

From the Catastrophic Objective Irreproducibility of Cancer Research and Unavoidable Failures of Molecular Targeted Therapies to the Sparkling Hope of Supramolecular Targeted Strategies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Fundamentally Low Reproducibility in Cancer Research
2. Heterogeneity within Cancer Cells and in Their Interactions with Microenvironment
3. Tumor Micro Environment (TME) and Its Immunological Components (TIME) Heterogeneity
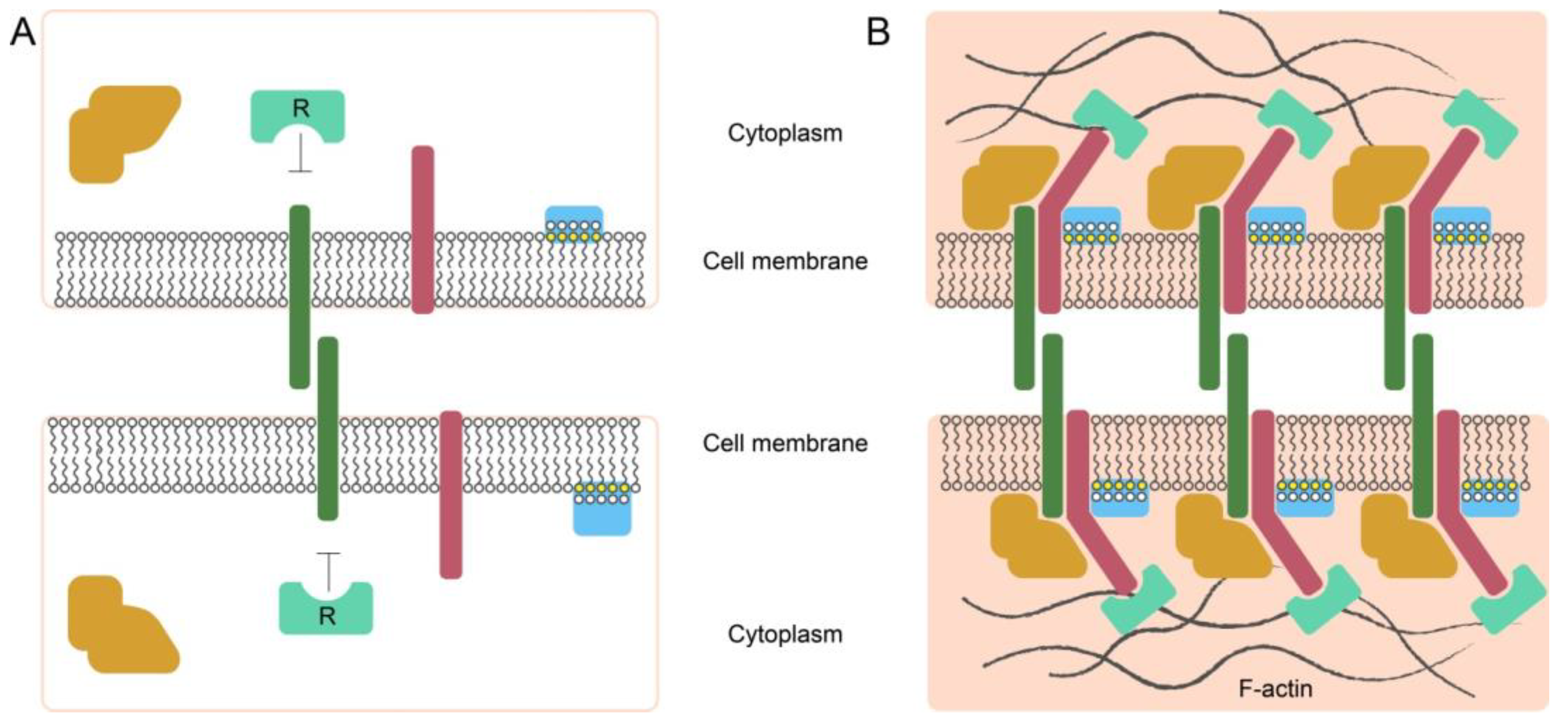
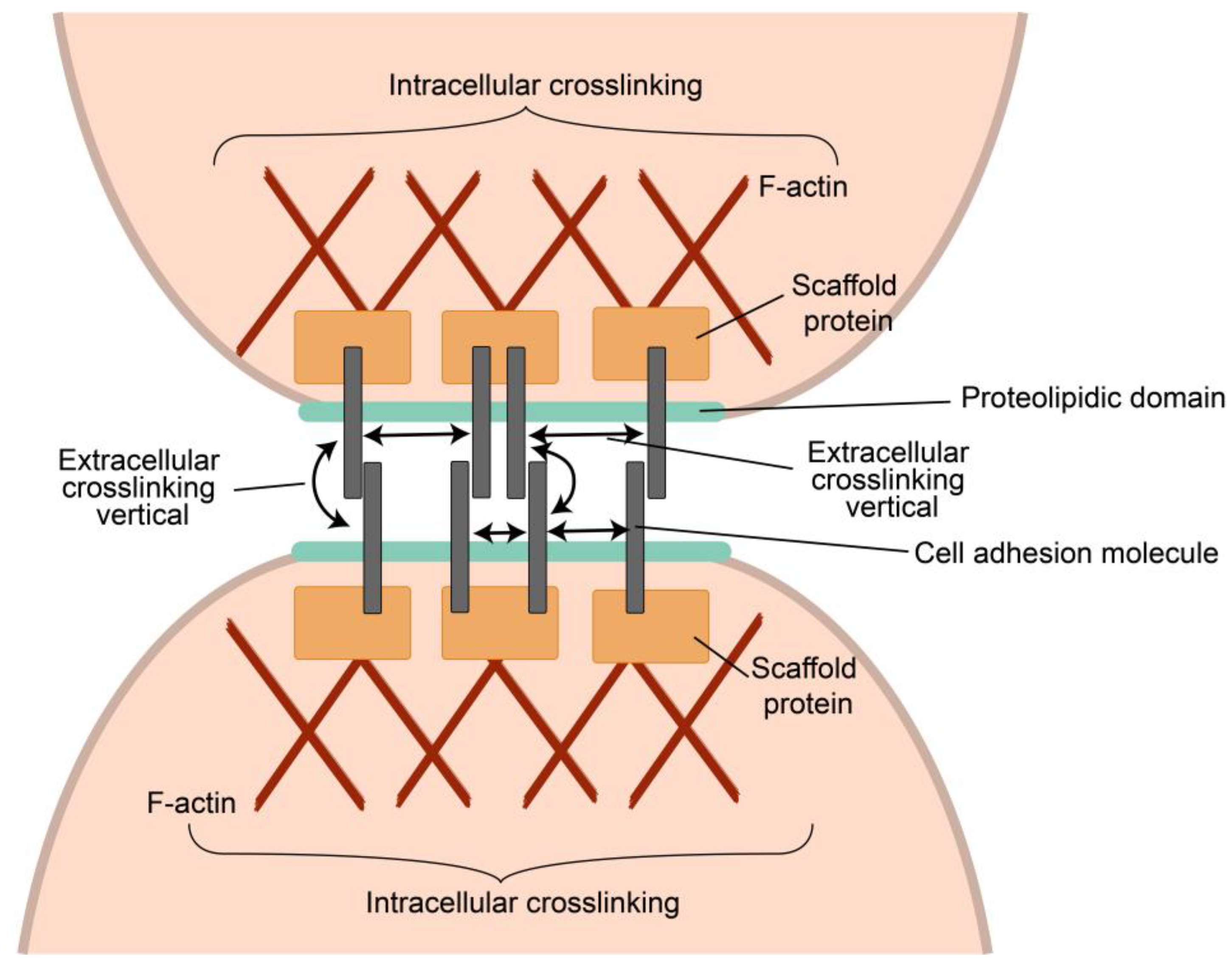
4. General Principles of Intercellular Interactions
4.1. Synapses as a Way of Communications of Tumor and Its TIME
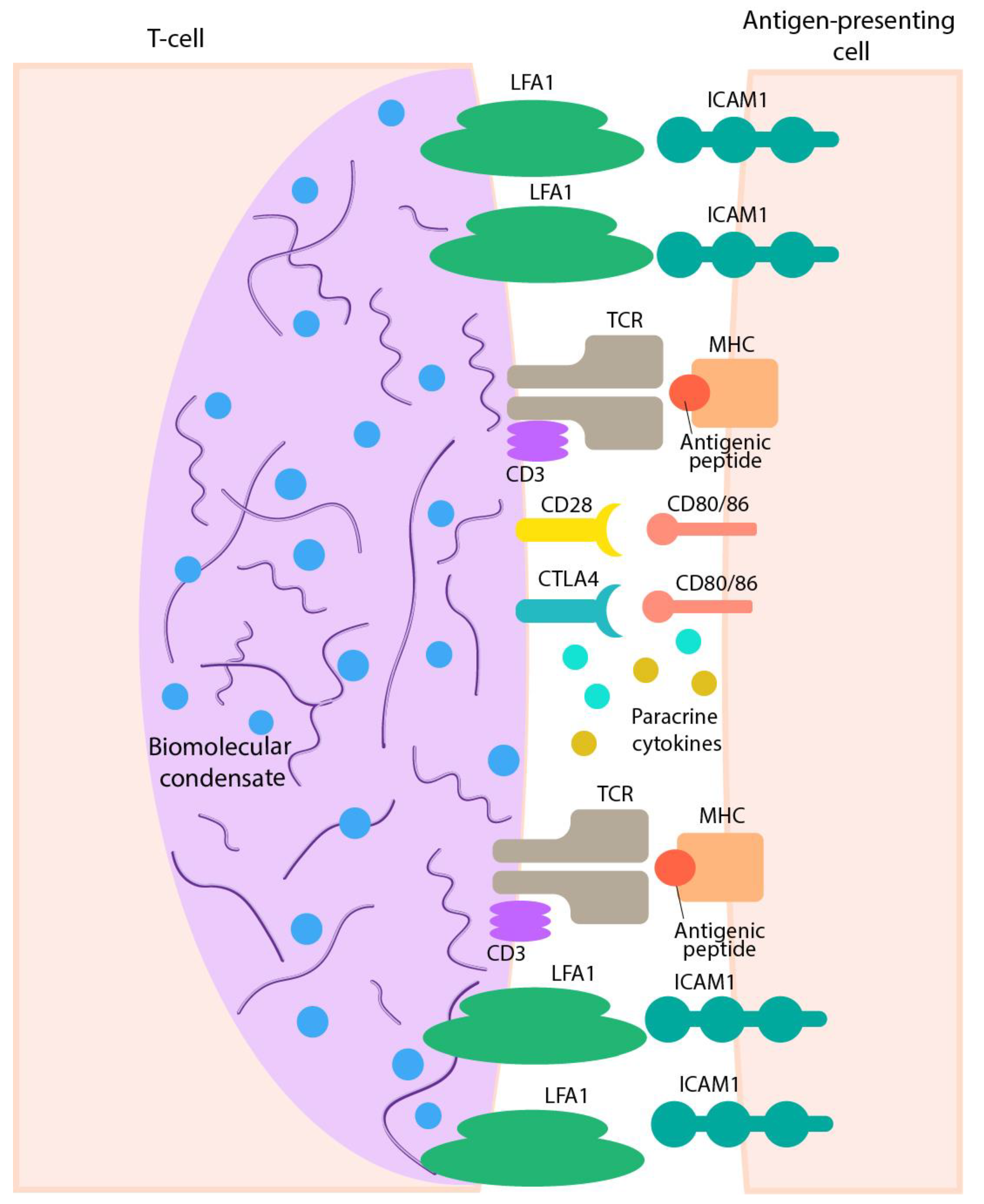
4.2. Immunological Synapse as a Classical Example of Cell–Cell Functionally Efficient Interactions

4.3. Other Systems also Use Synapse-like Contacts for Cell–Cell Communication
4.3.1. Ephrin Type-A Receptor 2 (EphA2)/EphrinA1 System
4.3.2. Phagocytic Synapse-like Behavior
4.3.3. Viral (Virological) Synapses
4.3.4. Gap Junctions
5. Metastasis
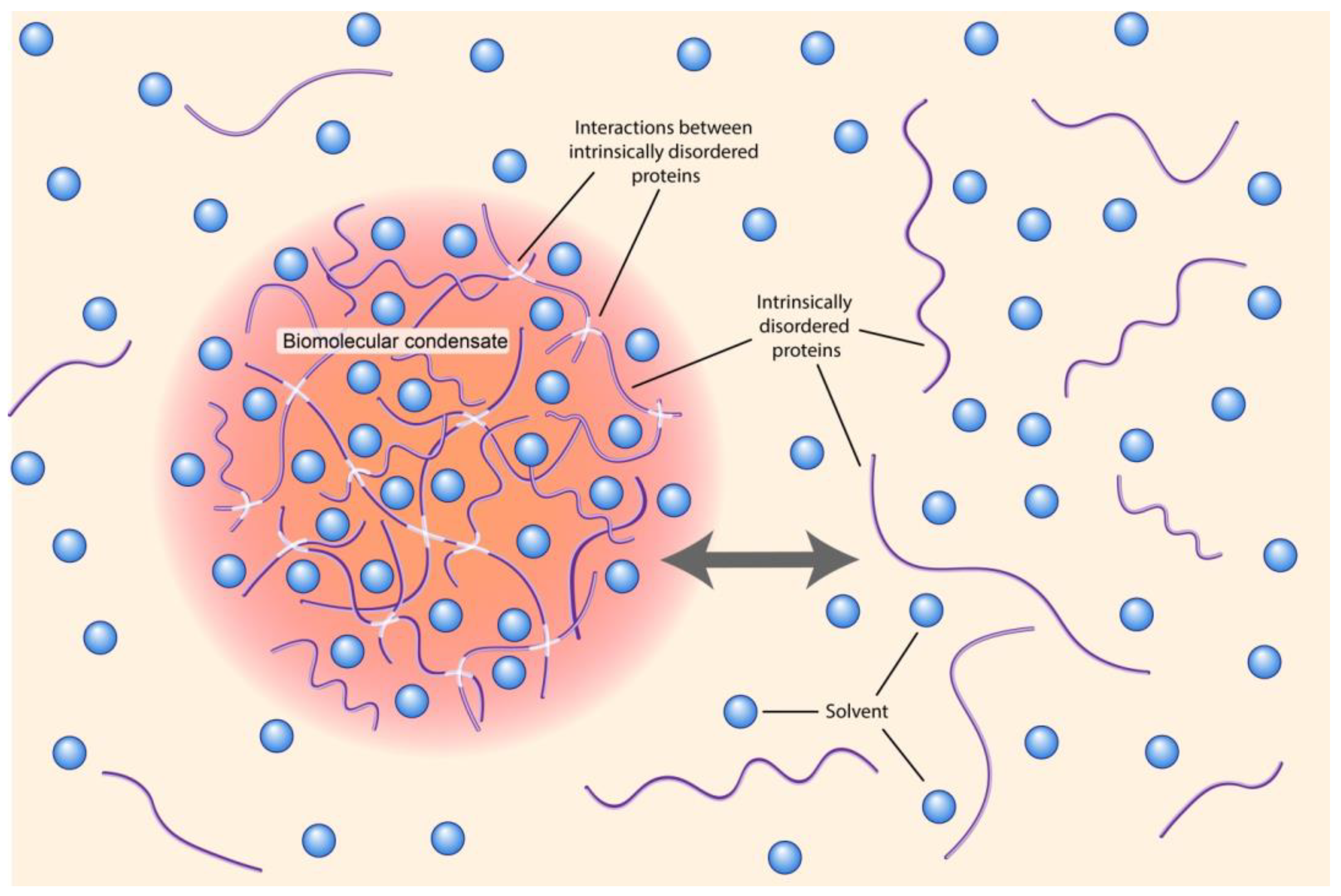
6. Biomolecular Condensates (Membrane Less Compartments)
7. P.S. Overcautious Fear
8. Instead of a Conclusion: Persistent Therapeutic Supramolecular Targets in Cancer’s Fickle Microcosm
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hannun, Y.A. Build a registry of results that students can replicate. Nature 2021, 600, 571. [Google Scholar] [CrossRef] [PubMed]
- Amaral, O.B.; Neves, K. Reproducibility: Expect less of the scientific paper. Nature 2021, 597, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Offord, C. Study Finds Reproducibility Issues in High-Impact Cancer Papers. Available online: https://www.the-scientist.com/news-opinion/study-finds-reproducibility-issues-in-high-impact-cancer-papers-69490 (accessed on 3 October 2022).
- Errington, T.M.; Mathur, M.; Soderberg, C.K.; Denis, A.; Perfito, N.; Iorns, E.; Nosek, B.A. Investigating the replicability of preclinical cancer biology. eLife 2021, 10, e71601. [Google Scholar] [CrossRef]
- Errington, T.M.; Denis, A.; Perfito, N.; Iorns, E.; Nosek, B.A. Challenges for assessing replicability in preclinical cancer biology. eLife 2021, 10, e67995. [Google Scholar] [CrossRef]
- Errington, T.M.; Denis, A.; Allison, A.B.; Araiza, R.; Aza-Blanc, P.; Bower, L.R.; Campos, J.; Chu, H.; Denson, S.; Donham, C.; et al. Experiments from unfinished Registered Reports in the Reproducibility Project: Cancer Biology. eLife 2021, 10, e73430. [Google Scholar] [CrossRef]
- Alekseenko, I.V.; Pleshkan, V.V.; Monastyrskaya, G.S.; Kuzmich, A.I.; Snezhkov, E.V.; Didych, D.A.; Sverdlov, E.D. Fundamentally low reproducibility in molecular genetic cancer research. Genetika 2016, 52, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ioannidis, J.P. Reproducibility in Science. Circ. Res. 2015, 116, 116–126. [Google Scholar] [CrossRef]
- Bero, L. Stamp out fake clinical data by working together. Nature 2022, 601, 167. [Google Scholar] [CrossRef]
- Mullard, A. Half of top cancer studies fail high-profile reproducibility effort. Nature 2021, 600, 368–369. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabási, A.-L. Interactome Networks and Human Disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Magrinelli, F.; Balint, B.; Bhatia, K.P. Challenges in Clinicogenetic Correlations: One Gene—Many Phenotypes. Mov. Disord. Clin. Pr. 2021, 8, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, E.D. Fundamental taboos of biology. Biochemistry 2009, 74, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, E.D. Genetic surgery—A right strategy to attack cancer. Curr. Gene Ther. 2011, 11, 501–531. [Google Scholar] [CrossRef] [PubMed]
- Gilson, P.; Merlin, J.-L.; Harlé, A. Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef]
- Jacquemin, V.; Antoine, M.; Dom, G.; Detours, V.; Maenhaut, C.; Dumont, J.E. Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications. Cancers 2022, 14, 280. [Google Scholar] [CrossRef]
- Gupta, R.G.; Somer, R.A. Intratumor Heterogeneity: Novel Approaches for Resolving Genomic Architecture and Clonal Evolution. Mol. Cancer Res. 2017, 15, 1127–1137. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The Genomic Landscapes of Human Breast and Colorectal Cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Hambley, T.W.; Hait, W.N. Is anticancer drug development heading in the right direction? Cancer Res. 2009, 69, 1259–1262. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Bielas, J.H.; Beckman, R.A. Cancers Exhibit a Mutator Phenotype: Clinical Implications. Cancer Res. 2008, 68, 3551–3557. [Google Scholar] [CrossRef] [PubMed]
- Natali, F.; Rancati, G. The Mutator Phenotype: Adapting Microbial Evolution to Cancer Biology. Front. Genet. 2019, 10, 713. [Google Scholar] [CrossRef] [PubMed]
- Bielas, J.H.; Loeb, K.R.; Rubin, B.P.; True, L.D.; Loeb, L.A. Human cancers express a mutator phenotype. Proc. Natl. Acad. Sci. USA 2006, 103, 18238–18242. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Antal, T.; Dingli, D.; Traulsen, A.; Kinzler, K.W.; Velculescu, V.; Vogelstein, B.; Nowak, M.A. Genetic Progression and the Waiting Time to Cancer. PLoS Comput. Biol. 2007, 3, e225. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Garg, N.; Sinha, D.; Yadav, B.; Gupta, B.; Gupta, S.; Miah, S. ML-Based Texture and Wavelet Features Extraction Technique to Predict Gastric Mesothelioma Cancer. BioMed Res. Int. 2022, 2022, 1012684. [Google Scholar] [CrossRef]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef]
- Bausch-Fluck, D.; Milani, E.S.; Wollscheid, B. Surfaceome nanoscale organization and extracellular interaction networks. Curr. Opin. Chem. Biol. 2018, 48, 26–33. [Google Scholar] [CrossRef]
- Lim, L.C.; Lim, Y.M. Proteome Heterogeneity in Colorectal Cancer. Proteomics 2018, 18, 1700169. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Doerr, A. Single-cell proteomics. Nat. Methods 2018, 16, 20. [Google Scholar] [CrossRef] [PubMed]
- Zenobi, R. Single-Cell Metabolomics: Analytical and Biological Perspectives. Science 2013, 342, 1243259. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.; Zhang, Y.; Liu, J. Advancing single-cell proteomics and metabolomics with microfluidic technologies. Analyst 2019, 144, 846–858. [Google Scholar] [CrossRef]
- Ju, Y.S. A large-scale snapshot of intratumor heterogeneity in human cancer. Cancer Cell 2021, 39, 463–465. [Google Scholar] [CrossRef]
- Gibson, G. Perspectives on rigor and reproducibility in single cell genomics. PLoS Genet. 2022, 18, e1010210. [Google Scholar] [CrossRef]
- Dance, A. Which single-cell analysis tool is best? Scientists offer advice. Nature 2022, 612, 577–579. [Google Scholar] [CrossRef]
- Deng, Y.; Finck, A.; Fan, R. Single-Cell Omics Analyses Enabled by Microchip Technologies. Annu. Rev. Biomed. Eng. 2019, 21, 365–393. [Google Scholar] [CrossRef]
- Kang, Y.; Thieffry, D.; Cantini, L. Evaluating the Reproducibility of Single-Cell Gene Regulatory Network Inference Algorithms. Front. Genet. 2021, 12, 362. [Google Scholar] [CrossRef]
- Sun, G.; Li, Z.; Rong, D.; Zhang, H.; Shi, X.; Yang, W.; Zheng, W.; Sun, G.; Wu, F.; Cao, H.; et al. Single-cell RNA sequencing in cancer: Applications, advances, and emerging challenges. Mol. Ther. Oncolytics 2021, 21, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Wendt, F.; Milani, E.S.; Wollscheid, B. Elucidation of host-virus surfaceome interactions using spatial proteotyping. Adv. Virus Res. 2021, 109, 105–134. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yuan, J.; Long, M.; Jiang, J.; Zhang, Y.; Zhang, T.; Xu, M.; Fan, Y.; Tanyi, J.L.; Montone, K.T.; et al. The Cancer Surfaceome Atlas integrates genomic, functional and drug response data to identify actionable targets. Nat. Cancer 2021, 2, 1406–1422. [Google Scholar] [CrossRef]
- Hegewisch-Solloa, E.; Seo, S.; Mundy-Bosse, B.L.; Mishra, A.; Waldman, E.H.; Maurrasse, S.; Grunstein, E.; Connors, T.J.; Freud, A.G.; Mace, E.M. Differential Integrin Adhesome Expression Defines Human NK Cell Residency and Developmental Stage. J. Immunol. 2021, 207, 950–965. [Google Scholar] [CrossRef]
- Zhong, X.; Drgonova, J.; Li, C.-Y.; Uhl, G.R. Human cell adhesion molecules: Annotated functional subtypes and overrepresentation of addiction-associated genes. Ann. N. Y. Acad. Sci. 2015, 1349, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chen, J.; Liu, H.; Chen, S.; Zhang, Y.; Li, P.; Thierry-Mieg, D.; Thierry-Mieg, J.; Mattes, W.; Ning, B.; et al. Comprehensive Identification and Characterization of Human Secretome Based on Integrative Proteomic and Transcriptomic Data. Front. Cell Dev. Biol. 2019, 7, 299. [Google Scholar] [CrossRef]
- Zullo, J.; Matsumoto, K.; Xavier, S.; Ratliff, B.; Goligorsky, M.S. The cell secretome, a mediator of cell-to-cell communication. Prostaglandins Other Lipid Mediat. 2015, 120, 17–20. [Google Scholar] [CrossRef]
- Gladilin, E. Graph-theoretical model of global human interactome reveals enhanced long-range communicability in cancer networks. PLoS ONE 2017, 12, e0170953. [Google Scholar] [CrossRef]
- Banerjee, A.; Chabria, Y.; Kanna NR, R.; Gopi, J.; Rowlo, P.; Sun, X.-F.; Pathak, S. Role of Tumor Specific niche in Colon Cancer Progression and Emerging Therapies by Targeting Tumor Microenvironment. In Cell Biology and Translational Medicine; Turksen, K., Ed.; Springer: Cham, Switzerland, 2019; pp. 177–192. [Google Scholar] [CrossRef]
- Alekseenko, I.V.; Chernov, I.P.; Kostrov, S.V.; Sverdlov, E.D. Are Synapse-Like Structures a Possible Way for Crosstalk of Cancer with Its Microenvironment? Cancers 2020, 12, 806. [Google Scholar] [CrossRef] [Green Version]
- Sverdlov, E. Missed Druggable Cancer Hallmark: Cancer-Stroma Symbiotic Crosstalk as Paradigm and Hypothesis for Cancer Therapy. Bioessays 2018, 40, e1800079. [Google Scholar] [CrossRef]
- Sverdlov, E.D.; Chernov, I.P. Cancer Stem Complex, Not a Cancer Stem Cell, Is the Driver of Cancer Evolution. Biochemistry 2019, 84, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/tumor-microenvironment (accessed on 20 September 2022).
- Bota-Rabassedas, N.; Banerjee, P.; Niu, Y.; Cao, W.; Luo, J.; Xi, Y.; Tan, X.; Sheng, K.; Ahn, Y.-H.; Lee, S.; et al. Contextual cues from cancer cells govern cancer-associated fibroblast heterogeneity. Cell Rep. 2021, 35, 109009. [Google Scholar] [CrossRef] [PubMed]
- Conod, A.; Silvano, M.; Ruiz i Altaba, A. On the origin of metastases: Induction of pro-metastatic states after impending cell death via ER stress, reprogramming, and a cytokine storm. Cell Rep. 2022, 38, 110490. [Google Scholar] [CrossRef]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C. Cancer-associated fibroblasts: How do they contribute to metastasis? Clin. Exp. Metastasis 2019, 36, 71–86. [Google Scholar] [CrossRef]
- Juan, B.P.S.; Garcia-Leon, M.J.; Rangel, L.; Goetz, J.G.; Chaffer, C.L. The Complexities of Metastasis. Cancers 2019, 11, 1575. [Google Scholar] [CrossRef] [PubMed]
- Satcher, R.L.; Zhang, X.H.-F. Evolving cancer–niche interactions and therapeutic targets during bone metastasis. Nat. Rev. Cancer 2021, 22, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Williams, S. New Understanding of Metastasis Could Lead to Better Treatments. The Scientist 2021, 1 April 2021, 587. Available online: https://www.the-scientist.com/features/new-understanding-of-metastasis-could-lead-to-better-treatments-68572 (accessed on 22 January 2023).
- Xiong, S.; Dong, L.; Cheng, L. Neutrophils in cancer carcinogenesis and metastasis. J. Hematol. Oncol. 2021, 14, 173. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A. Tumor and the Microenvironment: A Chance to Reframe the Paradigm of Carcinogenesis? BioMed Res. Int. 2014, 2014, 934038. [Google Scholar] [CrossRef] [Green Version]
- Perrimon, N.; Pitsouli, C.; Shilo, B.-Z. Signaling Mechanisms Controlling Cell Fate and Embryonic Patterning. Cold Spring Harb. Perspect. Biol. 2012, 4, a005975. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9, e57243. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Aspeslagh, S.; Garaud, S.; Dupont, F.; Solinas, C.; Kok, M.; Routy, B.; Sotiriou, C.; Stagg, J.; Buisseret, L. Immuno-oncology-101: Overview of major concepts and translational perspectives. Semin. Cancer Biol. 2018, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Huang, X.; Carlos, C.J.J.; Lu, X. Research progress of tumor microenvironment and tumor-associated macrophages. Clin. Transl. Oncol. 2020, 22, 2141–2152. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; Canè, S.; De Sanctis, F.; Bronte, V. Monocytes in the Tumor Microenvironment. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 93–122. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Collier, A.I.; Beatty, G.L. Macrophages: Key orchestrators of a tumor microenvironment defined by therapeutic resistance. Mol. Immunol. 2017, 110, 3–12. [Google Scholar] [CrossRef]
- Rad, H.S.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 2020, 41, 1474–1498. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Yamamoto, Y.; Akasaki, Y.; Sasaki, H. Dual role of macrophage in tumor immunity. Immunotherapy 2018, 10, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.E.; Youn, H.; Kang, K.W. Imaging in Tumor Immunology. Nucl. Med. Mol. Imaging 2021, 55, 225–236. [Google Scholar] [CrossRef]
- Zhuang, X.; Zhang, H.; Hu, G. Cancer and Microenvironment Plasticity: Double-Edged Swords in Metastasis. Trends Pharmacol. Sci. 2019, 40, 419–429. [Google Scholar] [CrossRef]
- Yang, X.; Annaert, W. The Nanoscopic Organization of Synapse Structures: A Common Basis for Cell Communication. Membranes 2021, 11, 248. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of immune cell and tumor microenvironment imaging in the new era of immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 89. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Mhaidly, R.; Mechta-Grigoriou, F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol. Rev. 2021, 302, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, X.H.-F. Tumor-Associated Neutrophils and Macrophages—Heterogenous but Not Chaotic. Front. Immunol. 2020, 11, 3117. [Google Scholar] [CrossRef] [PubMed]
- Peña-Romero, A.C.; Orenes-Piñero, E. Dual Effect of Immune Cells within Tumour Microenvironment: Pro- and Anti-Tumour Effects and Their Triggers. Cancers 2022, 14, 1681. [Google Scholar] [CrossRef] [PubMed]
- Kalafati, L.; Mitroulis, I.; Verginis, P.; Chavakis, T.; Kourtzelis, I. Neutrophils as Orchestrators in Tumor Development and Metastasis Formation. Front. Oncol. 2020, 10, 2799. [Google Scholar] [CrossRef]
- Nielsen, S.R.; Schmid, M.C. Macrophages as Key Drivers of Cancer Progression and Metastasis. Mediat. Inflamm. 2017, 2017, 9624760. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Zhu, L.; Yakoub, M.; Reißfelder, C.; Loges, S.; Schölch, S. Cell–Cell Interactions Drive Metastasis of Circulating Tumor Microemboli. Cancer Res 2022, 82, 2661–2671. [Google Scholar] [CrossRef]
- Duits, D.E.; de Visser, K.E. Impact of cancer cell-intrinsic features on neutrophil behavior. Semin. Immunol. 2021, 57, 101546. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.J. Signal initiation in biological systems: The properties and detection of transient extracellular protein interactions. Mol. Biosyst. 2009, 5, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Jang, H. Dynamic multiprotein assemblies shape the spatial structure of cell signaling. Prog. Biophys. Mol. Biol. 2014, 116, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Bongrand, P. Is There a Need for a More Precise Description of Biomolecule Interactions to Understand Cell Function? Curr. Issues Mol. Biol. 2022, 44, 505–525. [Google Scholar] [CrossRef] [PubMed]
- Mège, R.M.; Ishiyama, N. Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions. Cold Spring Harb. Perspect. Biol. 2017, 9, a028738. [Google Scholar] [CrossRef]
- Garcia-Parajo, M.F.; Cambi, A.; Torreno-Pina, J.A.; Thompson, N.; Jacobson, K. Nanoclustering as a dominant feature of plasma membrane organization. J. Cell Sci. 2014, 127, 4995–5005. [Google Scholar] [CrossRef]
- Cebecauer, M.; Spitaler, M.; Sergé, A.; Magee, A.I. Signalling complexes and clusters: Functional advantages and methodological hurdles. J. Cell Sci. 2010, 123, 309–320. [Google Scholar] [CrossRef]
- Baluška, F.; Mancuso, S. Synaptic view of eukaryotic cell. Int. J. Gen. Syst. 2014, 43, 740–756. [Google Scholar] [CrossRef]
- Baluška, F.; Miller, W.B.; Reber, A.S. Cellular and evolutionary perspectives on organismal cognition: From unicellular to multicellular organisms. Biol. J. Linn. Soc. 2022. [Google Scholar] [CrossRef]
- Yamada, S.; Nelson, W.J. Synapses: Sites of Cell Recognition, Adhesion, and Functional Specification. Annu. Rev. Biochem. 2007, 76, 267–294. [Google Scholar] [CrossRef]
- Angus, K.L.; Griffiths, G.M. Cell polarisation and the immunological synapse. Curr. Opin. Cell Biol. 2013, 25, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Calvo, V.; Izquierdo, M. Imaging Polarized Secretory Traffic at the Immune Synapse in Living T Lymphocytes. Front. Immunol. 2018, 9, 684. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. A dynamic view of the immunological synapse. Semin. Immunol. 2005, 17, 400–410. [Google Scholar] [CrossRef]
- Dustin, M.L. T-cell activation through immunological synapses and kinapses. Immunol. Rev. 2008, 221, 77–89. [Google Scholar] [CrossRef]
- Dustin, M.L. Modular Design of Immunological Synapses and Kinapses. Cold Spring Harb. Perspect. Biol. 2009, 1, a002873. [Google Scholar] [CrossRef]
- Dustin, M.L. The Immunological Synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef]
- Dustin, M.L.; Baldari, C.T. The Immune Synapse: Past, Present, and Future. In The Immune Synapse; Humana Press: New York, NY, USA, 2017; Volume 1584, pp. 1–5. [Google Scholar] [CrossRef]
- Dustin, M.L.; Choudhuri, K. Signaling and Polarized Communication across the T Cell Immunological Synapse. Annu. Rev. Cell Dev. Biol. 2016, 32, 303–325. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.T.; Angus, K.L.; Griffiths, G.M. The role of the cytoskeleton at the immunological synapse. Immunol. Rev. 2013, 256, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Tato, C.M.; Davis, M.M. How the immune system talks to itself: The varied role of synapses. Immunol. Rev. 2012, 251, 65–79. [Google Scholar] [CrossRef]
- Ortega-Carrion, A.; Vicente-Manzanares, M. Concerning immune synapses: A spatiotemporal timeline. F1000Research 2016, 5, 418. [Google Scholar] [CrossRef]
- Shi, Y. To forge a solid immune recognition. Protein Cell 2012, 3, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Hartman, N.C.; Groves, J.T. Signaling clusters in the cell membrane. Curr. Opin. Cell Biol. 2011, 23, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Yap, A.S.; Gomez, G.A.; Parton, R.G. Adherens Junctions Revisualized: Organizing Cadherins as Nanoassemblies. Dev. Cell 2015, 35, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.M.; Sikorski, A.F.; Nawara, T.; Grzesiak, J.; Marycz, K.; Bogusławska, D.M.; Michalczyk, I.; LeComte, M.-C.; Machnicka, B. αII-spectrin in T cells is involved in the regulation of cell-cell contact leading to immunological synapse formation? PLoS ONE 2017, 12, e0189545. [Google Scholar] [CrossRef]
- Hatherley, D.; Lea, S.M.; Johnson, S.; Barclay, A.N. Structures of CD200/CD200 Receptor Family and Implications for Topology, Regulation, and Evolution. Structure 2013, 21, 820–832. [Google Scholar] [CrossRef]
- Wright, G.J.; Puklavec, M.J.; Willis, A.C.; Hoek, R.M.; Sedgwick, J.D.; Brown, M.H.; Barclay, A. Lymphoid/Neuronal Cell Surface OX2 Glycoprotein Recognizes a Novel Receptor on Macrophages Implicated in the Control of Their Function. Immunity 2000, 13, 233–242. [Google Scholar] [CrossRef]
- Graham, D.B.; Cella, M.; Giurisato, E.; Fujikawa, K.; Miletic, A.V.; Kloeppel, T.; Brim, K.; Takai, T.; Shaw, A.S.; Colonna, M.; et al. Vav1 Controls DAP10-Mediated Natural Cytotoxicity by Regulating Actin and Microtubule Dynamics. J. Immunol. 2006, 177, 2349–2355. [Google Scholar] [CrossRef]
- Giurisato, E.; Cella, M.; Takai, T.; Kurosaki, T.; Feng, Y.; Longmore, G.D.; Colonna, M.; Shaw, A.S. Phosphatidylinositol 3-Kinase Activation Is Required To Form the NKG2D Immunological Synapse. Mol. Cell. Biol. 2007, 27, 8583–8599. [Google Scholar] [CrossRef] [Green Version]
- Wilton, K.M.; Overlee, B.L.; Billadeau, D.D. NKG2D/DAP10 Signaling recruits EVL to the cytotoxic synapse to generate F-actin and promote NK cell cytotoxicity. J. Cell Sci. 2019, 133, jcs230508. [Google Scholar] [CrossRef]
- Cohen, D.J.; Nelson, W.J. Secret handshakes: Cell–cell interactions and cellular mimics. Curr. Opin. Cell Biol. 2018, 50, 14–19. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Seiradake, E.; Jones, E.Y.; Klein, R. Structural Perspectives on Axon Guidance. Annu. Rev. Cell Dev. Biol. 2016, 32, 577–608. [Google Scholar] [CrossRef] [PubMed]
- Ojosnegros, S.; Cutrale, F.; Rodríguez, D.; Otterstrom, J.J.; Chiu, C.L.; Hortigüela, V.; Tarantino, C.; Seriola, A.; Mieruszynski, S.; Martínez, E.; et al. Eph-ephrin signaling modulated by polymerization and condensation of receptors. Proc. Natl. Acad. Sci. USA 2017, 114, 13188–13193. [Google Scholar] [CrossRef] [PubMed]
- Rozbesky, D.; Jones, E.Y. Cell guidance ligands, receptors and complexes – orchestrating signalling in time and space. Curr. Opin. Struct. Biol. 2019, 61, 79–85. [Google Scholar] [CrossRef]
- Sergé, A. The Molecular Architecture of Cell Adhesion: Dynamic Remodeling Revealed by Videonanoscopy. Front. Cell Dev. Biol. 2016, 4, 36. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, X.; Huang, L. Macrophage-Mediated Tumor Cell Phagocytosis: Opportunity for Nanomedicine Intervention. Adv. Funct. Mater. 2020, 31, 2006220. [Google Scholar] [CrossRef]
- Niedergang, F.; Di Bartolo, V.; Alcover, A. Comparative Anatomy of Phagocytic and Immunological Synapses. Front. Immunol. 2016, 7, 18. [Google Scholar] [CrossRef]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. BioMed Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef] [Green Version]
- Barth, N.D.; Marwick, J.A.; Vendrell, M.; Rossi, A.G.; Dransfield, I. The “Phagocytic Synapse” and Clearance of Apoptotic Cells. Front. Immunol. 2017, 8, 1708. [Google Scholar] [CrossRef]
- Gordon, S. Phagocytosis: An Immunobiologic Process. Immunity 2016, 44, 463–475. [Google Scholar] [CrossRef]
- Röszer, T. Understanding the Biology of Self-Renewing Macrophages. Cells 2018, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Filiberti, S.; Russo, M.; Lonardi, S.; Bugatti, M.; Vermi, W.; Tournier, C.; Giurisato, E. Self-Renewal of Macrophages: Tumor-Released Factors and Signaling Pathways. Biomedicines 2022, 10, 2709. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, Y. Innate immune receptor clustering and its role in immune regulation. J. Cell Sci. 2021, 134, jcs249318. [Google Scholar] [CrossRef] [PubMed]
- Lagrue, K.; Carisey, A.; Oszmiana, A.; Kennedy, P.R.; Williamson, D.J.; Cartwright, A.; Barthen, C.; Davis, D.M. The central role of the cytoskeleton in mechanisms and functions of the NK cell immune synapse. Immunol. Rev. 2013, 256, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, R.J.; Piguet, V. Masters of manipulation: Viral modulation of the immunological synapse. Cell. Microbiol. 2018, 20, e12944. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Evans, J.P.; King, T.; Zheng, Y.-M.; Oltz, E.M.; Whelan, S.P.J.; Saif, L.J.; Peeples, M.E.; Liu, S.-L. SARS-CoV-2 spreads through cell-to-cell transmission. Proc. Natl. Acad. Sci. USA 2021, 119, e2111400119. [Google Scholar] [CrossRef] [PubMed]
- Real, F.; Sennepin, A.; Ganor, Y.; Schmitt, A.; Bomsel, M. Live Imaging of HIV-1 Transfer across T Cell Virological Synapse to Epithelial Cells that Promotes Stromal Macrophage Infection. Cell Rep. 2018, 23, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Rossello, R.A.; Kohn, D.H. Gap junction intercellular communication: A review of a potential platform to modulate craniofacial tissue engineering. J. Biomed. Mater. Res. Part B Appl. Biomater. 2008, 88B, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Jolly, M.K. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition in Cancer Metastasis. Cold Spring Harb. Perspect. Med. 2020, 10, a036905. [Google Scholar] [CrossRef]
- Beckmann, A.; Hainz, N.; Tschernig, T.; Meier, C. Facets of Communication: Gap Junction Ultrastructure and Function in Cancer Stem Cells and Tumor Cells. Cancers 2019, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- Zefferino, R.; Piccoli, C.; Di Gioia, S.; Capitanio, N.; Conese, M. How Cells Communicate with Each Other in the Tumor Microenvironment: Suggestions to Design Novel Therapeutic Strategies in Cancer Disease. Int. J. Mol. Sci. 2021, 22, 2550. [Google Scholar] [CrossRef]
- Tittarelli, A.; Navarrete, M.; Gleisner, M.A.; Gebicke-Haerter, P.; Salazar-Onfray, F. Connexin-Mediated Signaling at the Immunological Synapse. Int. J. Mol. Sci. 2020, 21, 3736. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Naranjo, A.; Bouma, G.; Pereda, C.; Ramírez, M.; Webb, K.F.; Tittarelli, A.; López, M.N.; Kalergis, A.M.; Thrasher, A.J.; Becker, D.L.; et al. Functional Gap Junctions Accumulate at the Immunological Synapse and Contribute to T Cell Activation. J. Immunol. 2011, 187, 3121–3132. [Google Scholar] [CrossRef]
- Hofmann, F.; Navarrete, M.; Álvarez, J.; Guerrero, I.; Gleisner, M.A.; Tittarelli, A.; Salazar-Onfray, F. Cx43-Gap Junctions Accumulate at the Cytotoxic Immunological Synapse Enabling Cytotoxic T Lymphocyte Melanoma Cell Killing. Int. J. Mol. Sci. 2019, 20, 4509. [Google Scholar] [CrossRef]
- Belardi, B.; Son, S.; Felce, J.H.; Dustin, M.L.; Fletcher, D.A. Cell–cell interfaces as specialized compartments directing cell function. Nat. Rev. Mol. Cell Biol. 2020, 21, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Adil, M.S.; Narayanan, S.P.; Somanath, P.R. Cell-cell junctions: Structure and regulation in physiology and pathology. Tissue Barriers 2020, 9, 1848212. [Google Scholar] [CrossRef]
- Xu, Q.-R.; Du, X.-H.; Huang, T.-T.; Zheng, Y.-C.; Li, Y.-L.; Huang, D.-Y.; Dai, H.-Q.; Li, E.-M.; Fang, W.-K. Role of Cell-Cell Junctions in Oesophageal Squamous Cell Carcinoma. Biomolecules 2022, 12, 1378. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. Steps in metastasis: An updated review. Med Oncol. 2021, 38, 3. [Google Scholar] [CrossRef]
- Zhu, K.; Li, P.; Mo, Y.; Wang, J.; Jiang, X.; Ge, J.; Huang, W.; Liu, Y.; Tang, Y.; Gong, Z.; et al. Neutrophils: Accomplices in metastasis. Cancer Lett. 2020, 492, 11–20. [Google Scholar] [CrossRef]
- Sutton, T.L.; Patel, R.K.; Anderson, A.N.; Bowden, S.G.; Whalen, R.; Giske, N.R.; Wong, M.H. Circulating Cells with Macrophage-like Characteristics in Cancer: The Importance of Circulating Neoplastic-Immune Hybrid Cells in Cancer. Cancers 2022, 14, 3871. [Google Scholar] [CrossRef]
- Cao, M.-F.; Chen, L.; Dang, W.-Q.; Zhang, X.-C.; Zhang, X.; Shi, Y.; Yao, X.-H.; Li, Q.; Zhu, J.; Lin, Y.; et al. Hybrids by tumor-associated macrophages × glioblastoma cells entail nuclear reprogramming and glioblastoma invasion. Cancer Lett. 2018, 442, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef]
- Leonard, F.; Curtis, L.T.; Ware, M.J.; Nosrat, T.; Liu, X.; Yokoi, K.; Frieboes, H.B.; Godin, B. Macrophage Polarization Contributes to the Anti-Tumoral Efficacy of Mesoporous Nanovectors Loaded with Albumin-Bound Paclitaxel. Front. Immunol. 2017, 8, 693. [Google Scholar] [CrossRef] [PubMed]
- Quan, Q.; Wang, X.; Lu, C.; Ma, W.; Wang, Y.; Xia, G.; Wang, C.; Yang, G. Cancer stem-like cells with hybrid epithelial/mesenchymal phenotype leading the collective invasion. Cancer Sci. 2019, 111, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Mercedes, S.A.V.; Bocci, F.; Levine, H.; Onuchic, J.N.; Jolly, M.K.; Wong, P.K. Decoding leader cells in collective cancer invasion. Nat. Rev. Cancer 2021, 21, 592–604. [Google Scholar] [CrossRef]
- Swierczak, A.; Pollard, J.W. Myeloid cells in metastasis. Cold Spring Harbor Perspect. Med. 2020, 10, a038026. [Google Scholar] [CrossRef]
- Bailey, P.C.; Martin, S.S. Insights on CTC Biology and Clinical Impact Emerging from Advances in Capture Technology. Cells 2019, 8, 553. [Google Scholar] [CrossRef] [Green Version]
- Burr, R.; Gilles, C.; Thompson, E.W.; Maheswaran, S. Epithelial-Mesenchymal Plasticity in Circulating Tumor Cells, the Precursors of Metastasis. Circ. Tumor Cells Breast Cancer Metastatic Dis. 2020, 1220, 11–34. [Google Scholar] [CrossRef]
- Imodoye, S.O.; Adedokun, K.A.; Muhammed, A.O.; Bello, I.O.; Muhibi, M.A.; Oduola, T.; Oyenike, M.A. Understanding the Complex Milieu of Epithelial-Mesenchymal Transition in Cancer Metastasis: New Insight Into the Roles of Transcription Factors. Front. Oncol. 2021, 11, 4360. [Google Scholar] [CrossRef]
- Wang, W.-M.; Shen, H.; Liu, Z.-N.; Chen, Y.-Y.; Hou, L.-J.; Ding, Y. Interaction between tumor microenvironment, autophagy, and epithelial-mesenchymal transition in tumor progression. Cancer Treat. Res. Commun. 2022, 32, 100592. [Google Scholar] [CrossRef]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Jolly, M.K.; Woodward, W.; Levine, H.; Deem, M.W. Analysis of Hierarchical Organization in Gene Expression Networks Reveals Underlying Principles of Collective Tumor Cell Dissemination and Metastatic Aggressiveness of Inflammatory Breast Cancer. Front. Oncol. 2018, 8, 244. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Becker, T.M.; Po, J.W.; Chua, W.; Ma, Y. Single-Circulating Tumor Cell Whole Genome Amplification to Unravel Cancer Heterogeneity and Actionable Biomarkers. Int. J. Mol. Sci. 2022, 23, 8386. [Google Scholar] [CrossRef] [PubMed]
- Paizal, J.P.; Au, S.H.; Bakal, C. Squeezing through the microcirculation: Survival adaptations of circulating tumour cells to seed metastasis. Br. J. Cancer 2020, 124, 58–65. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Jiao, X.; Upadhyaya, C.; Zhang, Z.; Zhao, J.; Li, Z.; Patel, V.I.; Pestell, R.G. Assays for the Spectrum of Circulating Tumor Cells. In Stem Cell Assays; Humana: New York, NY, USA, 2022; pp. 533–545. [Google Scholar] [CrossRef]
- Micalizzi, D.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Wrenn, E.; Huang, Y.; Cheung, K. Collective metastasis: Coordinating the multicellular voyage. Clin. Exp. Metastasis 2021, 38, 373–399. [Google Scholar] [CrossRef]
- Tang, S.C.; Vijayakumar, U.; Zhang, Y.; Fullwood, M.J. Super-Enhancers, Phase-Separated Condensates, and 3D Genome Organization in Cancer. Cancers 2022, 14, 2866. [Google Scholar] [CrossRef]
- Chen, Q.; Zou, J.; He, Y.; Pan, Y.; Yang, G.; Zhao, H.; Huang, Y.; Zhao, Y.; Wang, A.; Chen, W.; et al. A narrative review of circulating tumor cells clusters: A key morphology of cancer cells in circulation promote hematogenous metastasis. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef]
- Castro-Giner, F.; Aceto, N. Tracking cancer progression: From circulating tumor cells to metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef]
- Amintas, S.; Bedel, A.; Moreau-Gaudry, F.; Boutin, J.; Buscail, L.; Merlio, J.-P.; Vendrely, V.; Dabernat, S.; Buscail, E. Circulating Tumor Cell Clusters: United We Stand Divided We Fall. Int. J. Mol. Sci. 2020, 21, 2653. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N. Bring along your friends: Homotypic and heterotypic circulating tumor cell clustering to accelerate metastasis. Biomed. J. 2020, 43, 18–23. [Google Scholar] [CrossRef] [PubMed]
- López-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-C.; Zhang, X.-F.; Peng, J.; Li, X.-F.; Wang, A.-L.; Bie, Y.-Q.; Shi, L.-H.; Lin, M.-B.; Zhang, X.-F. Survival Mechanisms and Influence Factors of Circulating Tumor Cells. BioMed Res. Int. 2018, 2018, 6304701. [Google Scholar] [CrossRef]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.-F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019, 9, 96–113. [Google Scholar] [CrossRef]
- Rodrigues, P.; Vanharanta, S. Circulating Tumor Cells: Come Together, Right Now, Over Metastasis. Cancer Discov. 2019, 9, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef]
- Marsh, T.; Pietras, K.; McAllister, S.S. Fibroblasts as architects of cancer pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1070–1078. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Schuster, E.; Taftaf, R.; Reduzzi, C.; Albert, M.K.; Romero-Calvo, I.; Liu, H. Better together: Circulating tumor cell clustering in metastatic cancer. Trends Cancer 2021, 7, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Herath, S.; Bazaz, S.R.; Monkman, J.; Warkiani, M.E.; Richard, D.; O’Byrne, K.; Kulasinghe, A. Circulating tumor cell clusters: Insights into tumour dissemination and metastasis. Expert Rev. Mol. Diagn. 2020, 20, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hu, W.; Liu, B.; Yang, T. Insights into Circulating Tumor Cell Clusters: A Barometer for Treatment Effects and Prognosis for Prostate Cancer Patients. Cancers 2022, 14, 3985. [Google Scholar] [CrossRef] [PubMed]
- Jaqaman, K.; Ditlev, J.A. Biomolecular condensates in membrane receptor signaling. Curr. Opin. Cell Biol. 2021, 69, 48–54. [Google Scholar] [CrossRef]
- Smith, H.A.; Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 2013, 91, 411–429. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Hänggi, K.; Ruffell, B. The Immune Microenvironment and Cancer Metastasis. Cold Spring Harb. Perspect. Med. 2019, 10, a037424. [Google Scholar] [CrossRef]
- Smietanka, U.; Szostakowska-Rodzos, M.; Tabor, S.; Fabisiewicz, A.; Grzybowska, E. Clusters, Assemblies and Aggregates of Tumor Cells in the Blood of Breast Cancer Patients; Composition, Mode of Action, Detection and Impact on Metastasis and Survival. Int. J. Transl. Med. 2021, 1, 55–68. [Google Scholar] [CrossRef]
- Khalil, A.A.; Ilina, O.; Vasaturo, A.; Venhuizen, J.-H.; Vullings, M.; Venhuizen, V.; Bilos, A.; Figdor, C.G.; Span, P.N.; Friedl, P. Collective invasion induced by an autocrine purinergic loop through connexin-43 hemichannels. J. Cell Biol. 2020, 219, e201911120. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Phase Separation in Membrane Biology: The Interplay between Membrane-Bound Organelles and Membraneless Condensates. Dev. Cell 2020, 55, 30–44. [Google Scholar] [CrossRef]
- Lu, J.; Qian, J.; Xu, Z.; Yin, S.; Zhou, L.; Zheng, S.; Zhang, W. Emerging Roles of Liquid–Liquid Phase Separation in Cancer: From Protein Aggregation to Immune-Associated Signaling. Front. Cell Dev. Biol. 2021, 9, 631486. [Google Scholar] [CrossRef] [PubMed]
- Strilic, B.; Offermanns, S. Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell 2017, 32, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Sluka, P.; Davis, I.D. Cell mates: Paracrine and stromal targets for prostate cancer therapy. Nat. Rev. Urol. 2013, 10, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Bergonzini, C.; Kroese, K.; Zweemer, A.J.M.; Danen, E.H.J. Targeting Integrins for Cancer Therapy—Disappointments and Opportunities. Front. Cell Dev. Biol. 2022, 10, 863850. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Mittasch, M.; Gomes, B.F.; Klein, I.A.; Murcko, M.A. Modulating biomolecular condensates: A novel approach to drug discovery. Nat. Rev. Drug Discov. 2022, 21, 841–862. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; McGrail, D.J.; Lozanoski, T.M.; Dunn, B.S.; Shih, D.J.; Cirillo, K.M.; Cetinkaya, S.H.; Zheng, W.J.; Mills, G.B.; Yi, S.S.; et al. Biomolecular Condensation: A New Phase in Cancer Research. Cancer Discov. 2022, 12, 2031–2043. [Google Scholar] [CrossRef]
- Taniue, K.; Akimitsu, N. Aberrant phase separation and cancer. FEBS J. 2021, 289, 17–39. [Google Scholar] [CrossRef]
- Orti, F.; Navarro, A.M.; Rabinovich, A.; Wodak, S.J.; Marino-Buslje, C. Insight into membraneless organelles and their associated proteins: Drivers, Clients and Regulators. Comput. Struct. Biotechnol. J. 2021, 19, 3964–3977. [Google Scholar] [CrossRef]
- Abyzov, A.; Blackledge, M.; Zweckstetter, M. Conformational Dynamics of Intrinsically Disordered Proteins Regulate Biomolecular Condensate Chemistry. Chem. Rev. 2022, 122, 6719–6748. [Google Scholar] [CrossRef]
- Deniz, A.A. Networking and Dynamic Switches in Biological Condensates. Cell 2020, 181, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Fare, C.M.; Villani, A.; Drake, L.E.; Shorter, J. Higher-order organization of biomolecular condensates. Open Biol. 2021, 11, 210137. [Google Scholar] [CrossRef] [PubMed]
- Ramella, M.; Ribolla, L.M.; de Curtis, I. Liquid–Liquid Phase Separation at the Plasma Membrane–Cytosol Interface: Common Players in Adhesion, Motility, and Synaptic Function. J. Mol. Biol. 2021, 434, 167228. [Google Scholar] [CrossRef]
- Li, W.; Jiang, C.; Zhang, E. Advances in the phase separation-organized membraneless organelles in cells: A narrative review. Transl. Cancer Res. 2021, 10, 4929–4946. [Google Scholar] [CrossRef]
- Garcia-Cabau, C.; Salvatella, X. Regulation of biomolecular condensate dynamics by signaling. Curr. Opin. Cell Biol. 2021, 69, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- André, A.A.M.; Spruijt, E. Liquid–Liquid Phase Separation in Crowded Environments. Int. J. Mol. Sci. 2020, 21, 5908. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Young, R.A. Biomolecular Condensates and Cancer. Cancer Cell 2021, 39, 174–192. [Google Scholar] [CrossRef]
- Laghmach, R.; Di Pierro, M.; Potoyan, D. A Liquid State Perspective on Dynamics of Chromatin Compartments. Front. Mol. Biosci. 2022, 8, 1370. [Google Scholar] [CrossRef]
- Laflamme, G.; Mekhail, K. Biomolecular condensates as arbiters of biochemical reactions inside the nucleus. Commun. Biol. 2020, 3, 773. [Google Scholar] [CrossRef]
- Serebreni, L.; Stark, A. Insights into gene regulation: From regulatory genomic elements to DNA-protein and protein-protein interactions. Curr. Opin. Cell Biol. 2020, 70, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Liu, Z.; Lippincott-Schwartz, J. Biomolecular Condensates and Their Links to Cancer Progression. Trends Biochem. Sci. 2021, 46, 535–549. [Google Scholar] [CrossRef] [PubMed]
- André, K.M.; Sipos, E.H.; Soutourina, J. Mediator Roles Going Beyond Transcription. Trends Genet. 2020, 37, 224–234. [Google Scholar] [CrossRef]
- Wei, M.-T.; Chang, Y.-C.; Shimobayashi, S.F.; Shin, Y.; Strom, A.R.; Brangwynne, C.P. Nucleated transcriptional condensates amplify gene expression. Nature 2020, 22, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Rippe, K.; Papantonis, A. RNA polymerase II transcription compartments: From multivalent chromatin binding to liquid droplet formation? Nat. Rev. Mol. Cell Biol. 2021, 22, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jiang, H. Nuclear Protein Condensates and Their Properties in Regulation of Gene Expression. J. Mol. Biol. 2022, 434, 167151. [Google Scholar] [CrossRef]
- Palacio, M.; Taatjes, D.J. Merging Established Mechanisms with New Insights: Condensates, Hubs, and the Regulation of RNA Polymerase II Transcription. J. Mol. Biol. 2021, 434, 167216. [Google Scholar] [CrossRef]
- Xiao, Q.; McAtee, C.K.; Su, X. Phase separation in immune signalling. Nat. Rev. Immunol. 2021, 22, 188–199. [Google Scholar] [CrossRef]
- Tulpule, A.; Guan, J.; Neel, D.S.; Allegakoen, H.R.; Lin, Y.P.; Brown, D.; Chou, Y.-T.; Heslin, A.; Chatterjee, N.; Perati, S.; et al. Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell 2021, 184, 2649–2664.e18. [Google Scholar] [CrossRef]
- Imperial, R.; Toor, O.M.; Hussain, A.; Subramanian, J.; Masood, A. Comprehensive pancancer genomic analysis reveals (RTK)-RAS-RAF-MEK as a key dysregulated pathway in cancer: Its clinical implications. Semin. Cancer Biol. 2019, 54, 14–28. [Google Scholar] [CrossRef]
- Guillén-Boixet, J.; Kopach, A.; Holehouse, A.S.; Wittmann, S.; Jahnel, M.; Schlüßler, R.; Kim, K.; Trussina, I.R.; Wang, J.; Mateju, D.; et al. RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell 2020, 181, 346–361.e17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C. The Molecular and Functional Interaction Between Membrane-Bound Organelles and Membrane-Less Condensates. Front. Cell Dev. Biol. 2022, 10, 896305. [Google Scholar] [CrossRef] [PubMed]
- Peran, I.; Mittag, T. Molecular structure in biomolecular condensates. Curr. Opin. Struct. Biol. 2019, 60, 17–26. [Google Scholar] [CrossRef]
- Forman-Kay, J.D.; Ditlev, J.A.; Nosella, M.L.; Lee, H.O. What are the distinguishing features and size requirements of biomolecular condensates and their implications for RNA-containing condensates? RNA 2021, 28, 36–47. [Google Scholar] [CrossRef]
- Espinosa, J.R.; Joseph, J.A.; Sanchez-Burgos, I.; Garaizar, A.; Frenkel, D.; Collepardo-Guevara, R. Liquid network connectivity regulates the stability and composition of biomolecular condensates with many components. Proc. Natl. Acad. Sci. USA 2020, 117, 13238–13247. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Zwicker, D.; Sohrabi-Jahromi, S.; Boehning, M.; Kirschbaum, J. Mechanisms for Active Regulation of Biomolecular Condensates. Trends Cell Biol. 2019, 30, 4–14. [Google Scholar] [CrossRef]
- Huang, N.; Dong, H.; Shao, B. Phase separation in immune regulation and immune-related diseases. J. Mol. Med. 2022, 100, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, A.; Palomino-Schätzlein, M.; de Molina, P.M.; Arbe, A.; Pierattelli, R.; Rizzuti, B.; Iovanna, J.L.; Neira, J.L. Crowding Effects on the Structure and Dynamics of the Intrinsically Disordered Nuclear Chromatin Protein NUPR1. Front. Mol. Biosci. 2021, 8, 684622. [Google Scholar] [CrossRef]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.; Darling, A.L.; Zaslavsky, B.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem. Sci. 2019, 44, 716–728. [Google Scholar] [CrossRef]
- Kilgore, H.R.; Young, R.A. Learning the chemical grammar of biomolecular condensates. Nat. Chem. Biol. 2022, 18, 1298–1306. [Google Scholar] [CrossRef]
- McSwiggen, D.T.; Mir, M.; Darzacq, X.; Tjian, R. Evaluating phase separation in live cells: Diagnosis, caveats, and functional consequences. Genes Dev. 2019, 33, 1619–1634. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.C. Evidence for and against Liquid-Liquid Phase Separation in the Nucleus. Non-Coding RNA 2019, 5, 50. [Google Scholar] [CrossRef]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2020, 22, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.J.; Fox, E.J.; Loeb, L.A. Mutational Heterogeneity in Human Cancers: Origin and Consequences. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 51–75. [Google Scholar] [CrossRef] [PubMed]
- Sundar, R.; Chénard-Poirier, M.; Collins, D.C.; Yap, T.A. Imprecision in the Era of Precision Medicine in Non-Small Cell Lung Cancer. Front. Med. 2017, 4, 39. [Google Scholar] [CrossRef]
- Hou, J.; He, Z.; Liu, T.; Chen, D.; Wang, B.; Wen, Q.; Zheng, X. Evolution of Molecular Targeted Cancer Therapy: Mechanisms of Drug Resistance and Novel Opportunities Identified by CRISPR-Cas9 Screening. Front. Oncol. 2022, 12, 755053. [Google Scholar] [CrossRef]
- Crimini, E.; Repetto, M.; Tarantino, P.; Ascione, L.; Antonarelli, G.; Rocco, E.G.; Barberis, M.; Mazzarella, L.; Curigliano, G. Challenges and Obstacles in Applying Therapeutical Indications Formulated in Molecular Tumor Boards. Cancers 2022, 14, 3193. [Google Scholar] [CrossRef]
- Min, H.-Y.; Lee, H.-Y. Molecular targeted therapy for anticancer treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2011, 31, 195–208. [Google Scholar] [CrossRef]
- Lee, J.B.; Kim, H.R.; Ha, S.-J. Immune Checkpoint Inhibitors in 10 Years: Contribution of Basic Research and Clinical Application in Cancer Immunotherapy. Immune Netw. 2022, 22, e2. [Google Scholar] [CrossRef]
- Peng, S.; Bao, Y. A narrative review of immune checkpoint mechanisms and current immune checkpoint therapy. Ann. Blood 2022, 7, 33. [Google Scholar] [CrossRef]
- Calabrese, L.; Velcheti, V. Checkpoint immunotherapy: Good for cancer therapy, bad for rheumatic diseases. Ann. Rheum. Dis. 2016, 76, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Conroy, M.; Naidoo, J. Immune-related adverse events and the balancing act of immunotherapy. Nat. Commun. 2022, 13, 392. [Google Scholar] [CrossRef]
- Madden, D.L. From a Patient Advocate’s Perspective: Does Cancer Immunotherapy Represent a Paradigm Shift? Curr. Oncol. Rep. 2018, 20, 8. [Google Scholar] [CrossRef]
- Dempke, W.C.M.; Fenchel, K.; Uciechowski, P.; Dale, S.P. Second- and third-generation drugs for immuno-oncology treatment—The more the better? Eur. J. Cancer 2017, 74, 55–72. [Google Scholar] [CrossRef]
- Van Deventer, S.; Arp, A.B.; van Spriel, A.B. Dynamic Plasma Membrane Organization: A Complex Symphony. Trends Cell Biol. 2020, 31, 119–129. [Google Scholar] [CrossRef]
- Lezoualc’H, F.; Jockers, R. Bivalent Ligands as Specific Pharmacological Tools for G Protein-Coupled Receptor Dimers. Curr. Cancer Drug Targets 2008, 5, 312–318. [Google Scholar] [CrossRef]
- Jonas, K.; Hanyaloglu, A. Impact of G protein-coupled receptor heteromers in endocrine systems. Mol. Cell. Endocrinol. 2017, 449, 21–27. [Google Scholar] [CrossRef]
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L.F. Receptor-Receptor Interactions as a Widespread Phenomenon: Novel Targets for Drug Development? Front. Endocrinol. 2019, 10, 53. [Google Scholar] [CrossRef]
- Shonberg, J.; Scammells, P.J.; Capuano, B. Design Strategies for Bivalent Ligands Targeting GPCRs. Chemmedchem 2011, 6, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Onge, C.M.S.; Ma, H.; Zhang, Y. Design of bivalent ligands targeting putative GPCR dimers. Drug Discov. Today 2020, 26, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.J. Therapeutics—how to treat phase separation-associated diseases. Emerg. Top. Life Sci. 2020, 4, 331–342. [Google Scholar] [CrossRef]
- Mehta, S.; Zhang, J. Liquid–liquid phase separation drives cellular function and dysfunction in cancer. Nat. Rev. Cancer 2022, 22, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Tan, S.; Xia, L.; Wu, N.; Oyang, L.; Tang, Y.; Su, M.; Luo, X.; Wang, Y.; Sheng, X.; et al. Phase separation in Cancer: From the Impacts and Mechanisms to Treatment potentials. Int. J. Biol. Sci. 2022, 18, 5103–5122. [Google Scholar] [CrossRef]
- Igelmann, S.; Lessard, F.; Ferbeyre, G. Liquid–Liquid Phase Separation in Cancer Signaling, Metabolism and Anticancer Therapy. Cancers 2022, 14, 1830. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhuang, A.; Yu, J.; Chai, P.; Jia, R.; Ruan, J. Phase separation drives tumor pathogenesis and evolution: All roads lead to Rome. Oncogene 2022, 41, 1527–1535. [Google Scholar] [CrossRef]
- Swain, P.; Weber, S.C. Dissecting the complexity of biomolecular condensates. Biochem. Soc. Trans. 2020, 48, 2591–2602. [Google Scholar] [CrossRef]
- Jiang, S.; Fagman, J.B.; Chen, C.; Alberti, S.; Liu, B. Protein phase separation and its role in tumorigenesis. eLife 2020, 9, e60264. [Google Scholar] [CrossRef]
- Zhang, J.; Yue, W.; Zhou, Y.; Liao, M.; Chen, X.; Hua, J. Super enhancers—Functional cores under the 3D genome. Cell Prolif. 2020, 54, e12970. [Google Scholar] [CrossRef]
- Klein, I.A.; Boija, A.; Afeyan, L.K.; Hawken, S.W.; Fan, M.; Dall’Agnese, A.; Oksuz, O.; Henninger, J.E.; Shrinivas, K.; Sabari, B.R.; et al. Partitioning of cancer therapeutics in nuclear condensates. Science 2020, 368, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Lemos, C.; Schulze, L.; Weiske, J.; Meyer, H.; Braeuer, N.; Barak, N.; Eberspächer, U.; Werbeck, N.; Stresemann, C.; Lange, M.; et al. Identification of Small Molecules that Modulate Mutant p53 Condensation. Iscience 2020, 23, 101517. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alekseenko, I.; Kondratyeva, L.; Chernov, I.; Sverdlov, E. From the Catastrophic Objective Irreproducibility of Cancer Research and Unavoidable Failures of Molecular Targeted Therapies to the Sparkling Hope of Supramolecular Targeted Strategies. Int. J. Mol. Sci. 2023, 24, 2796. https://doi.org/10.3390/ijms24032796
Alekseenko I, Kondratyeva L, Chernov I, Sverdlov E. From the Catastrophic Objective Irreproducibility of Cancer Research and Unavoidable Failures of Molecular Targeted Therapies to the Sparkling Hope of Supramolecular Targeted Strategies. International Journal of Molecular Sciences. 2023; 24(3):2796. https://doi.org/10.3390/ijms24032796
Chicago/Turabian StyleAlekseenko, Irina, Liya Kondratyeva, Igor Chernov, and Eugene Sverdlov. 2023. "From the Catastrophic Objective Irreproducibility of Cancer Research and Unavoidable Failures of Molecular Targeted Therapies to the Sparkling Hope of Supramolecular Targeted Strategies" International Journal of Molecular Sciences 24, no. 3: 2796. https://doi.org/10.3390/ijms24032796