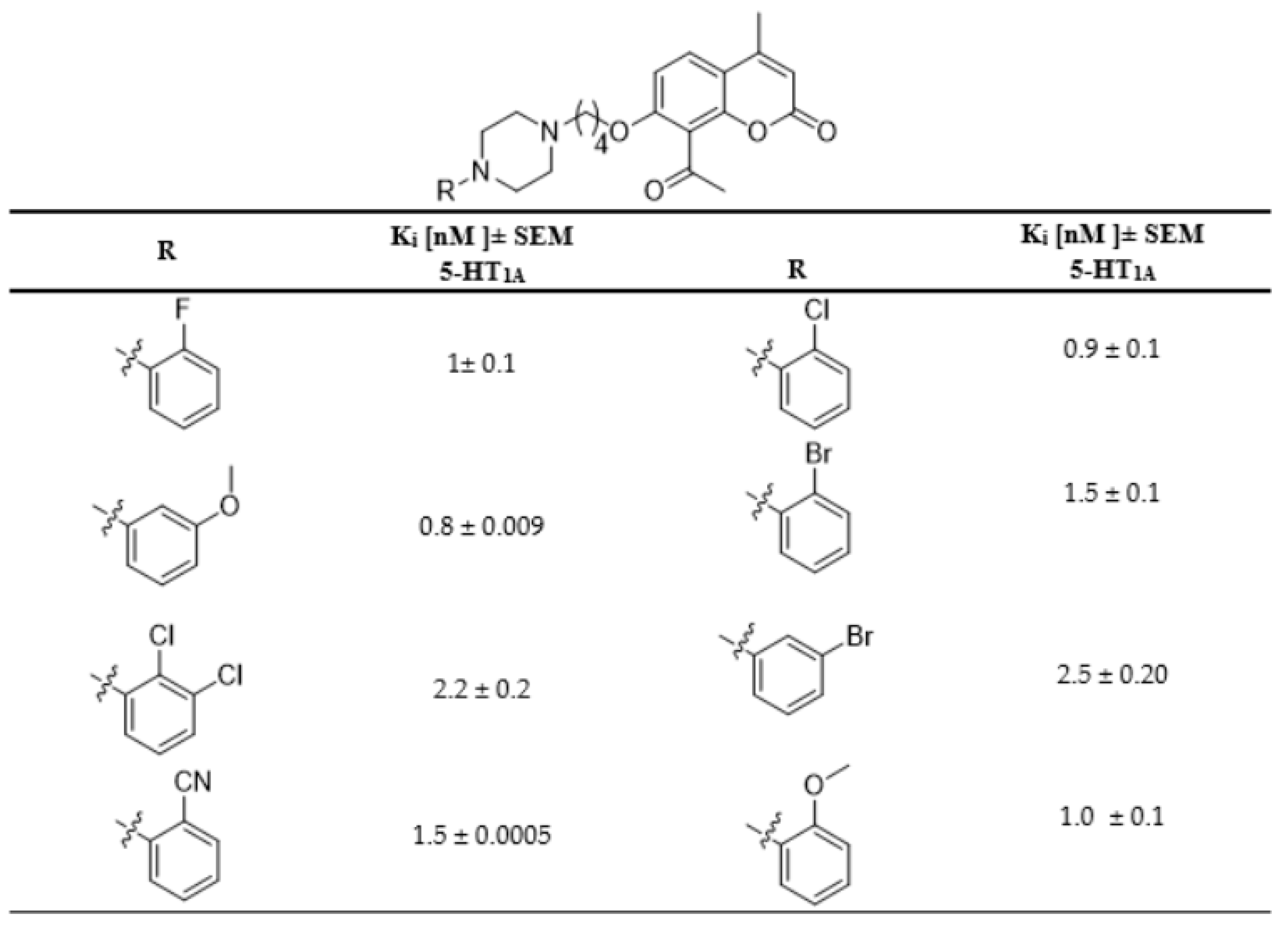
2.4. Biological Evaluation
3. Materials and Methods
3.1. Chemistry
- 6-Acetyl-7-(4-bromobutoxy)-4-methylchromen-2-one (A)
- 6-Acetyl-7-{4-[4-(2-methoxyphenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (1)
- 6-Acetyl-7-{4-[4-(3-methoxyphenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (2)
- 6-Acetyl-7-{4-[4-(2-bromophenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (3)
- 6-Acetyl-7-{4-[4-(3-bromophenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (4)
- 6-Acetyl-7-{4-[4-(4-bromophenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (5)
- 6-Acetyl-7-{4-[4-(2-fluorophenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (6)
- 6-Acetyl-7-{4-[4-(2-chlorophenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (7)
- 6-Acetyl-7-{4-[4-(2-cyanophenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (8)
- 6-Acetyl-7-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (9)
- 6-Acetyl-7-{4-[4-(3,5-dimethylphenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (10)
- 6-Acetyl-7-{4-[4-(2,5-dimethylphenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (11)
- 6-Acetyl-7-{4-[4-(4-nitrophenyl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (12)
- 6-Acetyl-7-[4-(morpholin-4-yl)butoxy]-4-methylchromen-2-one (13)
- 6-Acetyl-7-{4-(4-piridin)piperazin-1-yl}butoxy}-4-methylchromen-2-one (14)
- 6-Acetyl-7-{4-[4-(pyrazin-2-yl)piperazin-1-yl]butoxy}-4-methylchromen-2-one (15)
3.2. X-ray Crystallography
3.3. Biological Evaluation
3.3.1. Membrane Preparation
3.3.2. Competitive 5-HT1A and 5-HT2A Binding Assays
3.3.3. 5-HT1A Receptor Activation in the [35S]GTP-γ-S Assay
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corvino, A.; Fiorino, F.; Severino, B.; Saccone, I.; Frecentese, F.; Perissutti, E.; Di Vaio, P.; Santagada, V.; Caliendo, G.; Magli, E. The Role of 5-HT1A Receptor in Cancer as a New Opportunity in Medicinal Chemistry. Curr. Med. Chem. 2018, 25, 3214–3227. [Google Scholar] [CrossRef]
- Nichols, D.E.; Nichols, C.D. Serotonin Receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef] [PubMed]
- Amidfar, M.; Colic, L.; Walter, M.; Kim, Y.K. Biomarkers of Major Depression Related to Serotonin Receptors. Curr. Psychiatry Rev. 2018, 14, 239–244. [Google Scholar] [CrossRef]
- Rojas, P.; Fiedler, J.L. What Do We Really Know About 5-HT1A Receptor Signaling in Neuronal Cells? Front. Cell. Neurosci. 2016, 10, 272. [Google Scholar] [CrossRef]
- Asarch, K.B.; Ransom, R.W.; Shih, J.C. 5-HT-la and 5-HT-lb selectivity of two phenylpiperazine derivatives: Evidence for 5-HT-lb heterogeneity. Life Sci. 1985, 36, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Maj, J.; Chojnacka-Wójcik, E.; Kłodzińska, A.; Dereń, A.; Moryl, E. Hypothermia induced by m-trifluoromethylphenylpiperazine or m-chlorophenylpiperazine: An effect mediated by 5-HT1B receptors? J. Neural Transm. 1988, 73, 43–55. [Google Scholar] [CrossRef]
- Sylte, I.; Chilmończyk, Z.; Dahl, S.G.; Cybulski, J.; Edvardsen, O. The Ligand-binding Site of Buspirone Analogues at the 5-HT1A Receptor. J. Pharm. Pharmacol. 1997, 49, 698–705. [Google Scholar] [CrossRef]
- Ostrowska, K. Coumarin-piperazine derivatives as biologically active compounds. Saudi Pharm. J. 2020, 28, 220–232. [Google Scholar] [CrossRef]
- Ostrowska, K.; Młodzikowska, K.; Głuch-Lutwin, M.; Gryboś, A.; Siwek, A. Synthesis of a new series of aryl/heteroarylpiperazinyl derivatives of 8-acetyl-7-hydroxy-4-methylcoumarin with low nanomolar 5-HT1A affinities. Eur. J. Med. Chem. 2017, 137, 108–116. [Google Scholar] [CrossRef]
- Ostrowska, K.; Grzeszczuk, D.; Głuch-Lutwin, M.; Gryboś, A.; Siwek, A.; Leśniak, A.; Sacharczuk, M.; Trzaskowski, B. 5-HT1A and 5-HT2A receptors affinity, docking studies and pharmacological evaluation of a series of 8-acetyl-7-hydroxy-4-methylcoumarin derivatives. Bioorg. Med. Chem. 2018, 26, 527–535. [Google Scholar] [CrossRef]
- Ostrowska, K.; Grzeszczuk, D.; Głuch-Lutwin, M.; Gryboś, A.; Siwek, A.; Dobrzycki, Ł.; Trzaskowski, B. Development of selective agents targeting serotonin 5-HT1A receptors with subnanomolar activities based on a coumarin core. MedChemComm 2017, 8, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Żołek, T.; Enyedy, E.A.; Ostrowska, K.; Posa, V.; Maciejewska, D. Drug likeness prediction of 5-hydroxy-substituted coumarins with high affinity for5-HT1A and 5-HT2A receptors. Eur. J. Pharm. Sci. 2018, 115, 25–36. [Google Scholar] [CrossRef]
- Żołek, T.; Domotor, O.; Ostrowska, K.; Enyedy, E.A.; Maciejewska, D. Evaluation of blood-brain barrier penetration and examination of binding to human serum albumin of 7-O-arylpiperazinylcoumarins as potential antipsychotic agents. Bioorg. Chem. 2019, 84, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, K.; Leśniak, A.; Karczyńska, U.; Jeleniewicz, P.; Głuch-Lutwin, M.; Mordyl, B.; Siwek, A.; Trzaskowski, B.; Sacharczuk, M.; Bujalska-Zadrozny, M. 6-Acetyl-5-hydroxy-4,7-dimethylcoumarin derivatives: Design, synthesis, modeling studies, 5-HT1A, 5-HT2A and D2 receptors affinity. Bioorg. Chem. 2020, 100, 103912. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, K.; Grzeszczuk, D.; Maciejewska, D.; Młynarczuk-Biały, I.; Czajkowska, A.; Sztokfisz, A.; Dobrzycki, L.; Kruszewska, H. Synthesis and biological screening of a new series of 5-[4-(4-aryl-1-piperazinyl)butoxy]coumarins. Monats. Chem. 2016, 147, 1615–1627. [Google Scholar] [CrossRef]
- Ostrowska, K.; Leśniak, A.; Czarnocka, Z.; Chmiel, J.; Bujalska-Zadrożny, M.; Trzaskowski, B. Design, Synthesis and Biological Evaluation of a Series of 5- and 7-Hydroxycoumarin Derivatives as 5-HT1A Serotonin Receptor Antagonists. Pharmaceuticals 2021, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Hejchman, E.; Kruszewska, H.; Maciejwska, D.; Sowirka-Taciak, B.; Tomczyk, M.; Sztokfisz-Ignasiak, A.; Jankowski, J.; Mlynarczuk-Biały, I. Design, synthesis, and biological activity of Schiff bases bearing salicyl and 7-hydroxycoumarinyl moieties. Monats. Chem. 2019, 150, 255–266. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Matsson, P.; Doak, B.C.; Over, B.; Kihlber, J. Cell permeability beyond the rule of 5. Adv. Drug. Deliver. Rev. 2016, 101, 42–61. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: Prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.-X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, A Novel Atypical Antipsychotic Drug with a Unique and Robust Pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Keck, P.E., Jr.; McElroy, S.L. Aripiprazole: A partial dopamine D2 receptor agonist antipsychotic. Expert Opin. Investig. Drug. 2003, 12, 655–662. [Google Scholar]
- Kroeze, W.K.; Hufeisen, S.J.; Popadak, B.A.; Renock, S.M.; Steinberg, S.; Ernsberger, P.; Jayathilake, K.; Meltzer, H.Y.; Roth, B.L. H1-Histamine Receptor Affinity Predicts Short-Term Weight Gain for Typical and Atypical Antipsychotic Drugs. Neuropsychopharmacology 2003, 28, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.B.; Thompson, D.C.; Rai, B.K.; Baber, J.C.; Fan, K.Y.; Hu, Y.; Humbler, C. Comparison of Several Molecular Docking Programs: Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2009, 49, 1455–1474. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef]
- Bruker Nano Analitycs. APEX3 V2019; Bruker Nano, Inc.: Billerica, MA, USA, 2019. [Google Scholar]
- Bruker Nano Analitycs. SAINT V8.40A; Bruker Nano, Inc.: Billerica, MA, USA, 2019. [Google Scholar]
- Bruker Nano Analitycs. SADABS V2016/2; Bruker Nano, Inc.: Billerica, MA, USA, 2019. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Wilson, A.J.C. (Ed.) International Tables for Crystallography; Kluwer: Dordrecht, The Netherlands, 1992; Volume C. [Google Scholar]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar]
- Xu, P.; Huang, S.; Zhang, H.; Mao, C.; Zhou, X.E.; Cheng, X.; Simon, I.A.; Shen, D.-D.; Yen, H.-Y.; Robinson, C.V.; et al. Structural insights into the lipid and ligand regulation of serotonin receptors. Nature 2021, 592, 469–486. [Google Scholar] [CrossRef]
- Cao, D.; Yu, J.; Wang, H.; Luo, Z.; Liu, X.; He, L.; Qi, J.; Fan, L.; Tang, L.; Chen, Z.; et al. Structure-based discovery of nonhallucinogenic psychedelic analogs. Science 2022, 375, 403–411. [Google Scholar] [CrossRef]
- Chen, Z.; Fan, L.; Wang, H.; Yu, J.; Lu, D.; Qi, J.; Nie, F.; Luo, Z.; Liu, Z.; Cheng, J.; et al. Structure-based design of a novel third-generation antipsychotic drug lead with potential antidepressant properties. Nat. Neurosci. 2022, 25, 39–49. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | C27H32N2O5 |
|---|---|
| Mx/g mol−1 | 464.54 |
| T/K | 130.0(5) |
| λ/Å | 0.71073 |
| Crystal size | 0.098 mm × 0.334 mm × 0.607 mm |
| Space group | P21/c |
| Unit cell dimensions | a = 24.3110(12) Å b = 12.0631(6) Å, β = 96.000(2) c = 8.0498(4) Å |
| V/Å3, Z | 2347.8(2), 4 |
| Dx/g cm−3 | 1.314 |
| μ/mm−1 | 0.091 |
| F(000) | 992 |
| θmin, θmax | 2.53°, 28.50° |
| Index ranges | −32 ≤ h ≤ 32, −16 ≤ k ≤ 16, −10 ≤ l ≤ 10 |
| Reflections collected/independent | 62369/5948 (Rint = 0.0278) |
| Completeness | 99.9% |
| Absorption correction | Multi-Scan |
| Tmax, Tmin | 0.991, 0.947 |
| Refinement method | Full-matrix LSQ on F2 |
| Data/restraints/parameters | 5948/0/311 |
| GOF on F2 | 1.031 |
| Final R indices | 5240 data; I > 2σ(I) R1 = 0.0368, wR2 = 0.1005 all data R1 = 0.0425, wR2 = 0.1058 |
| Extinction coefficient | 0.0014(4) |
| Δρmax, Δρmin | 0.355 eÅ−3, −0.217 eÅ−3 |
| Compound | MW | Dipole | SASA | Volume | dHB | aHB | logP | metab | Ro5 | Ro3 | pKa |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 464.6 | 4.0 | 799.3 | 1465.5 | 0 | 9 | 3.37 | 5 | 0 | 0 | 7.55 |
| 2 | 464.6 | 4.4 | 787.7 | 1457.4 | 0 | 9 | 3.29 | 4 | 0 | 0 | 7.55 |
| 3 | 513.4 | 4.8 | 796.9 | 1455.2 | 0 | 8.25 | 3.87 | 4 | 1 | 0 | 7.38 |
| 4 | 513.4 | 4.8 | 790.0 | 1446.9 | 0 | 8.25 | 3.90 | 3 | 1 | 0 | 7.39 |
| 5 | 513.4 | 4.2 | 789.7 | 1446.5 | 0 | 8.25 | 3.89 | 3 | 1 | 0 | 7.49 |
| 6 | 452.5 | 4.7 | 762.8 | 1400.4 | 0 | 8.25 | 3.44 | 3 | 0 | 0 | 7.55 |
| 7 | 469.0 | 4.9 | 798.5 | 1452.8 | 0 | 8.25 | 3.84 | 4 | 0 | 0 | 7.39 |
| 8 | 459.5 | 10.2 | 796.6 | 1469.0 | 0 | 9.75 | 2.72 | 3 | 0 | 0 | 7.55 |
| 9 | 503.4 | 5.6 | 818.5 | 1492.5 | 0 | 8.25 | 4.28 | 4 | 1 | 0 | 7.39 |
| 10 | 462.6 | 3.6 | 826.2 | 1516.0 | 0 | 8.25 | 3.98 | 6 | 0 | 0 | 7.63 |
| 11 | 462.6 | 3.8 | 811.1 | 1502.4 | 0 | 8.25 | 3.92 | 5 | 0 | 0 | 7.63 |
| 12 | 479.5 | 13.1 | 801.6 | 1469.4 | 0 | 9.25 | 2.55 | 4 | 0 | 1 | 7.27 |
| 13 | 359.4 | 3.6 | 642.4 | 1157.3 | 0 | 8.95 | 1.07 | 4 | 0 | 0 | 7.37 |
| 14 | 435.5 | 4.5 | 754.7 | 1382.4 | 0 | 9.75 | 2.27 | 5 | 0 | 0 | 6.86 |
| 15 | 436.5 | 3.6 | 754.3 | 1378.5 | 0 | 10.25 | 1.91 | 6 | 0 | 0 | 7.44 |
| aripiprazole | 448.4 | 7.8 | 709.0 | 1318.3 | 1 | 6.25 | 4.43 | 5 | 0 | 0 | 7.39 |
| ketanserin | 395.4 | 7.8 | 699.4 | 1235.5 | 1 | 7.5 | 2.93 | 3 | 0 | 0 | 6.45 |
| 8-OH-DPAT | 247.4 | 1.0 | 556.1 | 959.5 | 1 | 2.75 | 3.46 | 4 | 0 | 0 | 9.40 |
| WAY 100635 | 422.6 | 3.9 | 761.8 | 1413.2 | 0 | 8.25 | 3.95 | 6 | 0 | 0 | 6.34 |
| 5-HT1A Affinity | 5-HT2A Affinity | |||
|---|---|---|---|---|
| Compound | Ki (nM) Local Search | Ki (nM) Flexible Docking | Ki (nM) Local Search | Ki (nM) Flexible Docking |
| 1 | 25.00 | 3.91 | 0.76 | 98.68 |
| 2 | 50.71 | 1.83 | 0.87 | 12.41 |
| 3 | 26.75 | 4.90 | 0.49 | 54.84 |
| 4 | 21.88 | 1.28 | 0.31 | 36.10 |
| 5 | 70.79 | 0.62 | 0.74 | 26.48 |
| 6 | 66.33 | 0.67 | 0.91 | 244.04 |
| 7 | 31.06 | 0.67 | 0.52 | 9.18 |
| 8 | 26.33 | 4.43 | 0.40 | 10.55 |
| 9 | 23.60 | 0.73 | 0.34 | 2.85 |
| 10 | 18.43 | 0.76 | 0.67 | 10.41 |
| 11 | 18.83 | 0.69 | 0.30 | 7.49 |
| 12 | 176.44 | 1.08 | 3.01 | 48.36 |
| 13 | 3190 | 66.64 | 36.17 | 374.04 |
| 14 | 114.61 | 0.66 | 1.28 | 66.48 |
| 15 | 193.72 | 6.00 | 2.65 | 38.20 |
| aripiprazole | 45.58 | - | 0.85 | - |
| 5-HT1A Affinity | 5-HT2A Affinity | |||
|---|---|---|---|---|
| Compound | pKi (nM ± SEM) | Ki (nM, 95% CI) | pKi | Ki (nM, 95% CI) |
| 1 | 8.24 ± 0.13 | 5.75 (3.1–10.7) | 5.85 ± 0.07 | 1422 (1007–2008) |
| 2 | 7.89 ± 0.1 | 12.9 (8.0–20.8) | 5.93 ± 0.07 | 1160 (819–1644) |
| 3 | 8.71 ± 0.18 | 1.96 (0.8–4.7) | 5.99 ± 0.06 | 1018 (752–1380) |
| 4 | 9.12 ± 0.12 | 0.78 (0.4–1.4) | 5.93 ± 0.13 | 1164 (613–2213) |
| 5 | 8.85 ± 0.2 | 1.40 (0.5–3.7) | 5.87 ± 0.1 | 1347 (775–2339) |
| 6 | 8.97 ± 0.22 | 1.04 (0.3–3.1) | 5.83 ± 0.14 | 1467 (755–2848) |
| 7 | 9.26 ± 0.17 | 0.57 (0.2–1.3) | 5.68 ± 0.14 | 2079 (1005–4302) |
| 8 | 8.03 ± 0.11 | 9.44 (5.4–17.9) | 6.15 ± 0.09 | 705 (453–1099) |
| 9 | 8.20 ± 0.17 | 6.30 (1.8–21.2) | 5.91 ± 0.13 | 1286 (732–2260) |
| 10 | 7.65 ± 0.15 | 22.37 (9.8–51.3) | 6.29 ± 0.08 | 516 (355–7501) |
| 11 | 6.76 ± 0.16 | 173.6 (79.2–393.2) | 5.40 ± 0.09 | 3712 (2350–5903) |
| 12 | 5.75 ± 0.13 | 1800 (949.8–3535) | 5.81 ± 0.13 | 1503 (810–2787) |
| 13 | 5.02 ± 0.18 | 9617 (4172–21590) | 5.2 ± 0.15 | 6393 (3103–13170) |
| 14 | 5.78 ± 0.10 | 1658 (972.3–2880) | 5.47 ± 0.10 | 3359 (2019–5588) |
| 15 | 7.6 ± 0.12 | 25 (12.1–51.0) | 5.2 ± 0.17 | 6597 (2855–15240) |
| 8-OH-DPAT | 9.59 ± 0.12 | 0.25 (0.097–0.66) | - | - |
| ketanserin | - | - | 8.87 ± 0.07 | 1.33 (0.57–3.1) |
| 5-HT1A Receptor Inhibition | 5-HT1A Receptor Stimulation | ||||
|---|---|---|---|---|---|
| Compound | pIC50 (nM ± SEM) | IC50 (nM, 95% CI) | pEC50 (nM ± SEM) | EC50 (nM, 95% CI) | Emax (%) |
| 1 | 6.5 ± 0.08 | 301 (206–442) | - | - | - |
| 2 | - | - | 6.7 ± 0.05 | 181 (140–234) | 146 ± 1.1 |
| 3 | 5.3 ± 0.09 | 5181 (3823–7021) | - | - | - |
| 4 | - | - | 6.95 ± 0.04 | 113 (94–135) | 176 ± 1.2 |
| 5 | 4.2 ± 0.08 | 63,200 (43,770–91,250) | - | - | - |
| 6 | - | - | 6.99 ± 0.06 | 101 (78–133) | 139 ± 1.1 |
| 7 | - | - | 7.3 ± 0.11 | 49 (29–82) | 130 ± 1.4 |
| 8 | - | - | 7.2 ± 0.11 | 59 (35–99) | 124 ± 1.1 |
| 9 | - | - | 6.8 ± 0.08 | 149 (102–218) | 133 ± 1.3 |
| 10 | - | - | 6.3 ± 0.07 | 475 (334–677) | 139 ± 1.4 |
| 11 | 4.7 ± 0.05 | 21,860 (16,950–28,190) | - | - | - |
| 12 | 4.27 ± 0.7 | 53,360 (32,150–88,570) | - | - | - |
| 13 | 4.03 ± 0.85 | 93,380 (70,480–123,700) | - | - | - |
| 14 | 4.6 ± 0.13 | 25,020 (13,790–45,400) | - | - | - |
| 15 | - | - | 5.9 ± 0.03 | 1295 (1102–1522) | 170 ± 1.3 |
| 8-OH-DPAT | - | - | 7.4 ± 0.04 | 38 (31–45) | 177 ± 1.2 |
| WAY 100635 | 7.8 ± 0.15 | 15 (7–31) | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostrowska, K.; Leśniak, A.; Gryczka, W.; Dobrzycki, Ł.; Bujalska-Zadrożny, M.; Trzaskowski, B. New Piperazine Derivatives of 6-Acetyl-7-hydroxy-4-methylcoumarin as 5-HT1A Receptor Agents. Int. J. Mol. Sci. 2023, 24, 2779. https://doi.org/10.3390/ijms24032779
Ostrowska K, Leśniak A, Gryczka W, Dobrzycki Ł, Bujalska-Zadrożny M, Trzaskowski B. New Piperazine Derivatives of 6-Acetyl-7-hydroxy-4-methylcoumarin as 5-HT1A Receptor Agents. International Journal of Molecular Sciences. 2023; 24(3):2779. https://doi.org/10.3390/ijms24032779
Chicago/Turabian StyleOstrowska, Kinga, Anna Leśniak, Weronika Gryczka, Łukasz Dobrzycki, Magdalena Bujalska-Zadrożny, and Bartosz Trzaskowski. 2023. "New Piperazine Derivatives of 6-Acetyl-7-hydroxy-4-methylcoumarin as 5-HT1A Receptor Agents" International Journal of Molecular Sciences 24, no. 3: 2779. https://doi.org/10.3390/ijms24032779