
Cell Immunotherapy against Melanoma: Clinical Trials Review
 ,
,  , , and
, , and
Abstract
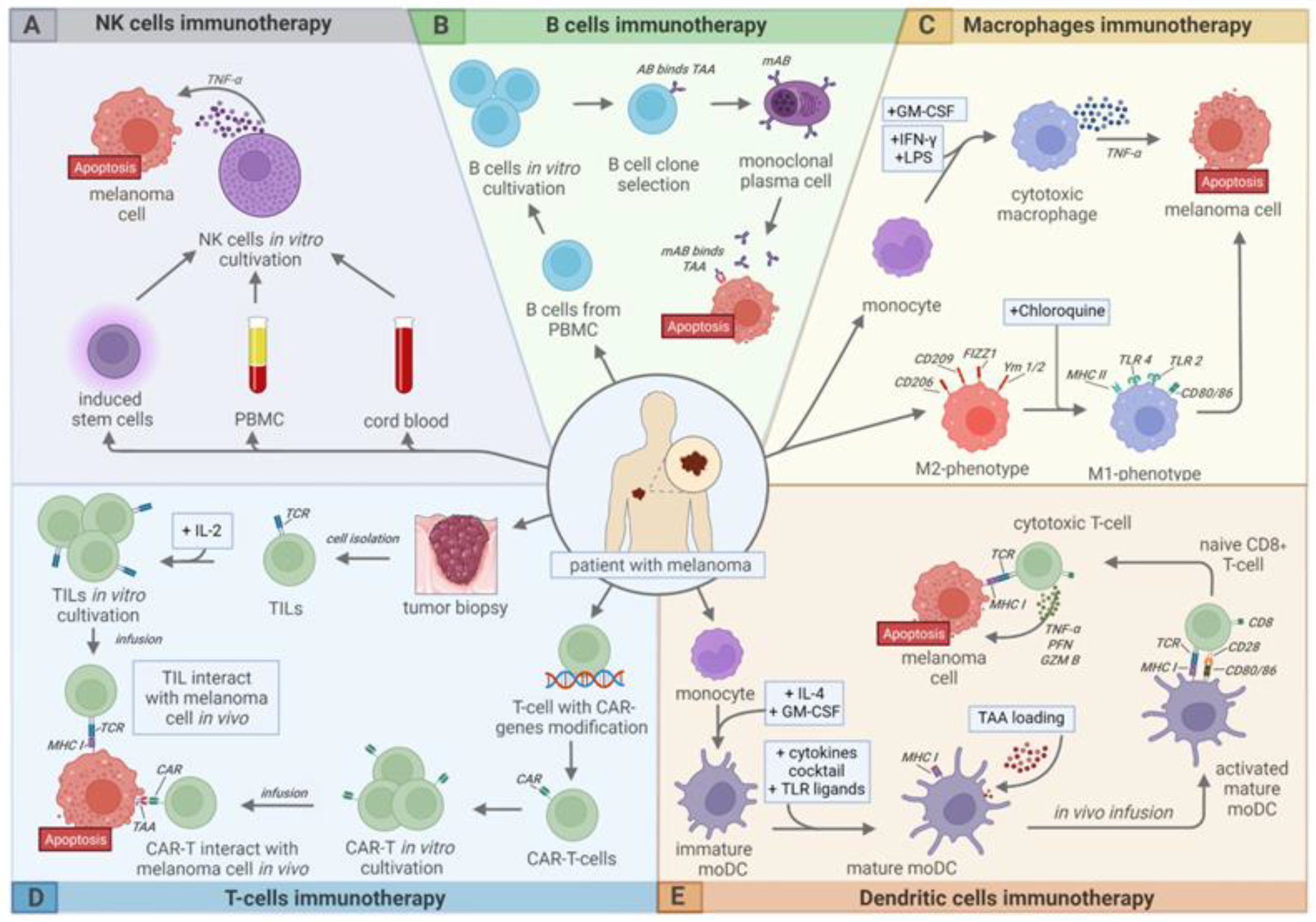
:1. Introduction
2. Melanoma
3. T-Cell Therapy
3.1. TIL Therapy
3.2. TCR Therapy
3.3. CAR T-Cell Therapy
3.4. NKT-Cell Therapy
4. B Cell Therapy
5. NK Cell Therapy
6. Dendritic Cell Therapy
6.1. Autologous DC-Based Therapy
6.2. Therapy Based on DC-Derived Exosomes
7. Macrophage-Based Cell Therapy
7.1. Cultivation of Cytotoxic Macrophages Ex Vivo
7.2. CAR-M Therapy
7.3. M2-to-M1 Repolarization
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular markers and targets in melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [Green Version]
- Premi, S. Role of melanin chemiexcitation in melanoma progression and drug resistance. Front. Oncol. 2020, 10, 1305. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA. Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Sondak, V.K.; Smalley, K.S.M. A brief history of melanoma: From mummies to mutations. Melanoma Res. 2012, 22, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasono, K.; Sato, K.; Han, D.C.; Fujii, Y.; Tsushima, T.; Shizume, K. Stimulation of alkaline phosphatase activity by thyroid hormone in mouse osteoblast-like cells (MC3T3-E1): A possible mechanism of hyperalkaline phosphatasia in hyperthyroidism. Bone Miner. 1988, 4, 355–363. [Google Scholar] [PubMed]
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo 2014, 28, 1005–1011. [Google Scholar]
- Bevona, C.; Goggins, W.; Quinn, T.; Fullerton, J.; Tsao, H. Cutaneous melanomas associated with nevi. Arch. Dermatol. 2003, 139, 1620–1624. [Google Scholar] [CrossRef]
- Isola, A.L.; Eddy, K.; Chen, S. Biology, therapy and implications of tumor exosomes in the progression of melanoma. Cancers 2016, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, J.E.; Truong, A.; Meyer, L.J. Genetic predisposition to melanoma. Semin. Oncol. 2016, 43, 591–597. [Google Scholar] [CrossRef]
- Bradford, P.T.; Goldstein, A.M.; McMaster, M.L.; Tucker, M.A. Acral lentiginous melanoma: Incidence and survival patterns in the United States, 1986–2005. Arch. Dermatol. 2009, 145, 427–434. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Overcoming immune evasion in melanoma. Int. J. Mol. Sci. 2020, 21, 8984. [Google Scholar] [CrossRef]
- Wang, Y.; Qiu, F.; Xu, Y.; Hou, X.; Zhang, Z.; Huang, L.; Wang, H.; Xing, H.; Wu, S. Stem cell-like memory T cells: The generation and application. J. Leukoc. Biol. 2021, 110, 1209–1223. [Google Scholar] [CrossRef]
- Krishna, S.; Lowery, F.J.; Copeland, A.R.; Bahadiroglu, E.; Mukherjee, R.; Jia, L.; Anibal, J.T.; Sachs, A.; Adebola, S.O.; Gurusamy, D.; et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 2020, 370, 1328–1334. [Google Scholar] [CrossRef]
- Li, Y.; Wu, D.; Yang, X.; Zhou, S. Immunotherapeutic potential of T memory stem cells. Front. Oncol. 2021, 11, 723888. [Google Scholar] [CrossRef]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Patterson, A.; Johnson, C.B.; Neitzke, D.J.; Mehrotra, S.; Denlinger, C.E.; Paulos, C.M.; Li, Z.; Cole, D.J.; Rubinstein, M.P. IL-2 and beyond in cancer immunotherapy. J. Interferon Cytokine Res. 2018, 38, 45–68. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the treatment of cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Rohaan, M.W.; Van Den Berg, J.H.; Kvistborg, P.; Haanen, J.B.A.G. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [CrossRef]
- Maibach, F.; Sadozai, H.; Seyed Jafari, S.M.; Hunger, R.E.; Schenk, M. Tumor-infiltrating lymphocytes and their prognostic value in cutaneous melanoma. Front. Immunol. 2020, 11, 2105. [Google Scholar] [CrossRef]
- Fu, Q.; Chen, N.; Ge, C.; Li, R.; Li, Z.; Zeng, B.; Li, C.; Wang, Y.; Xue, Y.; Song, X.; et al. Prognostic value of tumor-infiltrating lymphocytes in melanoma: A systematic review and meta-analysis. Oncoimmunology 2019, 8, 1593806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.E.; Jiang, Y.; Lorigan, P.C.; Thistlethwaite, F.C.; Pillai, M.; Thomas, M.; Kirilova, N.; Bridgeman, J.S.; Kueberuwa, G.; Guest, R.S.; et al. Clinical feasibility and treatment outcomes with unselected autologous tumor infiltrating lymphocyte therapy in patients with advanced cutaneous melanoma. Cancer Res. 2021, 81, LB150. [Google Scholar] [CrossRef]
- Khattar, M.; Burga, R.; Pedro, K.; Foley, C.; Lajoie, S.; Ocando, A.V.; Tremblay, J.; Thornton, D.; Tam, S.; Nabulsi, F.; et al. 1008P CytoTIL15: A novel TIL therapy for melanoma with superior potency and enhanced persistence without IL2 to improve safety & efficacy and expand patient eligibility. Ann. Oncol. 2021, 32, S852. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar] [CrossRef]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous Tumor-Infiltrating Lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef]
- Hopewell, E.L.; Cox, C.; Pilon-Thomas, S.; Kelley, L.L. Tumor-infiltrating lymphocytes: Streamlining a complex manufacturing process. Cytotherapy 2019, 21, 307–314. [Google Scholar] [CrossRef]
- Wolf, B.; Zimmermann, S.; Arber, C.; Irving, M.; Trueb, L.; Coukos, G. Safety and tolerability of adoptive cell therapy in cancer. Drug Saf. 2019, 42, 315–334. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.M.; Marincola, F.M.; Leitman, S.F.; Seipp, C.A.; et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J. Immunother. 2002, 25, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Tsimberidou, A.M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 1–22. [Google Scholar] [CrossRef]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer therapy with TCR-engineered T cells: Current strategies, challenges, and prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Rath, J.A.; Arber, C. Engineering strategies to enhance TCR-based adoptive T cell therapy. Cells 2020, 9, 1485. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T cell therapy for cancer in the clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Ping, Y.; Liu, C.; Zhang, Y. T-cell receptor-engineered T cells for cancer treatment: Current status and future directions. Protein Cell 2018, 9, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, F.; Sonnemann, H.; Jackson, K.R.; Talukder, A.H.; Katailiha, A.S.; Lizee, G. Evolution of CD8+ T Cell Receptor (TCR) engineered therapies for the treatment of cancer. Cells 2021, 10, 2379. [Google Scholar] [CrossRef]
- Hayes, C. Cellular immunotherapies for cancer. Ir. J. Med. Sci. 2021, 190, 41–57. [Google Scholar] [CrossRef]
- Yang, J.C. Toxicities associated with adoptive T-cell transfer for cancer. Cancer J. Sudbury Mass 2015, 21, 506–509. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yan, X.; Zhang, F.; Zhang, X.; Tang, F.; Han, Z.; Li, Y. TCR-T Immunotherapy: The challenges and solutions. Front. Oncol. 2022, 11, 794183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Wei, W.; Li, Y. TCR Engineered T cells for solid tumor immunotherapy. Exp. Hematol. Oncol. 2022, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. mAbs 2022, 14, 2014296. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.W. CAR-T cell therapy: Mechanism, management, and mitigation of inflammatory toxicities. Front. Immunol. 2021, 12, 2423. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Zhang, Q.; Ping, J.; Huang, Z.; Zhang, X.; Zhou, J.; Wang, G.; Liu, S.; Ma, J. CAR-T cell therapy in cancer: Tribulations and road ahead. J. Immunol. Res. 2020, 2020, 1924379. [Google Scholar] [CrossRef] [Green Version]
- Chandran, S.S.; Klebanoff, C.A. T Cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Soltantoyeh, T.; Akbari, B.; Karimi, A.; Chalbatani, G.M.; Ghahri-saremi, N.; Hadjati, J.; Hamblin, M.R.; Mirzaei, H.R. Chimeric Antigen Receptor (CAR) T cell therapy for metastatic melanoma: Challenges and road ahead. Cells 2021, 10, 1450. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Prudent, V.; Breitbart, W.S. Chimeric antigen receptor T-cell neuropsychiatric toxicity in acute lymphoblastic leukemia. Palliat. Support. Care 2017, 15, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Leyfman, Y. Chimeric antigen receptors: Unleashing a new age of anti-cancer therapy. Cancer Cell Int. 2018, 18, 182. [Google Scholar] [CrossRef]
- Zhang, Y.; Springfield, R.; Chen, S.; Li, X.; Feng, X.; Moshirian, R.; Yang, R.; Yuan, W. α-GalCer and INKT cell-based cancer immunotherapy: Realizing the therapeutic potentials. Front. Immunol. 2019, 10, 1126. [Google Scholar] [CrossRef] [Green Version]
- Webb, T.J.; Yuan, W.; Meyer, E.; Dellabona, P. Editorial: NKT cells in cancer immunotherapy. Front. Immunol. 2020, 11, 1314. [Google Scholar] [CrossRef]
- Morita, M.; Motoki, K.; Akimoto, K.; Natori, T.; Sakai, T.; Sawa, E.; Yamaji, K.; Koezuka, Y.; Kobayashi, E.; Fukushima, H. Structure-activity relationship of alpha-galactosylceramides against B16-bearing mice. J. Med. Chem. 1995, 38, 2176–2187. [Google Scholar] [CrossRef]
- Exley, M.A.; Friedlander, P.; Alatrakchi, N.; Vriend, L.; Yue, S.; Sasada, T.; Zeng, W.; Mizukami, Y.; Clark, J.; Nemer, D.; et al. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: A phase I clinical trial. Clin. Cancer Res. 2017, 23, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Kriegsmann, K.; Kriegsmann, M.; von Bergwelt-Baildon, M.; Cremer, M.; Witzens-Harig, M. NKT cells—New players in CAR cell immunotherapy? Eur. J. Haematol. 2018, 101, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.L.; Khan, N.; Basu, S.; Soni, V. B-cell-based immunotherapy: A promising new alternative. Vaccines 2022, 10, 879. [Google Scholar] [CrossRef]
- Thomas, A.; Smitha, T. Can B- cell based immunotherapy be our new perspective to exit cancer? J. Oral Maxillofac. Pathol. JOMFP 2020, 24, 15–17. [Google Scholar] [CrossRef]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandaru, M.; Rotte, A. Monoclonal antibodies for the treatment of melanoma: Present and future strategies. Methods Mol. Biol. 2019, 1904, 83–108. [Google Scholar] [CrossRef] [PubMed]
- Wennhold, K.; Shimabukuro-Vornhagen, A.; Von Bergwelt-Baildon, M. B cell-based cancer immunotherapy. Transfus. Med. Hemother. 2019, 46, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Natkanski, E.; Lee, W.Y.; Mistry, B.; Casal, A.; Molloy, J.E.; Tolar, P. B cells use mechanical energy to discriminate antigen affinities. Science 2013, 340, 1587–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, L.N.; Jiang, W.; Bhamidipati, K.; Millican, M.; Macaubas, C.; Hung, S.-C.; Mellins, E.D. The other function: Class II-restricted antigen presentation by B cells. Front. Immunol. 2017, 8, 319. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Pinto, D. B cells as antigen presenting cells. Cell. Immunol. 2005, 238, 67–75. [Google Scholar] [CrossRef]
- Lee, J.; Dollins, C.M.; Boczkowski, D.; Sullenger, B.A.; Nair, S. Activated B cells modified by electroporation of multiple MRNAs encoding immune stimulatory molecules are comparable to mature dendritic cells in inducing in vitro antigen-specific T-cell responses. Immunology 2008, 125, 229–240. [Google Scholar] [CrossRef]
- Wennhold, K.; Weber, T.M.; Klein-Gonzalez, N.; Thelen, M.; Garcia-Marquez, M.; Chakupurakal, G.; Fiedler, A.; Schlösser, H.A.; Fischer, R.; Theurich, S.; et al. CD40-activated B cells induce anti-tumor immunity in vivo. Oncotarget 2017, 8, 27740–27753. [Google Scholar] [CrossRef] [Green Version]
- Trefzer, U.; Weingart, G.; Chen, Y.; Herberth, G.; Adrian, K.; Winter, H.; Audring, H.; Guo, Y.; Sterry, W.; Walden, P. Hybrid cell vaccination for cancer immune therapy: First clinical trial with metastatic melanoma. J. Cancer 2000, 85, 618–626. [Google Scholar] [CrossRef]
- Van Vliet, A.A.; Georgoudaki, A.M.; Raimo, M.; de Gruijl, T.D.; Spanholtz, J. Adoptive NK cell therapy: A promising treatment prospect for metastatic melanoma. Cancers 2021, 13, 4722. [Google Scholar] [CrossRef]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10, 92. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, S.; Pal, P.; Lal, G. Key activating and inhibitory ligands involved in the mobilization of natural killer cells for cancer immunotherapies. ImmunoTargets Ther. 2021, 10, 387–407. [Google Scholar] [CrossRef]
- Urbonas, V.; Smailyte, G.; Urbonaite, G.V.; Dulskas, A.; Burokiene, N.; Kasiulevicius, V. Natural killer cell-based immunotherapy: A new fighter against melanoma? Melanoma Res. 2019, 29, 208–211. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.F.; et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Hallett, W.H.D.; Ames, E.; Motarjemi, M.; Barao, I.; Shanker, A.; Tamang, D.L.; Sayers, T.J.; Hudig, D.; Murphy, W.J. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J. Immunol. 2008, 180, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Galat, V.; Galat4, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. J. Hematol Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Rowley, D.A.; Fitch, F.W. The road to the discovery of dendritic cells, a tribute to Ralph Steinman. Cell. Immunol. 2012, 273, 95–98. [Google Scholar] [CrossRef]
- van Spriel, A.B.; de Jong, E.C. Dendritic cell science: More than 40 years of history. J. Leukoc. Biol. 2013, 93, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciscano, A.E.; Anandasabapathy, N. The role of dendritic cells in cancer and anti-tumor immunity. Semin. Immunol. 2021, 52, 101481. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic cells and their role in immunotherapy. Front. Immunol. 2020, 11, 1361. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.G.J.V.; De Goeje, P.L.; Cornelissen, R.; Kaijen-Lambers, M.E.H.; Bezemer, K.; Van Der Leest, C.H.; Mahaweni, N.M.; Kunert, A.; Eskens, F.A.L.M.; Waasdorp, C.; et al. Autologous dendritic cells pulsed with allogeneic tumor cell lysate in mesothelioma: From mouse to human. Clin. Cancer Res. 2018, 24, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.A.; Aamdal, S.; Dueland, S.; Sæbøe-Larsen, S.; Inderberg, E.M.; Madsbu, U.E.; Skovlund, E.; Gaudernack, G.; Kvalheim, G. Immune response and long-term clinical outcome in advanced melanoma patients vaccinated with tumor-MRNA-transfected dendritic cells. Oncoimmunology 2016, 5, e1232237. [Google Scholar] [CrossRef]
- Palucka, A.K.; Ueno, H.; Connolly, J.; Kerneis-Norvell, F.; Blanck, J.P.; Johnston, D.A.; Fay, J.; Banchereau, J. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J. Immunother. 2006, 29, 545–557. [Google Scholar] [CrossRef]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.J.G.; Duiveman-De Boer, T.; Van De Rakt, M.W.M.M.; Scharenborg, N.M.; De Boer, A.J.; Pots, J.M.; et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [Green Version]
- Markov, O.; Oshchepkova, A.; Mironova, N. Immunotherapy based on dendritic cell-targeted/-derived extracellular vesicles-a novel strategy for enhancement of the anti-tumor immune response. Front. Pharmacol. 2019, 10, 1152. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, L.H.; Vujanovic, L.; Santos, P.M.; Maurer, D.M.; Gambotto, A.; Lohr, J.; Li, C.; Waldman, J.; Chandran, U.; Lin, Y.; et al. Multiple antigen-engineered DC vaccines with or without IFNα to promote antitumor immunity in melanoma. J. Immunother. Cancer 2019, 7, 113. [Google Scholar] [CrossRef]
- Gross, S.; Erdmann, M.; Haendle, I.; Voland, S.; Berger, T.; Schultz, E.; Strasser, E.; Dankerl, P.; Janka, R.; Schliep, S.; et al. Twelve-year survival and immune correlates in dendritic cell-vaccinated melanoma patients. JCI Insight 2017, 2, e91438. [Google Scholar] [CrossRef] [Green Version]
- Kowal, J.; Tkach, M. Dendritic cell extracellular vesicles. Int. Rev. Cell Mol. Biol. 2019, 349, 213–249. [Google Scholar] [CrossRef]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Fernández-Delgado, I.; Calzada-Fraile, D.; Sánchez-Madrid, F. Immune regulation by dendritic cell extracellular vesicles in cancer immunotherapy and vaccines. Cancers 2020, 12, 3558. [Google Scholar] [CrossRef]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; André, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Ralpha. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef]
- Franken, L.; Schiwon, M.; Kurts, C. Macrophages: Sentinels and regulators of the immune system. Cell. Microbiol. 2016, 18, 475–487. [Google Scholar] [CrossRef]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Targeting macrophages in cancer: From bench to bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Poltavets, A.S.; Vishnyakova, P.A.; Elchaninov, A.V.; Sukhikh, G.T.; Fatkhudinov, T.K. Macrophage modification strategies for efficient cell therapy. Cells 2020, 9, 1535. [Google Scholar] [CrossRef]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The good, the bad, and the gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in M1(Lps+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Quillien, V.; Moisan, A.; Lesimple, T.; Leberre, C.; Toujas, L. Biodistribution of 111indium-labeled macrophages infused intravenously in patients with renal carcinoma. Cancer Immunol. Immunother. CII 2001, 50, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kivimäe, S.; Dolor, A.; Szoka, F.C. Macrophage-based cell therapies: The long and winding road. J. Control. Release 2016, 240, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatziioannou, E.; Aydin, S.A.; Forchhammer, S.; Sinnberg, T.; Eigentler, T. Melanoma-associated macrophages-from molecular signals to therapeutic application. Dermatol. Heidelb. Ger. 2022, 73, 915–928. [Google Scholar] [CrossRef]
- Hennemann, B.; Rehm, A.; Kottke, A.; Meidenbauer, N.; Andreesen, R. Adoptive immunotherapy with tumor-cytotoxic macrophages derived from recombinant human granulocyte-macrophage colony-stimulating factor (RhuGM-CSF) mobilized peripheral blood monocytes. J. Immunother. 1997, 20, 365–371. [Google Scholar] [CrossRef]
- Hennemann, B.; Beckmann, G.; Eichelmann, A.; Rehm, A.; Andreesen, R. Phase I trial of adoptive immunotherapy of cancer patients using monocyte-derived macrophages activated with interferon gamma and lipopolysaccharide. Cancer Immunol. Immunother. CII 1998, 45, 250–256. [Google Scholar] [CrossRef]
- Schwamberger, G.; Ferber, E.; Flesch, I. Characterization and partial purification of a high molecular weight tumoricidal activity secreted by murine bone marrow macrophages. Int. Immunol. 1992, 4, 253–264. [Google Scholar] [CrossRef]
- Su, S.; Lei, A.; Wang, X.; Lu, H.; Wang, S.; Yang, Y.; Li, N.; Zhang, Y.; Zhang, J. Induced CAR-macrophages as a novel therapeutic cell type for cancer immune cell therapies. Cells 2022, 11, 1652. [Google Scholar] [CrossRef]
- Sloas, C.; Gill, S.; Klichinsky, M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front. Immunol. 2021, 12, 783305. [Google Scholar] [CrossRef]
- Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal biology and function: Modern view of cellular debris bin. Cells 2020, 9, 1131. [Google Scholar] [CrossRef]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 873. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Therapy Type | Drug | Participants | Efficiency, % | References |
|---|---|---|---|---|
| TIL therapy entry 2 | Prior IL-2 Cyclophosphamide IL-2 Fludarabine 1200 total body irradiation | 26 (18-have not received prior IL-2) | CR-38.88 NAE-100 | NCT00314106 |
| (8-received prior IL-2) | CR-37.5 NAE-100 | |||
| Aldesleukin CD8+ enriched Young TIL | 12 (7, Aldesleukin) | CR-0; PR-14.28 PD-85.71; SD-0 NAE-100 | NCT01118091 | |
| 12 (5, Adoptive Cell Therapy) | CR-0; PR-20 PD-80; SD-0 NAE-100 | |||
| Prior IL-2 200cGy of total body irradiation TIL IL-2 | 23 | CR-4.34 PR-39.13 NAE-100 ACM-4.34 | NCT00096382 | |
| No prior IL-2 200cGy of total body irradiation TIL IL-2 | 3 | CR-33.33 PR-66.66 NAE-100 | ||
| TCR therapy | Tebentafusp (IMC10gp) | 252 | NAE-100 ACM-34,285 | NCT03070392 |
| Decarbazine Ipilimumab Pembrolizumab | 110 | NAE-99.09 ACM-51.35 | ||
| CAR-T therapy | Anti-VEGFR2 CAR CD8 plus PBL Cyclophosphamide Aldesleukin Fludarabine | 24 | Terminated (No objective responses were observed.) NAE-100 | NCT01218867 |
| NK therapy | FT500 Nivolumab Pembrolizumab Atezolizumab Cyclophosphamide Fludarabine IL-2 | 37 | ORR-24 ACM-<3 | NCT03841110 |
| Autologous NK-cells, Lymphodepleting chemotherapy | 8 | No clinical response | [76] | |
| B cell therapy | Hybrid cell vaccination Chemotherapy Immunotherapy IFN-α and/or IL-2Irradiation | 16 | CR-6; PR-6; SD-31; NAE-88 | [69] |
| DC therapy | AdVTMM2/DC Vaccination IFN | 35 | ORR-29 CR-12 NAE-51 NED-36.36 | NCT01622933 |
| Recombinant CD40-ligand Therapeutic autologous dendritic cells | 62 | OS-19 | NCT00053391 | |
| Exosomes from DCs DEX-based vaccine | 15 | ORR-50 | PMC2657211 | |
| Tumor lysate, particle-loaded, DC Placebo | 184 | OS (63-experimental group; 35-placebo group) | NCT02301611 | |
| Macrophage therapy | IFN-γ activated Monocyte-derived tumor-cytotoxic macrophages infusion Recombinant human GM-CSF | 12 | NAE I–II-100 SD-8.3 | [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filin, I.Y.; Mayasin, Y.P.; Kharisova, C.B.; Gorodilova, A.V.; Kitaeva, K.V.; Chulpanova, D.S.; Solovyeva, V.V.; Rizvanov, A.A. Cell Immunotherapy against Melanoma: Clinical Trials Review. Int. J. Mol. Sci. 2023, 24, 2413. https://doi.org/10.3390/ijms24032413
Filin IY, Mayasin YP, Kharisova CB, Gorodilova AV, Kitaeva KV, Chulpanova DS, Solovyeva VV, Rizvanov AA. Cell Immunotherapy against Melanoma: Clinical Trials Review. International Journal of Molecular Sciences. 2023; 24(3):2413. https://doi.org/10.3390/ijms24032413
Chicago/Turabian StyleFilin, Ivan Y., Yuri P. Mayasin, Chulpan B. Kharisova, Anna V. Gorodilova, Kristina V. Kitaeva, Daria S. Chulpanova, Valeriya V. Solovyeva, and Albert A. Rizvanov. 2023. "Cell Immunotherapy against Melanoma: Clinical Trials Review" International Journal of Molecular Sciences 24, no. 3: 2413. https://doi.org/10.3390/ijms24032413