
Cytokine Imbalance as a Biomarker of Intervertebral Disk Degeneration

 , ,
, ,
Abstract
:1. Introduction
2. Pathogenetic Aspect of Inflammation in Intervertebral Disk Degeneration
3. Cytokines Alteration in Intervertebral Disk Degeneration
3.1. Pro-Inflammatory Cytokines
3.1.1. Interleukin 1β
3.1.2. Interleukin 2
3.1.3. Interleukin 8
3.1.4. Interleukin 12
3.1.5. Interleukin 17
3.1.6. Interleukin 18
3.1.7. Tumor Necrosis Factor Alpha
3.1.8. Interferons
3.2. Anti-Inflammatory Cytokines
3.2.1. Interleukin 4
3.2.2. Interleukin 6
3.2.3. Interleukin 10
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luttsik, A.A.; Sadova, M.A.; Krutko, A.V.; Epifantsev, A.G.; Bondarenko, G.Y. Clinical recommendations of the All-Russian public organization “Association of Traumatologists and Orthopedists of Russia” (ATOR)—1. Degenerative-dystrophic diseases of the spine: Texbook. Novosibirsk 2012, 1–264. [Google Scholar]
- Byvaltsev, V.A.; Belykh, E.G.; Stepanov, I.A.; Morgan, G.; Mark, C.P. Cytokyne mechanism of intervertebral disk degeneration. Sib. Med. J. (Irkutsk) 2015, 6, 5–11. [Google Scholar]
- Li, Y.; Samartzis, D.; Campbell, D.D.; Cherny, S.S.; Cheung, K.M.; Luk, K.D.; Karppinen, J.; Song, Y.; Cheah, K.S.; Chan, D.; et al. Two subtypes of intervertebral disc degeneration distinguished by large-scale population-based study. Spine J. 2016, 9, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Byvaltsev, V.A.; Stepanov, I.A.; Bardonova, L.A.; Belykh, E.G. Degeneration of the intervertebral disc and the possibilities of tissue engineering. Surg. Spine 2017, 14, 60–67. [Google Scholar] [CrossRef]
- Zhang, Y.; Chee, A.; Thonar, E.J.; An, H.S. Intervertebral disk repair by protein, gene, or cell injection: A framework for rehabilitation-focused biologics in the spine. PM R. 2011, 6, 88–94. [Google Scholar] [CrossRef]
- Dowdell, J.; Erwin, M.; Choma, T.; Vaccaro, A.; Iatridis, J.; Cho, S.K. Intervertebral Disk Degeneration and Repair. Neurosurgery 2017, 80 (Suppl. 3), 46–54. [Google Scholar] [CrossRef]
- Kos, N.; Gradisnik, L.; Velnar, T. A Brief Review of the Degenerative Intervertebral Disc Disease. Med. Arch. 2019, 73, 421–424. [Google Scholar] [CrossRef]
- Hsieh, A.H.; Twomey, J.D. Cellular mechanobiology of the intervertebral disc: New directions and approaches. J. Biomech. 2010, 43, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Patil, P.; Niedernhofer, L.J.; Robbins, P.D.; Lee, J.; Sowa, G.; Vo, N. Cellular senescence in intervertebral disc aging and degeneration. Curr. Mol. Biol. Rep. 2018, 4, 180–190. [Google Scholar] [CrossRef]
- Brown, S.; Rodrigues, S.; Sharp, C.; Wade, K.; Broom, N.; McCall, I.W.; Roberts, S. Staying connected: Structural integration at the intervertebral disc-vertebra interface of human lumbar spines. Eur. Spine J. 2017, 26, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Grignon, B.; Grignon, Y.; Mainard, D.; Braun, M.; Netter, P.; Roland, J. The structure of the cartilaginous end-plates in elder people. Surg. Radiol. Anat. 2000, 22, 13–19. [Google Scholar] [CrossRef]
- Zieba, J.; Forlenza, K.N.; Khatra, J.S.; Sarukhanov, A.; Duran, I.; Rigueur, D.; Lyons, K.M.; Cohn, D.H.; Merrill, A.E.; Krakow, D. TGFβ and BMP Dependent Cell Fate Changes Due to Loss of Filamin B Produces Disc Degeneration and Progressive Vertebral Fusions. PLoS Genet. 2016, 12, e1005936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsingas, M.; Ottone, O.K.; Haseeb, A.; Barve, R.A.; Shapiro, I.M.; Lefebvre, V.; Risbud, M.V. Sox9 deletion causes severe intervertebral disc degeneration characterized by apoptosis; matrix remodeling; and compartment-specific transcriptomic changes. Matrix Biol. 2020, 94, 110–133. [Google Scholar] [CrossRef]
- Gkourogianni, A.; Andrew, M.; Tyzinski, L.; Crocker, M.; Douglas, J.; Dunbar, N.; Fairchild, J.; Funari, M.F.; Heath, K.E.; Jorge, A.A.; et al. Clinical Characterization of Patients With Autosomal Dominant Short Stature due to Aggrecan Mutations. J. Clin. Endocrinol. Metab. 2017, 102, 460–469. [Google Scholar] [CrossRef]
- Xue, J.; Song, Y.; Liu, H.; Liu, L.; Li, T.; Gong, Q. Vitamin D receptor gene polymorphisms and risk of intervertebral disc degeneration: An updated meta-analysis based on 23 studies. Medicine 2021, 100, e25922. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.Y.; Hong, T.; Li, C.Y.; Shi, C.L.; Liu, C.; Jiang, F.Y.; Li, J.; Fan, X.M.; Feng, S.B.; Wang, Y.F. Long non-coding RNA zinc finger antisense 1 expression associates with increased disease risk; elevated disease severity and higher inflammatory cytokines levels in patients with lumbar disc degeneration. Medicine 2019, 98, e18465. [Google Scholar] [CrossRef]
- Cazzanelli, P.; Wuertz-Kozak, K. MicroRNAs in Intervertebral Disc Degeneration; Apoptosis; Inflammation; and Mechanobiology. Int. J. Mol. Sci. 2020, 21, 3601. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Yang, C.; Yin, H.; Zhao, K.; Liu, W.; Hua, W.; Wang, K.; Song, Y.; Tu, J.; Li, S.; et al. MicroRNA-15b silencing inhibits IL-1β-induced extracellular matrix degradation by targeting SMAD3 in human nucleus pulposus cells. Biotechnol. Lett. 2017, 39, 623–632. [Google Scholar] [CrossRef]
- Hanaei, S.; Abdollahzade, S.; Sadr, M.; Mirbolouk, M.H.; Fattahi, E.; Khoshnevisan, A.; Rezaei, N. The role of interleukin 4 and IL-4RA in intervertebral disc degeneration: Investigation of single nucleotide polymorphisms in genes and a systematic review & meta-analysis of IL-4 expression level. Br. J. Neurosurg. 2020, 34, 66–71. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, S.; Wang, J.; Meng, D.; Wu, H.; Wei, Q.; Jiang, H. Gene polymorphisms and expression levels of interleukin-6 and interleukin-10 in lumbar disc disease: A meta-analysis and immunohistochemical study. J. Orthop. Surg. Res. 2020, 15, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Egan, B.; Wang, J. Genetic Factors in Intervertebral Disc Degeneration. Genes Dis. 2016, 3, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Q.; Ma, L.; Jain, A.; Crane, J.L.; Kebaish, K.H.; Wan, M.; Zhang, Z.; Edward Guo, X.; Sponseller, P.D.; Séguin, C.A.; et al. Mechanosignaling activation of TGF-β maintains intervertebral disc homeostasis. Bone Res. 2017, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Golubenko, E.O.; Silina, E.V.; Orlova, A.S. Personalized medicine in pain management. Modern science: Current problems of theory and practice. Ser. Nat. Tech. Sci. 2017, 7–8, 107–112. [Google Scholar]
- Shin, J.H.; Park, S.; Cho, H.; Kim, J.H.; Choi, H. Adipokine human Resistin promotes obesity-associated inflammatory intervertebral disc degeneration via pro-inflammatory cytokine cascade activation. Sci. Rep. 2022, 12, 8936. [Google Scholar] [CrossRef]
- Cannata, F.; Vadalà, G.; Ambrosio, L.; Fallucca, S.; Napoli, N.; Papalia, R.; Pozzilli, P.; Denaro, V. Intervertebral disc degeneration: A focus on obesity and type 2 diabetes. Diabetes Metab. Res. Rev. 2020, 36, e3224. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Zhang, L.L.; Mo, J.; Zhang, W.; Li, H.T.; Luo, Z.P.; Yang, H.L. Effect of Static Compression Loads on Intervertebral Disc: An in Vivo Bent Rat Tail Model. Orthop. Surg. 2018, 10, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Tian, Z.; Ma, X.; Yasen, M.; Mauck, R.L.; Qin, L.; Shofer, F.S.; Smith, L.J.; Pacifici, M.; Enomoto-Iwamoto, M.; Zhang, Y. Intervertebral Disc Degeneration in a Percutaneous Mouse Tail Injury Model. Am. J. Phys. Med. Rehabil. 2018, 97, 170–177. [Google Scholar] [CrossRef]
- Kiraz, M.; Demir, E. Relationship of lumbar disc degeneration with hemoglobin value and smoking. Neurochirurgie 2020, 66, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.K.; Moon, H.J.; Kwon, T.H.; Park, Y.K.; Kim, J.H. The Role of Hypoxia in Angiogenesis and Extracellular Matrix Regulation of Intervertebral Disc Cells During Inflammatory Reactions. Neurosurgery 2017, 81, 867–875. [Google Scholar] [CrossRef]
- Meng, X.; Zhuang, L.; Wang, J.; Liu, Z.; Wang, Y.; Xiao, D.; Zhang, X. Hypoxia-inducible factor (HIF)-1alpha knockout accelerates intervertebral disc degeneration in mice. Int. J. Clin. Exp. Pathol. 2018, 11, 548–557. [Google Scholar]
- Zhang, Y.; Si, M.; Li, C.; Liu, Y.; Han, Y.; Nie, L.; Wang, M. Effect of hyperlipidaemia to accelerate intervertebral disc degeneration in the injured rat caudal disc model. J. Orthop. Sci. 2019, 24, 42–49. [Google Scholar] [CrossRef]
- Beierfuß, A.; Hunjadi, M.; Ritsch, A.; Kremser, C.; Thomé, C.; Mern, D.S. APOE-knockout in rabbits causes loss of cells in nucleus pulposus and enhances the levels of inflammatory catabolic cytokines damaging the intervertebral disc matrix. PLoS ONE 2019, 14, e0225527. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, N.; Zhang, W.; Sun, T.; Zhang, D.; Chang, Y.; Li, J.; Li, Z. The Dual Effect of Abnormal Serum Uric Acid on Intervertebral Disc Degeneration. Oxid Med. Cell Longev. 2021, 2021, 2362799. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; González-Gay, M.Á.; Lago, F.; Karppinen, J.; Tervonen, O.; Mobasheri, A.; Gualillo, O. A new immunometabolic perspective of intervertebral disc degeneration. Nat. Rev. Rheumatol. 2022, 18, 47–60. [Google Scholar] [CrossRef]
- Natelson, D.M.; Lai, A.; Krishnamoorthy, D.; Hoy, R.C.; Iatridis, J.C.; Illien-Jünger, S. Leptin signaling and the intervertebral disc: Sex dependent effects of leptin receptor deficiency and Western diet on the spine in a type 2 diabetes mouse model. PLoS ONE 2020, 15, e0227527. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Jacobsen, T.D.; Maidhof, R.; Virojanapa, J.; Overby, C.; Bloom, O. Developments in intervertebral disc disease research: Pathophysiology; mechanobiology; and therapeutics. Curr. Rev. Musculoskelet. Med. 2015, 8, 18–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, T.; Akeda, K.; Yamada, J.; Takegami, N.; Sudo, T.; Sudo, A. Expression of the RANK/RANKL/OPG system in the human intervertebral disc: Implication for the pathogenesis of intervertebral disc degeneration. BMC Musculoskelet Disord. 2019, 20, 225. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Zhang, N.; Zhu, Z. Involvement of the G-Protein-Coupled Receptor 4 in the Increased Expression of RANK/RANKL/OPG System and Neurotrophins by Nucleus Pulposus Cells under the Degenerated Intervertebral Disc-Like Acidic Microenvironment. Biomed Res. Int. 2020, 2020, 1328436. [Google Scholar] [CrossRef]
- Han, Y.; Li, X.; Yan, M.; Yang, M.; Wang, S.; Pan, J.; Li, L.; Tan, J. Oxidative damage induces apoptosis and promotes calcification in disc cartilage endplate cell through ROS/MAPK/NF-κB pathway: Implications for disc degeneration. Biochem. Biophys. Res. Commun. 2019, 516, 1026–1032. [Google Scholar] [CrossRef]
- Grunhagen, T.; Shirazi-Adl, A.; Fairbank, J.C.; Urban, J.P. Intervertebral disk nutrition: A review of factors influencing concentrations of nutrients and metabolites. Orthop. Clin. North Am. 2011, 42, 465–477. [Google Scholar] [CrossRef]
- Zhao, K.; An, R.; Xiang, Q.; Li, G.; Wang, K.; Song, Y.; Liao, Z.; Li, S.; Hua, W.; Feng, X.; et al. Acid-sensing ion channels regulate nucleus pulposus cell inflammation and pyroptosis via the NLRP3 inflammasome in intervertebral disc degeneration. Cell Prolif. 2021, 54, e12941. [Google Scholar] [CrossRef]
- Wang, C.Y.; Hsieh, M.K.; Hu, Y.J.; Bit, A.; Lai, P.L. Monocarboxylate transporter 1-mediated lactate accumulation promotes nucleus pulposus degeneration under hypoxia in a 3D multilayered nucleus pulposus degeneration model. Eur. Cell Mater. 2022, 43, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.M.R.; Oliveira, L.Z.; Silva, M.B.R.D.; Accardo, C.M.; Giglio, A.B.D.; Pinhal, M.A.D.S. Inflammatory biomarkers in sera of patients with intervertebral disc degeneration. Einstein 2019, 17, eAO4637. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.Q.; Yong, Z.; Kuhn, A.; Riegger, J.; Goncalves, R.M.; Ruf, M.; Mauer, U.M.; Huber-Lang, M.; Ignatius, A.; Brenner, R.E.; et al. Interleukin-1β and cathepsin D modulate formation of the terminal complement complex in cultured human disc tissue. Eur. Spine J. 2021, 30, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Gullbrand, S.E.; Schaer, T.P.; Lau, Y.K.; Jiang, Z.; Dodge, G.R.; Elliott, D.M.; Mauck, R.L.; Malhotra, N.R.; Smith, L.J. Inflammatory cytokine and catabolic enzyme expression in a goat model of intervertebral disc degeneration. J. Orthop. Res. 2020, 38, 2521–2531. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, J.D.; Salehi, K.; Kanim, L.E.A.; NaPier, Z.; Kropf, M.A.; Cuéllar, J.M.; Perry, T.G.; Bae, H.W.; Sheyn, D. NF-κB inhibitor; NEMO-binding domain peptide attenuates intervertebral disc degeneration. Spine J. 2020, 20, 1480–1491. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Z.; Xie, Z.; Chen, Y.; Zheng, Z.; Wei, X.; Huang, B.; Shan, Z.; Liu, J.; Fan, S.; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 2020, 160, 552–565. [Google Scholar] [CrossRef]
- Shan, L.; Xu, X.; Zhang, J.; Cai, P.; Gao, H.; Lu, Y.; Shi, J.; Guo, Y.; Su, Y. Increased hemoglobin and heme in MALDI-TOF MS analysis induce ferroptosis and promote degeneration of herniated human nucleus pulposus. Mol. Med. 2021, 27, 103. [Google Scholar] [CrossRef]
- Gushcha, A.O.; Yusupova, A.R. Modern concepts of intervertebral disc degeneration. Zhurnal Vopr. Neirokhirurgii Im. N.N. Burdenko. 2020, 84, 112–117. [Google Scholar] [CrossRef]
- Sheng, B.; Li, X.; Zhou, L.; Zhou, J.; Guan, R.; Zhang, X. Targeting miR-10a-5p/IL-6R axis for reducing IL-6-induced cartilage cell ferroptosis. Exp. Mol. Pathol. 2021, 118, 104570. [Google Scholar] [CrossRef]
- Chao-Yang, G.; Peng, C.; Hai-Hong, Z. Roles of NLRP3 inflammasome in intervertebral disc degeneration. Osteoarthr. Cartil. 2021, 29, 793–801. [Google Scholar] [CrossRef]
- Terashima, Y.; Kakutani, K.; Yurube, T.; Takada, T.; Maeno, K.; Hirata, H.; Miyazaki, S.; Ito, M.; Kakiuchi, Y.; Takeoka, Y.; et al. Expression of adiponectin receptors in human and rat intervertebral disc cells and changes in receptor expression during disc degeneration using a rat tail temporary static compression model. J. Orthop. Surg. Res. 2016, 11, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuertz, K.; Haglund, L. Infl ammatory mediators in intervertebral disk degeneration and discogenic pain. Global. Spine J. 2013, 3, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekaran, S.; Soundararajan, D.C.R.; Tangavel, C.; Muthurajan, R.; Sri Vijay Anand, K.S.; Matchado, M.S.; Nayagam, S.M.; Shetty, A.P.; Kanna, R.M.; Dharmalingam, K. Human intervertebral discs harbour a unique microbiome and dysbiosis determines health and disease. Eur. Spine J. 2020, 29, 1621–1640. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lai, K.; Chopra, N.; Zheng, Z.; Das, A.; Diwan, A.D. Gut-disc axis: A cause of intervertebral disc degeneration and low back pain? Eur. Spine J. 2022, 31, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wang, Z.; Chen, J.; Zhang, Z.; Qian, H.; Chen, Y. Latent infection of low-virulence anaerobic bacteria in degenerated lumbar intervertebral discs. BMC Musculoskelet Disord. 2018, 19, 445. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, X.; Zhang, X.; Ren, H.; Huang, B.; Chen, J.; Liu, J.; Shan, Z.; Zhu, Z.; Zhao, F. Low virulence bacterial infections in cervical intervertebral discs: A prospective case series. Eur. Spine J. 2018, 27, 2496–2505. [Google Scholar] [CrossRef]
- Georgy, M.; Stern, M.; Murphy, K. What Is the Role of the Bacterium Propionibacterium acnes in Type 1 Modic Changes? A Review of the Literature. Can Assoc. Radiol. J. 2017, 68, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, S.; Tangavel, C.; Aiyer, S.N.; Nayagam, S.M.; Raveendran, M.; Demonte, N.L.; Subbaiah, P.; Kanna, R.; Shetty, A.P.; Dharmalingam, K. ISSLS PRIZE IN CLINICAL SCIENCE 2017: Is infection the possible initiator of disc disease? An insight from proteomic analysis. Eur. Spine J. 2017, 26, 1384–1400. [Google Scholar] [CrossRef]
- Lan, T.; Shen, Z.; Hu, Z.; Yan, B. Vitamin D/VDR in the pathogenesis of intervertebral disc degeneration: Does autophagy play a role? Biomed. Pharmacother. 2022, 148, 112739. [Google Scholar] [CrossRef]
- Biazzo, A.; Corriero, A.S.; Confalonieri, N. Intramuscular oxygen-ozone therapy in the treatment of low back pain. Acta Biomed. 2018, 89, 41–46. [Google Scholar] [CrossRef]
- Tchetina, E.V.; Markova, G.A. Regulation of energy metabolism in the growth plate and osteoarthritic chondrocytes. Rheumatol. Int. 2018, 38, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Wang, H.; Yang, X.; You, K.; Jiang, M.; Cai, F.; Zhang, Y.; Liu, L.; Liu, H.; Liu, X. Comprehensive Profile Analysis of Differentially Expressed circRNAs in Glucose Deprivation-Induced Human Nucleus Pulposus Cell Degeneration. Biomed. Res. Int. 2021, 2021, 4770792. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.Y.; Zhang, S.B.; Chen, H.; Xu, H.W.; Wang, S.J. Ascorbic acid promotes nucleus pulposus cell regeneration by regulating proliferation during intervertebral disc degeneration. J. Nutr. Biochem. 2022, 108, 109099. [Google Scholar] [CrossRef] [PubMed]
- Shnayder, N.A.; Petrova, M.M.; Popova, T.E.; Davidova, T.K.; Bobrova, O.P.; Trefilova, V.V.; Goncharova, P.S.; Balberova, O.V.; Petrov, K.V.; Gavrilyuk, O.A.; et al. Prospects for the Personalized Multimodal Therapy Approach to Pain Management via Action on NO and NOS. Molecules 2021, 26, 2431. [Google Scholar] [CrossRef]
- Xu, H.W.; Yi, Y.Y.; Zhang, S.B.; Hu, T.; Wang, S.J.; Zhao, W.D.; Wu, D.S. Does vitamin D status influence lumbar disc degeneration and low back pain in postmenopausal women? A retrospective single-center study. Menopause 2020, 27, 586–592. [Google Scholar] [CrossRef]
- Zhan, S.; Wang, K.; Song, Y.; Li, S.; Yin, H.; Luo, R.; Liao, Z.; Wu, X.; Zhang, Y.; Yang, C. Long non-coding RNA HOTAIR modulates intervertebral disc degenerative changes via Wnt/β-catenin pathway. Arthritis Res. Ther. 2019, 21, 201. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qu, Y.; Liu, L.; Zhao, H.; Ma, H.; Si, M.; Cheng, L.; Nie, L. PPAR-γ agonist pioglitazone protects against IL-17 induced intervertebral disc inflammation and degeneration via suppression of NF-κB signaling pathway. Int. Immunopharmacol. 2019, 72, 138–147. [Google Scholar] [CrossRef]
- He, D.; Zhou, M.; Bai, Z.; Wen, Y.; Shen, J.; Hu, Z. Propionibacterium acnes induces intervertebral disc degeneration by promoting nucleus pulposus cell pyroptosis via NLRP3-dependent pathway. Biochem. Biophys. Res. Commun. 2020, 526, 772–779. [Google Scholar] [CrossRef]
- Tang, G.; Han, X.; Lin, Z.; Qian, H.; Chen, B.; Zhou, C.; Chen, Y.; Jiang, W. Propionibacterium acnes Accelerates Intervertebral Disc Degeneration by Inducing Pyroptosis of Nucleus Pulposus Cells via the ROS-NLRP3 Pathway. Oxid. Med. Cell Longev. 2021, 2021, 4657014. [Google Scholar] [CrossRef]
- Bai, Z.; Liu, W.; He, D.; Wang, Y.; Yi, W.; Luo, C.; Shen, J.; Hu, Z. Protective effects of autophagy and NFE2L2 on reactive oxygen species-induced pyroptosis of human nucleus pulposus cells. Aging 2020, 12, 7534–7548. [Google Scholar] [CrossRef] [PubMed]
- He, W.S.; Zou, M.X.; Yan, Y.G.; Yao, N.Z.; Chen, W.K.; Li, Z.; Wang, W.J.; Ouyang, Z.H. Interleukin-17A Promotes Human Disc Degeneration by Inhibiting Autophagy Through the Activation of the Phosphatidylinositol 3-Kinase/Akt/Bcl2 Signaling Pathway. World Neurosurg. 2020, 143, 215–223. [Google Scholar] [CrossRef]
- Yang, M.; Peng, Y.; Liu, W.; Zhou, M.; Meng, Q.; Yuan, C. Sirtuin 2 expression suppresses oxidative stress and senescence of nucleus pulposus cells through inhibition of the p53/p21 pathway. Biochem. Biophys. Res. Commun. 2019, 513, 616–622. [Google Scholar] [CrossRef]
- Xie, J.; Li, B.; Zhang, P.; Wang, L.; Lu, H.; Song, X. Osteogenic protein-1 attenuates the inflammatory cytokine-induced NP cell senescence through regulating the ROS/NF-κB pathway. Biomed. Pharmacother. 2018, 99, 431–437. [Google Scholar] [CrossRef]
- Wang, K.; Chen, T.; Ying, X.; Zhang, Z.; Shao, Z.; Lin, J.; Xu, T.; Chen, Y.; Wang, X.; Chen, J.; et al. Ligustilide alleviated IL-1β induced apoptosis and extracellular matrix degradation of nucleus pulposus cells and attenuates intervertebral disc degeneration in vivo. Int. Immunopharmacol. 2019, 69, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Che, M.; Xin, J.; Zheng, Z.; Li, J.; Zhang, S. The role of IL-1β and TNF-α in intervertebral disc degeneration. Biomed. Pharmacother. 2020, 131, 110660. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, F.; Wu, Y.; Liu, N.; Wang, J.; Chen, R.; Lu, Z. In vitro resveratrol attenuates inflammation environment-induced nucleus pulposus cell senescence. Biosci. Rep. 2019, 39, BSR20190126. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, C.; Liang, A.; Peng, Y.; Sun, J.; Huang, D.; Xu, K.; Ye, W. Regulation of a disintegrins and metalloproteinase with thrombospondin motifs 7 during inflammation in nucleus pulposus (NP) cells: Role of AP-1; Sp1 and NF-κB signaling. Inflamm. Res. 2016, 65, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, K.; Li, X.; Pan, H.; Li, S.; Chen, F.; Zhang, J.; Zheng, Z.; Wang, J.; Liu, H. Wnt5a suppresses inflammation-driven intervertebral disc degeneration via a TNF-α/NF-κB-Wnt5a negative-feedback loop. Osteoarthr. Cartil. 2018, 26, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Peng, Y.; Sun, J.; Li, S.; Hong, J.; Zhou, J.; Chen, J.; Yan, J.; Huang, Z.; Wang, X.; et al. Nicotinamide phosphoribosyl transferase controls NLRP3 inflammasome activity through MAPK and NF-κB signaling in nucleus pulposus cells; as suppressed by melatonin. Inflammation 2020, 43, 796–809. [Google Scholar] [CrossRef]
- Navone, S.E.; Peroglio, M.; Guarnaccia, L.; Beretta, M.; Grad, S.; Paroni, M.; Cordiglieri, C.; Locatelli, M.; Pluderi, M.; Rampini, P.; et al. Mechanical loading of intervertebral disc modulates microglia proliferation; activation; and chemotaxis. Osteoarthr. Cartil. 2018, 26, 978–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omair, A.; Lie, B.A.; Reikeras, O.; Brox, J.I. An Association Study of Interleukin 18 Receptor Genes (IL18R1 and IL18RAP) in Lumbar Disc Degeneration. Open Orthop. J. 2012, 6, 164–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Liu, P.; Yang, D.; Wang, F.; Yuan, L.; Lin, Z.; Jiang, J. Interleukin-18-induced inflammatory responses in synoviocytes and chondrocytes from osteoarthritic patients. Int. J. Mol. Med. 2012, 30, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibin, M.K.; Harshitha, S.M.; Arati, S.; Chetan, G.K.; Shukla, P.D.; Bhat, I.D. IL18RAP polymorphisms and its plasma levels in patients with Lumbar disc degeneration. Clin. Neurol. Neurosurg. 2019, 184, 105374. [Google Scholar] [CrossRef]
- Millecamps, M.; Tajerian, M.; Naso, L.; Sage, E.H.; Stone, L.S. Lumbar intervertebral disc degeneration associated with axial and radiating low back pain in ageing SPARC-null mice. Pain 2012, 153, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Krock, E.; Millecamps, M.; Anderson, K.M.; Srivastava, A.; Reihsen, T.E.; Hari, P.; Stone, L.S. Interleukin-8 as a therapeutic target for chronic low back pain: Upregulation in human cerebrospinal fluid and pre-clinical validation with chronic reparixin in the SPARC-null mouse model. EBioMedicine 2019, 43, 487–500. [Google Scholar] [CrossRef] [Green Version]
- Krock, E.; Rosenzweig, D.H.; Currie, J.B.; Bisson, D.G.; Ouellet, J.A.; Haglund, L. Toll-like receptor activation induces degeneration of human intervertebral discs. Sci. Rep. 2017, 7, 17184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Likhitpanichkul, M.; Torre, O.M.; Gruen, J.; Walter, B.A.; Hecht, A.C.; Iatridis, J.C. Do mechanical strain and TNF-α interact to amplify pro-inflammatory cytokine production in human annulus fibrosus cells? J. Biomech. 2016, 49, 1214–1220. [Google Scholar] [CrossRef] [Green Version]
- Daniels, J.; Binch, A.A.L.; Le Maitre, C.L. Inhibiting IL-1 signaling pathways to inhibit catabolic processes in disc degeneration. J. Orthop. Res. 2017, 35, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.H.; Li, Z.P.; Liu, L.L.; Liu, H.; Xue, J.B. IL-17 in intervertebral disc degeneration: Mechanistic insights and therapeutic implications. Cell Biol. Int. 2022, 46, 535–547. [Google Scholar] [CrossRef]
- Cheng, L.; Fan, W.; Liu, B.; Wang, X.; Nie, L. Th17 Lymphocyte levels are higher in patients with ruptured than non-ruptured lumbar discs, and are correlated with pain intensity. Injury 2013, 44, 1805–1810. [Google Scholar] [CrossRef]
- Suyama, K.; Sakai, D.; Hirayama, N.; Nakamura, Y.; Matsushita, E.; Terayama, H.; Qu, N.; Tanaka, O.; Sakabe, K.; Watanabe, M. Effects of interleukin-17A in nucleus pulposus cells and its small-molecule inhibitors for intervertebral disc disease. J. Cell Mol. Med. 2019, 22, 5539–5551. [Google Scholar] [CrossRef] [PubMed]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; La Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Deng, Y.; Wang, T.; Ma, J.; Li, P.; Tian, P.; Han, C.; Ma, X. Interleukin-23 may contribute to the pathogenesis of lumbar disc herniation through the IL-23/IL-17 pathway. J. Orthop. Surg. Res. 2016, 11, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, K.; Hiyama, A.; Arai, F.; Nukaga, T.; Sakai, D.; Mochida, J. C-Fos regulation by the MAPK and PKC pathways in intervertebral disc cells. PloS ONE 2013, 8, e73210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suyama, K.; Sakai, D.; Watanabe, M. The role of IL-17-mediated inflammatory processes in the pathogenesis of intervertebral disc degeneration and herniation: A comprehensive review. Front. Cell Dev. Biol. 2022, 3, 857164. [Google Scholar] [CrossRef] [PubMed]
- Sunlong, L.; Chongan, H.; Jian, X.; Yuhao, W.; Zengjie, Z.; Yifei, Z.; Naifeng, T.; Yaosen, W.; Xiangyang, W.; Xiaolei, Z. The potential role of cytokines in diabetic intervertebral disc degeneration. Aging Dis. 2022, 13, 1323–1335. [Google Scholar] [CrossRef]
- Deng, X.; Zhao, F.; Kang, B.; Zhang, X. Elevated interleukin-6 expression levels are associated with intervertebral disc degeneration. Exp. Ther. Med. 2016, 11, 1425–1432. [Google Scholar] [CrossRef] [Green Version]
- Rigal, J.; Leglise, A.; Barnetche, T.; Cogniet, A.; Aunoble, S.; Le Huec, J.C. Meta-analysis of the effects of genetic polymorphisms on intervertebral disc degeneration. Eur. Spine J. 2017, 26, 2045–2052. [Google Scholar] [CrossRef]
- Huang, X.; Chen, F.; Zhao, J.; Wang, D.; Jing, S.; Li, H.; Meng, C. Interleukin 6 (IL-6) and IL-10 promoter region polymorphisms are associated with risk of lumbar disc herniation in a Northern Chinese Han population. Genet. Test. Mol. Biomark. 2017, 21, 17–23. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, H.; Suh, M.J.; Shin, J.H.; Hwang, M.H.; Lee, H.M. Effect of biphasic electrical current stimulation on IL-1β-stimulated annulus fibrosus cells using in vitro microcurrent generating chamber system. Spine 2013, 38, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Zhou, X.; Wang, J.; Xu, L.; Zhou, L.; Yu, W.; Tao, Y.; Zhu, J.; Hu, B.; Liang, C.; et al. Wogonin mitigates intervertebral disc degeneration through the Nrf2/ARE and MAPK signaling pathways. Int. Immunopharmacol. 2018, 65, 539–549. [Google Scholar] [CrossRef]
- Phillips, K.L.; Jordan-Mahy, N.; Nicklin, M.J.; Le Maitre, C.L. Interleukin-1 receptor antagonist deficient mice provide insights into pathogenesis of human intervertebral disc degeneration. Ann. Rheum. Dis. 2013, 72, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, L.; Zhu, L.; Zhang, C.; Li, Z.; Guo, Y.; Nie, Y.; Luo, Z. Aucubin inhibits IL-1β- or TNF-α-induced extracellular matrix degradation in nucleus pulposus cell through blocking the miR-140-5p/CREB1 axis. J. Cell. Physiol. 2019, 234, 13639–13648. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Ju, B.; Wang, H.; Lee, K.B. Bone morphogenetic protein-2 provokes interleukin-18-induced human intervertebral disc degeneration. Bone Jt. Res. 2016, 5, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Risbud, M.V.; Shapiro, I.M. Role of cytokines in intervertebral disc degeneration: Pain and disc content. Nat. Rev. Rheumatol. 2013, 10, 44–56. [Google Scholar] [CrossRef]
- Phillips, K.L.; Cullen, K.; Chiverton, N.; Michael, A.L.; Cole, A.A.; Breakwell, L.M.; Haddock, G.; Bunning, R.A.; Cross, A.K.; Le Maitre, C.; et al. Potential roles of cytokines and chemokines in human intervertebral disc degeneration: Interleukin-1 is a master regulator of catabolic processes. Osteoarthr. Cartil. 2015, 23, 1165–1177. [Google Scholar] [CrossRef] [Green Version]
- Studer, R.K.; Vo, N.; Sowa, G.; Ondeck, C.; Kang, J. Human nucleus pulposus cells react to IL-6: Independent actions and amplification of response to IL-1 and TNF-α. Spine 2011, 36, 593–599. [Google Scholar] [CrossRef]
- Dudli, S.; Haschtmann, D.; Ferguson, S.J. Fracture of the vertebral endplates; but not equienergetic impact load, promotes disc degeneration in vitro. J. Orthop. Res. 2012, 30, 809–816. [Google Scholar] [CrossRef]
- Sadowska, A.; Touli, E.; Hitzl, W.; Greutert, H.; Ferguson, S.J.; Wuertz-Kozak, K.; Hausmann, O.N. Inflammaging in cervical and lumbar degenerated intervertebral discs: Analysis of proinflammatory cytokine and TRP channel expression. Eur. Spine J. 2018, 27, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Gawri, R.; Rosenzweig, D.H.; Krock, E.; Ouellet, J.A.; Stone, L.S.; Quinn, T.M.; Haglund, L. High mechanical strain of primary intervertebral disc cells promotes secretion of inflammatory factors associated with disc degeneration and pain. Arthritis Res. Ther. 2014, 16, R21. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Nie, L.; Wang, Y.; Wang, X.P.; Zhao, H.; Dongol, S.; Maharjan, S.; Cheng, L. CCL20 Secretion from the nucleus pulposus improves the recruitment of CCR6-expressing Th17 cells to degenerated IDD tissues. PLoS ONE 2013, 8, e66286. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, L.; Wang, S.; Zhao, Y.; Liu, Y.; Li, J.; Nie, L.; Cheng, L. Production of CCL20 on nucleus pulposus cells recruits IL-17-producing cells to degenerated IDD tissues in rat models. J. Mol. Hist. 2016, 47, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, B.; Rosenzweig, D.H.; Krock, E.; Roughley, P.J.; Beckman, L.; Steffen, T.; Weber, M.H.; Ouellet, J.A.; Haglund, L. Acute mechanical injury of the human intervertebral disc: Link to degeneration and pain. Eur. Cell Mater. 2014, 28, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Li, X. Apigenin mitigates intervertebral disc degeneration through the amelioration of tumor necrosis factor α (TNF-α) signaling pathway. Med. Sci. Monit. 2020, 26, e924587. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fu, J.; Liu, Y.; Wu, Y.; Jiang, D. Osteogenic protein-1 inhibits nucleus pulposus cell apoptosis through regulating the NF-κB/ROS pathway in an inflammation environment. Biosci. Rep. 2018, 38, BSR20181530. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Li, X.; Jia, Z.; Lin, L.; Ying, J.; Wen, T.; Zhao, Y.; Guo, Z.; Zhao, X.; Li, D.; et al. The inflammatory cytokine TNF-α regulates the biological behavior of rat nucleus pulposus mesenchymal stem cells through the NF-κB signaling pathway in vitro. J. Cell Biochem. 2019, 120, 13664–13679. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xie, Z.; Yu, J.; Fu, L. Resveratrol inhibits IL-1β-mediated nucleus pulposus cell apoptosis through regulating the PI3K/Akt pathway. Biosci. Rep. 2019, 39, BSR20190043. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ding, W.; Yang, D.; Gu, T.; Yang, S.; Bai, Z. Different concentrations of 17β-estradiol modulates apoptosis induced by interleukin-1β in rat annulus fibrosus cells. Mol. Med. Rep. 2014, 10, 2745–2751. [Google Scholar] [CrossRef] [Green Version]
- Adamczak, S.E.; de Rivero Vaccari, J.P.; Dale, G.; Brand, F.J.; Nonner, D.; Bullock, M.R.; Dahl, G.P.; Dietrich, W.D.; Keane, R.W. Pyroptotic neuronal cell death mediated by the AIM2 in-flammasome. J. Cereb. Blood Flow Metab. 2014, 34, 621–629. [Google Scholar] [CrossRef]
- Ojala, J.O.; Sutinen, E.M. The role of interleukin-18, oxidative stress and metabolic syndrome in Alzheimer’s disease. J. Clin Med. 2017, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Gao, L.; Wang, H.X.; Ye, L.; Shi, Y.Y.; Yang, W.Y.; Li, Y.N.; Li, Y. Interleukin-18 inhibition protects against intervertebral disc degeneration via the inactivation of caspase-3/9 dependent apoptotic pathways. Immunol. Invest. 2022, 51, 1895–1907. [Google Scholar] [CrossRef] [PubMed]
- Sutinen, E.M.; Pirttila, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-beta production in human neuron-like cells. J. Neuroinflamm. 2012, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Zhou, F.; Deng, P.; Fan, Q.; Li, C.; Liu, Y.; Fu, X.; Zhou, Y.; Xu, X.; Sun, X. Interleukin 6 protects H(2)O(2)-induced cardiomyocytes injury through upregulation of prohibitin via STAT3 phosphorylation. Cell Biochem. Funct. 2012, 30, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Yu, Y.L.; Tang, C.L.; Lv, F.Z. Effects of TGF-β1 and IL-1β on expression of ADAMTS enzymes and TIMP-3 in human intervertebral disc degeneration. Exp. Ther. Med. 2013, 6, 1522–1526. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, L.; Xie, Z.Y.; Wang, F.; Sinkemani, A.; Zhang, C.; Wang, X.H.; Wang, K.; Hong, X.; Wu, X.T. Endoplasmic reticulum stress facilitates the survival and proliferation of nucleus pulposus cells in TNF-α stimulus by activating unfolded protein response DNA. Cell Biol. 2018, 37, 347–358. [Google Scholar] [CrossRef]
- Lin, X.; Lin, Q. MiRNA-495-3p attenuates TNF-α induced apoptosis and inflammation in human nucleus pulposus cells by targeting IL5RA. Inflammation 2020, 43, 1797–1805. [Google Scholar] [CrossRef]
- Hiyama, A.; Skubutyte, R.; Markova, D.; Anderson, D.G.; Yadla, S.; Sakai, D.; Mochida, J.; Albert, T.J.; Shapiro, I.M.; Risbud, M.V. Hypoxia activates the notch signaling pathway in cells of the intervertebral disc: Implications in degenerative disc disease. Arthritis Rheum. 2011, 63, 1355–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Tian, Y.; Wang, J.; Phillips, K.L.; Binch, A.L.; Dunn, S.; Cross, A.; Chiverton, N.; Zheng, Z.; Wang Shapiro, I.M.; et al. Inflammatory cytokines induce NOTCH signaling in nucleus pulposus cells: Implications in intervertebral disc degeneration. J. Biol. Chem. 2013, 288, 16761–16774. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Xie, Z.Y.; Liu, L.; Zhu, L.; Wang, F.; Fan, P.; Sinkemani, A.; Zhang, C.; Hong, X.; Wu, X.T. Nuclear factor-kappa B-dependent X-box binding protein 1 signalling promotes the proliferation of nucleus pulposus cells under tumour necrosis factor alpha stimulation. Cell Prolif. 2019, 52, e12542. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Lin, Q.; Ye, J.-J. Role of IL-17 in Nucleus Pulposus Cell Proliferation and Metabolism Cultured In Vitro. Asian Pac. J. Trop. Med. 2015, 8, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.B.; Yu, Y.B.; Wa, Q.B.; Zhou, J.W.; He, M.; Cen, Y. The role of quinazoline in ameliorating intervertebral disc degeneration by inhibiting oxidative stress and anti-inflammation via NF-κB/MAPKs signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Zuo, R.; Wang, Y.; Li, J.; Wu, J.; Wang, W.; Li, B.; Sun, C.; Wang, Z.; Shi, C.; Zhou, Y.; et al. Rapamycin Induced Autophagy Inhibits Inflammation-Mediated Endplate Degeneration by Enhancing Nrf2/Keap1 Signaling of Cartilage Endplate Stem Cells. Stem Cells 2019, 37, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gan, Y.; Xu, Y.; Wang, L.; Ouyang, B.; Zhang, C.; Luo, L.; Zhao, C.; Zhou, Q. 17beta-estradiol attenuates TNF-α-Induced premature senescence of nucleus pulposus cells through regulating the ROS/NF-κB pathway. Int. J. Biol. Sci. 2017, 13, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, A.; Han, C.; Chen, C.; Zhou, T.; Zhang, K.; Yang, X.; Chen, Z.; Qin, A.; Tian, H.; et al. Bone marrow-derived mesenchymal stem cells in three-dimensional co-culture attenuate degeneration of nucleus pulposus cells. Aging 2019, 11, 9167–9187. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, L.; Xie, Z.Y.; Wang, F.; Zhu, L.; Zhang, C.; Fan, P.; Sinkemani, A.; Hong, X.; Wu, X.T. Protein kinase RNA-like ER kinase/eukaryotic translation initiation factor 2α pathway attenuates tumor necrosis factor alpha-induced apoptosis in nucleus pulposus cells by activating autophagy. J. Cell Physiol. 2019, 234, 11631–11645. [Google Scholar] [CrossRef]
- Gruber, H.E.; Hoelscher, G.L.; Ingram, J.A.; Bethea, S.; Hanley, E.N. Autophagy in the degenerating human intervertebral disc: In vivo molecular and morphological evidence; and induction of autophagy in cultured annulus cells exposed to proinflammatory cytokines-implications for disc degeneration. Spine 2015, 40, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Ambjorn, M.; Ejlerskov, P.; Liu, Y.; Lees, M.; Jäättelä, M.; Issazadeh-Navikas, S. IFNB1/interferon-beta-induced autophagy in MCF-7 breast cancer cells counteracts its proapoptotic function. Autophagy 2013, 9, 287–302. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.W.; Kang, J.; Fan, X.D.; Sun, L.F. Expression and significance of VEGF and p53 in rat degenerated intervertebral disc tissues. Asian Pac. J. Trop. Med. 2013, 6, 404–406. [Google Scholar] [CrossRef]
- Moon, H.J.; Yurube, T.; Lozito, T.P.; Pohl, P.; Hartman, R.A.; Sowa, G.A.; Kang, J.D.; Vo, N.V. Effects of secreted factors in culture medium of annulus fibrosus cells on microvascular endothelial cells: Elucidating the possible pathomechanisms of matrix degradation and nerve in-growth in disc degeneration. Osteoarthr. Cartil. 2014, 22, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Isa, I.L.; Srivastava, A.; Tiernan, D.; Owens, P.; Rooney, P.; Dockery, P.; Pandit, A. Hyaluronic acid based hydrogels attenuate inflammatory receptors and neurotrophins in Interleukin-1β induced inflammation model of nucleus pulposus cells. Biomacromolecules 2015, 16, 1714–1725. [Google Scholar] [CrossRef]
- Miyagi, M.; Millecamps, M.; Danco, A.T.; Ohtori, S.; Takahashi, K.; Stone, L.S. ISSLS Prize winner: Increased innervation and sensory nervous system plasticity in a mouse model of low back pain due to intervertebral disc degeneration. Spine 2014, 39, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.M.; Shan, Z.Z.; Nakamura, H.; Masuko-Hongo, K.; Kato, T.; Nishioka, K.; Yudoh, K. Catabolic stress induces features of chondrocyte senescence through overexpression of caveolin 1: Possible involvement of caveolin 1-induced down-regulation of articular chondrocytes in the pathogenesis of osteoarthritis. Arthritis Rheum. 2006, 54, 818–831. [Google Scholar] [CrossRef]
- Shnayder, N.A.; Khasanova, A.K.; Strelnik, A.I.; Al-Zamil, M.; Otmakhov, A.P.; Neznanov, N.G.; Shipulin, G.A.; Petrova, M.M.; Garganeeva, N.P.; Nasyrova, R.F. Cytokine Imbalance as a Biomarker of Therapeutic Resistance to Antipsychotics. Int. J. Mol. Sci. 2022, 23, 11324. [Google Scholar] [CrossRef]
- Zádor, F.; Joca, S.; Nagy-Grócz, G.; Dvorácskó, S.; Szűcs, E.; Tömböly, C.; Benyhe, S.; Vécsei, L. Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression. Int. J. Mol. Sci. 2021, 22, 5903. [Google Scholar] [CrossRef]
- Migliorini, P.; Italiani, P.; Pratesi, F.; Puxeddu, I.; Boraschi, D. The IL-1 family cytokines and receptors in autoimmune diseases. Autoimmun. Rev. 2020, 19, 102617. [Google Scholar] [CrossRef]
- Dinarello, C.A. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 612–632. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, D.; Li, Y.; Wang, W.; Bei, W.; Guo, J. NLRP3 inflammasome and IL-1β pathway in type 2 diabetes and atherosclerosis: Friend or foe? Pharmacol Res. 2021, 173, 105885. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Cardona, A.E. The IL-1β phenomena in neuroinflammatory diseases. J. Neural Transm. 2018, 125, 781–795. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Wu, L.; Lin, W.; Cai, Y.; Zhang, Y.; Hu, B.; Gao, R.; Im, H.J.; Yuan, W.; Ye, X.; et al. MiR-202-3p regulates interleukin-1β-induced expression of matrix metalloproteinase 1 in human nucleus pulposus. Gene 2019, 687, 156–165. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Phillips, F.M.; An, H.S.; Ellman, M.; Thonar, E.J.; Wu, W.; Park, D.; Im, H.J. The action of resveratrol, a phytoestrogen found in grapes, on the intervertebral disc. Spine 2008, 33, 2586–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathy-Hartert, M.; Hogge, L.; Sanchez, C.; Deby-Dupont, G.; Crielaard, J.M.; Henrotin, Y. Interleukin-1beta and interleukin-6 disturb the antioxidant enzyme system in bovine chondrocytes: A possible explanation for oxidative stress generation. Osteoarthr. Cartil. 2008, 16, 756–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Online Mendelian Inheritance in Man (OMIM®); McKusick-Nathans Institute of Genetic Medicine; Johns Hopkins University (Baltimore). World Wide Web. 2020. Available online: https://omim.org/ (accessed on 12 November 2022).
- Teodorczyk-Injeyan, J.A.; Triano, J.J.; Injeyan, H.S. Nonspecific low Back pain: Inflammatory profiles of patients with acute and chronic pain. Clin. J Pain. 2019, 35, 818. [Google Scholar] [CrossRef]
- Weber, K.T.; Alipui, D.O.; Sison, C.P.; Bloom, O.; Quraishi, S.; Overby, M.C.; Levine, M.; Chahine, N.O. Serum levels of the proinflammatory cytokine interleukin-6 vary based on diagnoses in individuals with lumbar intervertebral disc diseases. Arthritis Res. Ther. 2016, 18, 3. [Google Scholar] [CrossRef] [Green Version]
- Qazi, B.S.; Tang, K.; Qazi, A. Recent advances in underlying pathologies provide insight into interleukin-8 expression-mediated infammation and angiogenesis. Int. J. Inflam. 2011, 2011, 908468. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, L.M.; Schistad, E.; Jacobsen, L.M.; Røe, C.; Gjerstad, J. Serum levels of the pro-infammatory interleukins 6 (IL-6) and -8 (IL-8) in patients with lumbar radicular pain due to disc herniation: A 12-month prospective study. Brain Behav. Immun. 2015, 46, 132–136. [Google Scholar] [CrossRef]
- Klawitter, M.; Hakozaki, M.; Kobayashi, H.; Krupkova, O.; Quero, L.; Ospelt, C.; Gay, S.; Hausmann, O.; Liebscher, T.; Meier, U.; et al. Expression and regulation of toll-like receptors (TLRs) in human intervertebral disc cells. Eur. Spine J. 2014, 23, 1878–1891. [Google Scholar] [CrossRef] [Green Version]
- Shamji, M.F.; Setton, L.A.; Jarvis, W.; So, S.; Chen, J.; Jing, L.; Bullock, R.; Isaacs, R.E.; Brown, C.; Richardson, W.J. Proinflammatory cytokine expression profile in degenerated and herniated human intervertebral disc tissues. Arthritis Rheum. 2010, 62, 1974–1982. [Google Scholar]
- Shamji, M.F.; Guha, D.; Paul, D.; Shcharinsky, A. Systemic inflammatory and Th17 immune activation among patients treated for lumbar radiculopathy exceeds that of patients treated for persistent postoperative neuropathic pain. Neurosurgery 2017, 81, 537–544. [Google Scholar] [CrossRef]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef]
- Xue, H.; Yao, Y.; Wang, X.; Zhang, F.; Jiang, X.; Liu, J.; Wang, H.; Li, Y.; Wang, X.; Li, H.; et al. Interleukin-21 Is Associated with the Pathogenesis of Lumbar Disc Herniation. Iran J. Allergy Asthma Immunol. 2015, 14, 509–518. [Google Scholar] [PubMed]
- Harper, E.G.; Guo, C.; Rizzo, H.; Lillis, J.V.; Kurtz, S.E.; Skorcheva, I.; Purdy, D.; Fitch, E.; Iordanov, M.; Blauvelt, A. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: Implications for psoriasis pathogenesis. J. Invest. Dermatol. 2009, 129, 2175–2183. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pène, J.; Chevalier, S.; Preisser, L.; Vénéreau, E.; Guilleux, M.-H.; Ghannam, S.; Molès, J.P.; Danger, Y.; Ravon, E.; Lesaux, S.; et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J. Immunol. 2008, 180, 7423–7430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Zhang, H.H.; Foley, J.F.; Hedrick, M.N.; Farber, J.M. Human T Cells that Are Able to Produce IL-17 Express the Chemokine Receptor CCR6. J. Immunol. 2008, 180, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Hiyama, A.; Suyama, K.; Sakai, D.; Tanaka, M.; Watanabe, M. Correlational analysis of chemokine and inflammatory cytokine expression in the intervertebral disc and blood in patients with lumbar disc disease. J. Orthop. Res. 2022, 40, 1213–1222. [Google Scholar] [CrossRef]
- Gu, C.; Wu, L.; Li, X. IL-17 Family: Cytokines; receptors and signaling. Cytokine 2013, 64, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor Necrosis Factor Receptor - Associated Factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef] [Green Version]
- Zepp, J.A.; Liu, C.; Qian, s.; Wu, L.; Gulen, M.F.; Kang, Z.; Li, X. Cutting edge: TNF receptor-associated factor 4 restricts IL-17-mediated pathology and Signaling processes. J. Immunol. 2012, 189, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Chen, X.; Zhao, J.; Martin, B.; Zepp, J.A.; Ko, J.S.; Gu, C.; Cai, G.; Ouyang, W.; Sen, G.; et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J. Exp. Med. 2015, 212, 1571–1587. [Google Scholar] [CrossRef]
- Zhu, S.; Pan, W.; Shi, P.; Gao, H.; Zhao, F.; Song, X.; Liu, Y.; Zhao, L.; Li, X.; Shi, Y.; et al. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin-17 receptor signaling. J. Exp. Med. 2010, 207, 2647–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.K.; Nie, L.; Zhao, Y.P.; Zhang, Y.Q.; Wang, X.; Wang, S.S.; Liu, Y.; Zhao, H.; Cheng, L. IL-17 mediates inflammatory reactions via p38/c-Fos and JNK/c-Jun activation in an AP-1-dependent manner in human nucleus pulposus cells. J. Transl. Med. 2016, 14, 77. [Google Scholar] [CrossRef] [Green Version]
- Musilli, C.; Paccosi, S.; Pala, L.; Gerlini, G.; Ledda, F.; Mugelli, A.; Rotella, C.M.; Parenti, A. Characterization of circulating and monocyte-derived dendritic cells in obese and diabetic patients. Mol. Immunol. 2011, 49, 234–238. [Google Scholar] [CrossRef]
- Sambrook, P.N.; MacGregor, A.J.; Spector, T.D. Genetic influences on cervical and lumbar disc degeneration: A magnetic resonance imaging study in twins. Arthritis Rheum. 1999, 42, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Zitting, P.; Rantakallio, P.; Vanharanta, H. Cumulative incidence of lumbar disc diseases leading to hospitalization up to the age of 28 years. Spine 1998, 23, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Cabal-Hierro, L.; Lazo, P.S. Signal transduction by tumor necrosis factor receptors. Cell Signal 2012, 24, 1297–1305. [Google Scholar] [CrossRef]
- Murata, Y.; Onda, A.; Rydevik, B.; Takahashi, I.; Takahashi, K.; Olmarker, K. Changes in pain behavior and histologic changes caused by application of tumor necrosis factor-alpha to the dorsal root ganglion in rats. Spine 2006, 31, 530–535. [Google Scholar] [CrossRef]
- Byvaltsev, V.A.; Garashchenko, N.E.; Stepanov, I.A.; Aldiyarova, N.T. Role of different signal-transductor systems in intervertebral discs degeneration. Genes Cells 2017, XII, 4. [Google Scholar] [CrossRef]
- Gabr, M.A.; Jing, L.; Helbling, A.R.; Sinclair, S.M.; Allen, K.D.; Shamji, M.F.; Richardson, W.J.; Fitch, R.D.; Setton, L.A.; Chen, J. Interleukin-17 synergizes with IFNγ or TNFα to promote inflammatory mediator release and intercellular adhesion molecule-1 (ICAM-1) expression in human intervertebral disc cells. J. Orthop. Res. 2011, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fei, H.D.; Sun, Z.Y.; Tian, J.W. Bioinformatic analysis of the microarray gene expression profile in degenerative intervertebral disc cells exposed to TNF-α. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3332–3339. [Google Scholar]
- Gruber, H.E.; Hoelscher, G.L.; Ingram, J.A.; Bethea, S.; Norton, H.J.; Hanley, E.N. Production and expression of RANTES (CCL5) by human disc cells and modulation by IL-1-β and TNF-α in 3D culture. Exp. Mol. Pathol. 2014, 96, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, Y.; Phillips, K.L.; Chiverton, N.; Haddock, G.; Bunning, R.A.; Cross, A.K.; Shapiro, I.M.; Le Maitre, C.L.; Risbud, M.V. Tumor necrosis factor α- and interleukin-1β-dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis Rheum. 2013, 65, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.E.; Hoelscher, G.L.; Ingram, J.A.; Bethea, S.; Cox, M.; Hanley, E.N. Proinflammatory cytokines modulate the chemokine CCL2 (MCP-1) in human annulus cells in vitro: CCL2 expression and production. Exp. Mol. Pathol. 2015, 98, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Moen, G.-H.; Moen, A.; Schistad, E.I.; Gjerstad, J. Local up-regulation of interferon-γ (IFN-γ) following disc herniation is involved in the inflammatory response underlying acute lumbar radicular pain. Cytokine 2017, 97, 181–186. [Google Scholar] [CrossRef]
- Kamieniak, P.; Bielewicz, J.; Grochowski, C.; Litak, J.; Bojarska-Junak, A.; Daniluk, B.; Trojanowski, T. The elevated serum level of IFN-γ in patients with failed Back surgery syndrome remains unchanged after spinal cord stimulation. Dis Markers 2019, 2019, 2606808. [Google Scholar] [CrossRef] [Green Version]
- Hanaei, S.; Abdollahzade, S.; Sadr, M.; Mirbolouk, M.H.; Fattahi, E.; Khoshnevisan, A.; Rezaei, N. Association of interleukin 2, interleukin 12, and interferon-γ with intervertebral disc degeneration in Iranian population. BMC Med. Genet. 2020, 21, 143. [Google Scholar] [CrossRef]
- Te Velde, A.A.; Klomp, J.P.; Yard, B.A.; de Vries, J.E.; Figdor, C.G. Modulation of phenotypic and functional properties of human peripheral blood monocytes by IL-4. J. Immunol. 1988, 140, 1548–1554. [Google Scholar] [CrossRef]
- Assirelli, E.; Pulsatelli, L.; Dolzani, P.; Platano, D.; Olivotto, E.; Filardo, G.; Trisolino, G.; Facchini, A.; Borzì, R.M.; Meliconi, R. Human osteoarthritic cartilage shows reduced in vivo expression of IL-4; a chondroprotective cytokine that differentially modulates IL-1beta-stimulated production of chemokines and matrixdegrading enzymes in vitro. PLoS ONE 2014, 9, e96925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.B.; Wong, H.L.; Costa, G.L.; Bienkowski, M.J.; Wahl, S.M. Suppression of monocyte function and differential regulation of IL-1 and IL1ra by IL-4 contribute to resolution of experimental arthritis. J. Immunol. 1993, 151, 4344–4351. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, M.; Nishida, K.; Shimizu, A.; Doi, H.; Miyazawa, S.; Komiyama, T.; Nasu, Y.; Yoshida, A.; Watanabe, S.; Ozaki, T. Intra-articular injection of interleukin-4 decreases nitric oxide production by chondrocytes and ameliorates subsequent destruction of cartilage in instability-induced osteoarthritis in rat knee joints. Osteoarthr. Cartil. 2008, 16, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Imagawa, T.; Boivin, G.P.; Gao, G.; Wilson, J.M.; Hirsch, R. Adeno-associated virus mediates long-term gene transfer and delivery of chondroprotective IL-4 to murine synovium. Mol. Ther. 2000, 2, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Kawalkowska, J.Z.; Hemmerle, T.; Pretto, F.; Matasci, M.; Neri, D.; Williams, R.O. Targeted IL-4 therapy synergizes with dexamethasone to induce a state of tolerance by promoting Treg cells and macrophages in mice with arthritis. Eur. J. Immunol. 2016, 46, 1246–1257. [Google Scholar] [CrossRef] [Green Version]
- Kedong, H.; Wang, D.; Sagaram, M.; An, H.S.; Chee, A. Anti-inflammatory effects of interleukin-4 on intervertebral disc cells. Spine J. 2020, 20, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowenthal, J.W.; Castle, B.E.; Christiansen, J.; Schreurs, J.; Rennick, D.; Arai, N.; Hoy, P.; Takebe, Y.; Howard, M. Expression of high affinity receptors for murine interleukin 4 (BSF-1) on hemopoietic and nonhemopoietic cells. J. Immunol. 1988, 140, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Physiological effects of modulating the interleukin-6 axis. Rheumatology 2018, 57, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Bao, J.P.; Yang, S.; Hong, X.; Liu, L.; Xie, X.H.; Wu, X.T. A cohort study comparing the serum levels of pro-or anti-inflammatory cytokines in patients with lumbar radicular pain and healthy subjects. Eur. Spine J. 2016, 25, 1428–1434. [Google Scholar] [CrossRef]
- Geiss, A.; Larsson, K.; Junevik, K.; Rydevik, B.; Olmarker, K. Autologous nucleus pulposus primes T cells to develop into interleukin-4-producing effector cells: An experimental study on the autoimmune properties of nucleus pulposus. J. Orthop. Res. 2009, 27, 97–103. [Google Scholar] [CrossRef]
- Akyol, S.; Eraslan, B.S.; Etyemez, H.; Tanriverdi, T.; Hanci, M. Catabolic cytokine expressions in patients with degenerative disc disease. Turk Neurosurg. 2010, 20, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Park, J.B.; Chang, H.; Kim, Y.S. The pattern of interleukin-12 and T-helper types 1 and 2 cytokine expression in herniated lumbar disc tissue. Spine 2002, 27, 2125–2128. [Google Scholar] [CrossRef] [PubMed]
- Capossela, S.; Pavlicek, D.; Bertolo, A.; Landmann, G.; Stoyanov, J.V. Unexpectedly decreased plasma cytokines in patients with chronic back pain. J. Pain Res. 2018, 11, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Taga, T.; Nakano, N.; Yasukawa, K.; Kashiwamura, S.; Shimizu, K.; Nakajima, K.; Pyun, K.H.; Kishimoto, T. Purification to homogeneity and characterization of human B-cell differentiation factor (BCDF or BSFp-2). Proc. Natl. Acad. Sci. USA 1985, 82, 5490–5494. [Google Scholar] [CrossRef] [Green Version]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Kishimoto, T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol. Rev. 1998, 127, 25–50. [Google Scholar] [CrossRef]
- Ishihara, K.; Hirano, T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev. 2002, 13, 357–368. [Google Scholar] [CrossRef]
- Sainoh, T.; Orita, S.; Miyagi, M.; Sakuma, Y.; Yamauchi, K.; Suzuki, M.; Kubota, G.; Oikawa, Y.; Inage, K.; Sato, J.; et al. Interleukin-6 and interleukin-6 receptor expression; localization; and involvement in pain-sensing neuron activation in a mouse intervertebral disc injury model. J. Orthop. Res. 2015, 95, 123–127. [Google Scholar] [CrossRef]
- Murata, Y.; Rydevik, B.; Nannmark, U.; Larsson, K.; Takahashi, K.; Kato, Y.; Olmarker, K. Local application of interleukin-6 to the dorsal root ganglion induces tumor necrosis factor-α in the dorsal root ganglion and results in apoptosis of the dorsal root ganglion cells. Spine 2011, 36, 926–932. [Google Scholar] [CrossRef]
- Galassetti, P.R.; Iwanaga, K.; Crisostomo, M.; Zaldivar, F.P.; Larson, J.; Pescatello, A. Inflammatory cytokine; growth factor and counterregulatory responses to exercise in children with type 1 diabetes and healthy controls. Pediatr Diabetes 2006, 7, 16–24. [Google Scholar] [CrossRef]
- Devaraj, S.; Venugopal, S.K.; Singh, U.; Jialal, I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-{alpha} and -{beta}. Diabetes 2005, 54, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Trehu, E.; Atkins, M.B.; Dinarello, C.A.; Mier, J.W. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: Induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood 1994, 83, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Fujita, N.; Fujii, T.; Watanabe, K.; Yagi, M.; Tsuji, T.; Ishii, K.; Miyamoto, T.; Horiuchi, K.; Nakamura, M.; et al. Potential Involvement of the IL-6/JAK/STAT3 Pathway in the Pathogenesis of Intervertebral Disc Degeneration. Spine 2017, 42, 817–824. [Google Scholar] [CrossRef]
- Phillips, K.L.; Chiverton, N.; Michael, A.L.; Cole, A.A.; Breakwell, L.M.; Haddock, G.; Bunning, R.A.; Cross, A.K.; Le Maitre, C.L. The cytokine and chemokine expression profile of nucleus pulposus cells: Implications for degeneration and regeneration of the intervertebral disc. Arthritis Res. Ther. 2013, 15, R213. [Google Scholar] [CrossRef] [Green Version]
- Takatalo, J.; Karppinen, J.; Niinimäki, J.; Taimela, S.; Näyhä, S.; Mutanen, P.; Sequeiros, R.B.; Kyllönen, E.; Tervonen, O. Does lumbar disc degeneration on magnetic resonance imaging associate with low back symptom severity in young Finnish adults? Spine 2011, 36, 2180–2189. [Google Scholar] [CrossRef]
- Fishman, D.; Faulds, G.; Jeffery, R.; Mohamed-Ali, V.; Yudkin, J.S.; Humphries, S.; Woo, P. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels; and an association with systemic-onset juvenile chronic arthritis. J. Clin. Invest. 1998, 102, 1369–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volk, H.; Asadullah, K.; Gallagher, G.; Sabat, R.; Grutz, G. IL-10 and its homologs: Important immune mediators and emerging immunotherapeutic targets. Trends Immunol. 2001, 22, 414–417. [Google Scholar] [CrossRef]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Sabat, R.; Grütz, G.; Warszawska, K. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 5, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Liu, W.; Ji, G.; Dai, Y. The essential role of p38 MAPK in mediating the interplay of oxLDL and IL-10 in regulating endothelial cell apoptosis. Eur. J. Cell Biol. 2013, 4–5, 150–159. [Google Scholar] [CrossRef]
- Epstein, J.; Docena, G.; Macdonald, T.T.; Sanderson, I.R. Curcumin suppresses p38 mitogen-activated protein kinase activation, reduces IL-1β and matrix metalloproteinase-3 and enhances IL-10 in the mucosa of children and adults with inflammatory bowel disease. Br. J. Nutr. 2010, 6, 824–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Bi, J.; Wang, D.; Fang, L.; Zhang, L.; Li, F.; Chen, H.; Xiao, S. Porcine reproductive and respiratory syndrome virus infection activates IL-10 production through NF-κB and p38 MAPK pathways in porcine alveolar macrophages. Dev. Comp. Immunol. 2013, 3, 265–272. [Google Scholar] [CrossRef]
- Mollazadeh, H.; Cicero, A.F.G.; Blesso, C.N.; Pirro, M.; Majeed, M.; Sahebkar, A. Immune modulation by curcumin: The role of interleukin-10. Crit. Rev. Food Sci. Nutr. 2019, 59, 89–101. [Google Scholar] [CrossRef]
- Tateiwa, D.; Yoshikawa, H.; Kaito, T. Cartilage and Bone Destruction in Arthritis: Pathogenesis and Treatment Strategy: A Literature Review. Cells 2019, 8, 818. [Google Scholar] [CrossRef] [Green Version]
- Schulze-Tanzil, G.; Zreiqat, H.; Sabat, R.; Kohl, B.; Halder, A.; Muller, R.D.; John, T. Interleukin-10 and articular cartilage: Experimental therapeutical approaches in cartilage disorders. Curr. Gene Ther. 2009, 9, 306–315. [Google Scholar] [CrossRef]
- Behrendt, P.; Hafelein, K.; Preusse-Prange, A.; Bayer, A.; Seekamp, A.; Kurz, B. IL-10 ameliorates TNF-alpha induced meniscus degeneration in mature meniscal tissue in vitro. BMC Musculoskelet Disord. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrendt, P.; Preusse-Prange, A.; Kluter, T.; Haake, M.; Rolauffs, B.; Grodzinsky, A.J.; Lippross, S.; Kurz, B. IL-10 reduces apoptosis and extracellular matrix degradation after injurious compression of mature articular cartilage. Osteoarthr. Cartil. 2016, 24, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.B.; Hu, Y.C.; Cheng, P.; Zhou, H.Y.; Chen, X.Y.; Wu, D.; Zhang, R.H.; Yu, D.C.; Gao, X.D.; Shi, J.T.; et al. Targeted therapy for intervertebral disc degeneration: Inhibiting apoptosis is a promising treatment strategy. Int. J. Med. Sci. 2021, 18, 2799–2813. [Google Scholar] [CrossRef]
- de Waal Malefyt, R.; Abrams, J.; Bennett, B.; Figdor, C.G.; De Vries, J.E. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef] [Green Version]
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Li, W.; Liu, T.; Wu, L.; Chen, C.; Jia, Z.; Bai, X.; Ruan, D. Blocking the function of inflammatory cytokines and mediators by using IL-10 and TGF-β: A potential biological immunotherapy for intervertebral disc degeneration in a beagle model. Int. J. Mol. Sci. 2014, 15, 17270–17283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.; Yan, Q.; Wang, Y.; Cheng, X.; Song, D.; Wu, C.; Yu, H.; Yang, H.; Zou, J. IL-10 delays the degeneration of intervertebral discs by suppressing the p38 MAPK signaling pathway. Free Radic. Biol. Med. 2020, 147, 262–270. [Google Scholar] [CrossRef]
- Lin, W.P.; Lin, J.H.; Chen, X.W.; Wu, C.Y.; Zhang, L.Q.; Huang, Z.D.; Lai, J.M. Interleukin-10 promoter polymorphisms associated with susceptibility to lumbar disc degeneration in a Chinese cohort. Genet Mol. Res. 2011, 10, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Hartvigsen, J.; Hancock, M.J.; Kongsted, A.; Louw, Q.; Ferreira, M.L.; Genevay, S.; Hoy, D.; Karppinen, J.; Pransky, G.; Sieper, J.; et al. Lancet Low Back Pain Series Working Group. What low back pain is and why we need to pay attention. Lancet 2018, 391, 2356–2367. [Google Scholar] [CrossRef] [Green Version]
- Frapin, L.; Clouet, J.; Delplace, V.; Fusellier, M.; Guicheux Visage, L. Lessons learned from intervertebral disc pathophysiology to guide rational design of sequential delivery systems for therapeutic biological factors. Adv. Drug. Deliv. Rev. 2019, 149–150, 49–71. [Google Scholar] [CrossRef]
- Ganko, R.; Rao, P.J.; Phan, K.; Mobbs, R.J. Can bacterial infection by low virulent organisms be a plausible cause for symptomatic disc degeneration? A systematic review. Spine (Phila Pa 1976) 2015, 40, E587–E592. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.; Underwood, M.; Buchbinder, R. Non-specific low back pain. Lancet 2017, 389, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Okada, E.; Daimon, K.; Fujiwara, H.; Nishiwaki, Y.; Nojiri, K.; Watanabe, M.; Katoh, H.; Ishihama, H.; Fujita, N.; Tsuji, T.; et al. Ten-year Longitudinal Follow-up MRI Study of Age-related Changes in Thoracic Intervertebral Discs in Asymptomatic Subjects. Spine 2019, 44, E1317–E1324. [Google Scholar] [CrossRef]
- Molinos, M.; Almeida, C.R.; Caldeira, J.; Cunha, C.; Gonçalves, R.M.; Barbosa, M.A. Inflammation in intervertebral disc degeneration and regeneration. J. R Soc. Interface 2015, 12, 20141191. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, X.S. Pre-operative blood test for antibody to nucleus pulposus may distinguish types of lumbar disc herniation. Med. Hypotheses 2010, 75, 464–465. [Google Scholar] [CrossRef]
- Kauppila, L.I. Ingrowth of blood vessels in disc degeneration. Angiographic and histological studies of cadaveric spines. J. Bone Jt. Surgery. Am. Vol. 1999, 77, 26–31. [Google Scholar] [CrossRef]
- Volz, M.; Elmasry, S.; Jackson, A.R.; Travascio, F. Computational modeling intervertebral disc pathophysiology: A Review. Front. Physiol. 2022, 12, 750668. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Shiyu-Hu Shen, Z.; Yan, B.; Chen, J. New insights into the interplay between miRNAs and autophagy in the aging of intervertebral discs. Ageing Res. Rev. 2021, 65, 101227. [Google Scholar] [CrossRef]
- Santos, S.; Lamghari, M.; Almeida, C.; Oliveira, M.; Neves, N.; Ribeiro, A.; Barbosa, J.; Barros, R.; Maciel, J.; Martins, M.; et al. Adsorbed fibrinogen leads to improved bone regeneration and correlates with differences in the systemic immune response. Acta Biomater. 2013, 9, 7209–7217. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, M.; Zhao, X.H.; Liu, Z.H.; Gao, Y.; Samartzis, D.; Wang, H.Q.; Luo, Z.J. Immune cascades in human intervertebral disc: The pros and cons. Int. J. Clin. Exp. Pathol. 2013, 6, 1009–1014, PMCID:PMC3657352. [Google Scholar] [PubMed]
- Yamamoto, J.; Maeno, K.; Takada, T.; Kakutani, K.; Yurube, T.; Zhang, Z.; Hirata, H.; Kurakawa, T.; Sakai, D.; Mochida, J.; et al. Fas ligand plays an important role for the production of pro-inflammatory cytokines in intervertebral disc nucleus pulposus cells. J. Orthop. Res. 2013, 31, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Purmessur, D.; Walter, B.; Roughley, P.; Laudier, D.; Hecht, A.; Iatridis, J. A role for TNFα in intervertebral disc degeneration: A non-recoverable catabolic shift. Biochem. Biophys. Res. Commun. 2013, 433, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Yan, J.; Jiang, L.S.; Dai, L.Y. Autophagy in rat annulus fibrosus cells: Evidence and possible implications. Arthritis Res. Ther. 2011, 13, R132. [Google Scholar] [CrossRef] [Green Version]
- Navone, S.E.; Marfia, G.; Giannoni, A.; Beretta, M.; Guarnaccia, L.; Gualtierotti, R.; Nicoli, D.; Rampini, P.; Campanella, R. Inflammatory mediators and signalling pathways controlling intervertebral disc degeneration. Histol. Histopathol. 2017, 32, 523–542. [Google Scholar] [CrossRef]
- Cunha, C.; Silva, A.J.; Pereira, P.; Vaz, R.; Gonçalves, R.M.; Barbosa, M.A. The inflammatory response in the regression of lumbar disc herniation. Arthritis Res. Ther. 2018, 20, 251. [Google Scholar] [CrossRef] [Green Version]
- Lyu, F.J.; Cui, H.; Pan, H.; Mc Cheung, K.; Cao, X.; Iatridis, J.C.; Zheng, Z. Painful intervertebral disc degeneration and inflammation: From laboratory evidence to clinical interventions. Bone Res. 2021, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Trefilova, V.V.; Shnayder, N.A.; Popova, T.E.; Balberova, O.V.; Nasyrova, R.F. The role of NO system in low back pain chronicity. Pers. Psychiatry Neurol. 2021, 1, 37–45. [Google Scholar] [CrossRef]
- Liu, M.; Saredy, J.; Zhang, R.; Shao, Y.; Sun, Y.; Yang, W.Y.; Wang, J.; Liu, L.; Drummer, C., 4th; Johnson, C.; et al. Approaching Inflammation Paradoxes-Proinflammatory Cytokine Blockages Induce Inflammatory Regulators. Front. Immunol. 2020, 11, 554301. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypothesis | Mechanism | References |
|---|---|---|
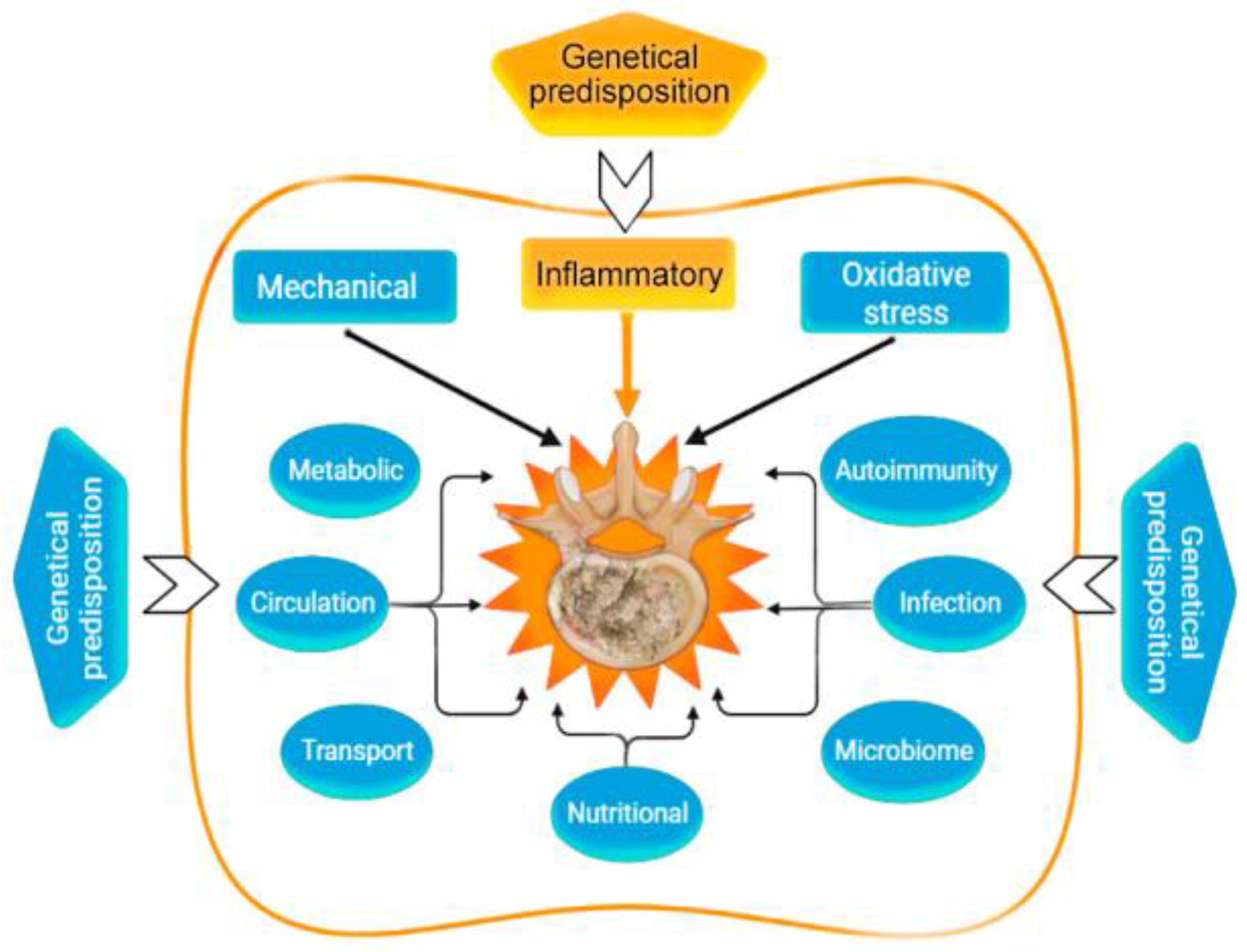
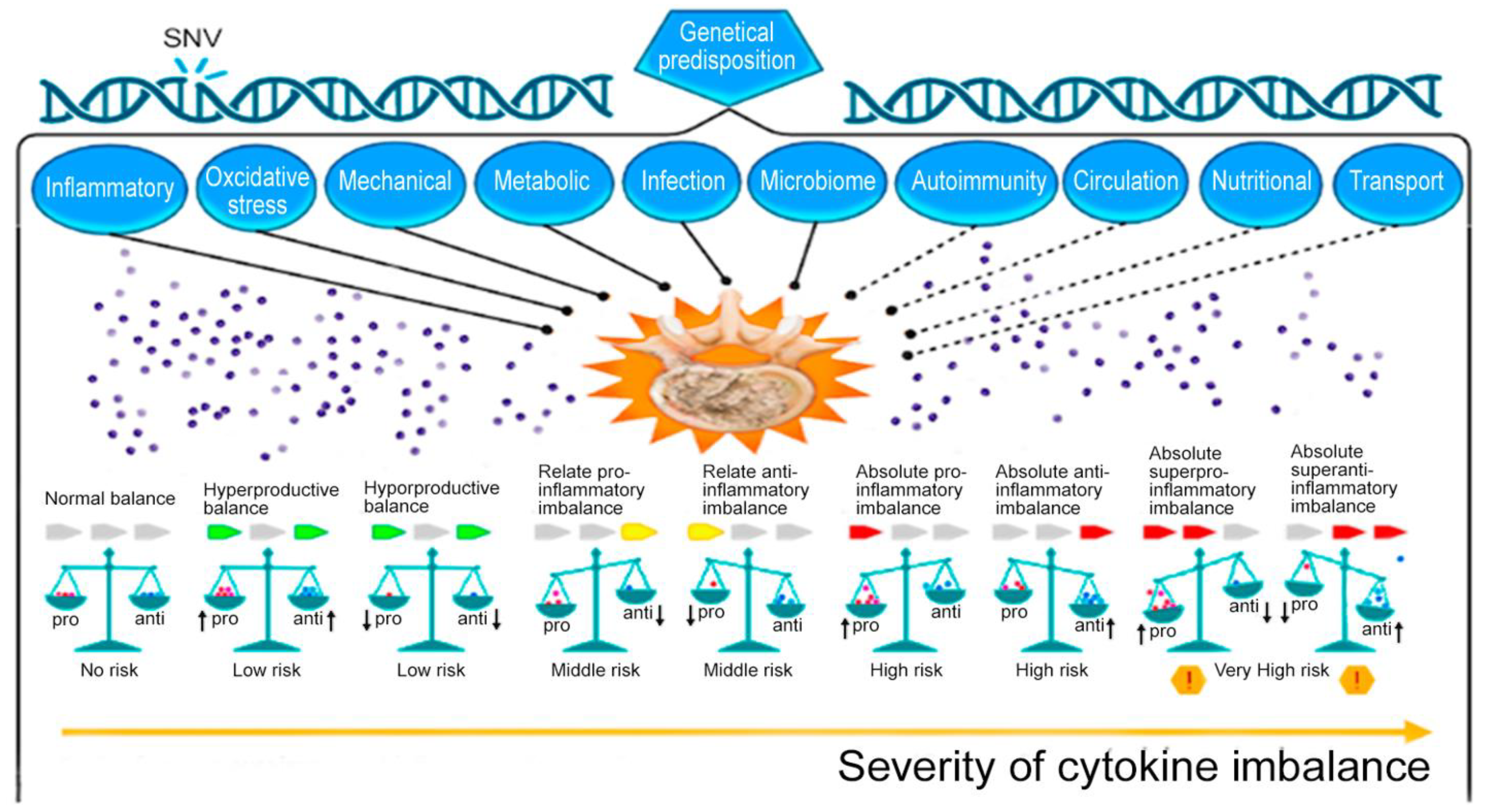
| Genetic | Congenital malformations IVD. Monogenic hereditary differentiated and undifferentiated connective tissue dysplasia. Genetic predisposition to impaired vitamin D metabolism ApaI (rs7975232), etc. Post-translational disorders (expression of zinc finger long non-coding RNA antisense 1). MicroRNAs as factors of degradation, inflammation, apoptosis, autophagy, or regulators of mechanosensory perception. Genetic predisposition to overproduction of pro-inflammatory cytokines, including SNVs of the genes: IL1A (rs1800587, rs2071375), IL1B (rs1143634), IL4 (rs2243250 rs2070874); IL-6 SNV in Exon 5 (rs13006435) other three SNVs of IL-6 (rs1800797, rs1800796, rs1800795, rs13006435). Genetic predisposition to impaired synthesis of IVD components, including genes: ACAN (rs1042631, rs1516797), COL1A1 (rs2075555, rs1007086, rs1800012, etc.), COL9A2 (rs137853213), COL9A3 (rs61734651), COL9A1 (rs696990, rs7533552), COL11A2 (rs1800587, rs1337185, rs1463035, rs2072915, rs9277933, rs2076311), HAPLN1 (rs179851), THBS2 (rs9406328), COMP (rs137852650), CD36 (rs3173798, rs3211892), CILP (rs2033711); ASPN (rs373444, rs13301537), GDF-5 (rs143383), etc. Genetic predisposition to increased oxidative processes in IDD: MMP1 (rs1799750), MMP2 (rs243865), MMP3 (rs3025058), MMP9 (rs17576), PARK2 (rs926849), RSMB9 (rs2187689, rs7767277), etc. | [12] [13,14] [15] [16] [17,18] [19,20] [21,22] [21,23] |
| Mechanical | Overweight and pathological obesity. Sedentary lifestyle (physical inactivity). Lifting weights, sharp turns, frequent bending, twisting, prolonged static load on the spine. Injury of the vertebral motor segment. | [24,25] [26] [26] [27] |
| Metabolic | Smoking. Hypoxia and high hemoglobin levels. Knockout of hypoxia-induced factor (HIF)-1alpha. High cholesterol and hyperlipidemia. Hyperglycemia. Hyperuricemia. Metabolic syndrome; Hypothyroidism. | [28] [28,29] [30] [31,32] [33] [25,34] [25,35] [36] |
| Circulation | Imbalance of bone homeostasis and osteoporosis. Violation of the blood supply (spasm of arterioles) of the end plate. Atherosclerosis of the arteries of the vertebral motor segment. Anemia, decompression sickness, Gaucher disease. | [37,38] [39] [32] [40] |
| Transport | Slow or insufficient outflow of lactate (acidification of the environment) IVD. | [41,42] |
| Oxidative stress | Activation of cathepsins in the acid environment of degenerating IVD. Activation of IVD matrix metalloproteinases. Homocysteine-induced oxidative stress and nucleus pulposus ferroptosis by increasing GPX4 methylation. Hemoglobin and heme-induced ferroptosis. | [43,44] [45,46] [47] [48] |
| Inflammatory | Absolute or relative overproduction of pro-inflammatory cytokines. Overexpression of ion channels of the TRP family (TRPV4). Overexpression of cytokine receptors. IL-6-induced ferroptosis. Abnormal activation of NLRP3 inflammasome (intracellular PRR). Adipokine resistin IVD degeneration associated with obesity. | [45,46] [49] [2] [50] [41,51] [24,52] |
| Autoimmunal | The APOE gene knockout and overexpression of catabolic cytokines in IVD. Autoimmune IVD degeneration due to FAS ligand hypo-expression. | [32] [53] |
| Microbiome | “Dysbacteriosis” (axis microbiome gut/skin/spine). Propionibacterium acnes and Staphylococcus epidermidis. | [54,55] |
| Infectional | Propionibacterium acnes, as a sluggish infection of IVD. Staphylococci (1% Staphylococcus epidermidis, 12% Staphylococcus auricularis, 12% Staphylococcus laminis, and 5% others). | [56,57] [58,59] |
| Nutritional | Autophagy. Increased nutritional requirements of IVD cells. Inadequate nutritional supply of IVD cells. Vitamin C deficiency in the elderly. Deficiency of proline, hydroxyproline. Vitamin D deficiency. | [60] [61,62] [63] [64] [65] [60,66] |
| Theory | Role of Cytokine | References |
|---|---|---|
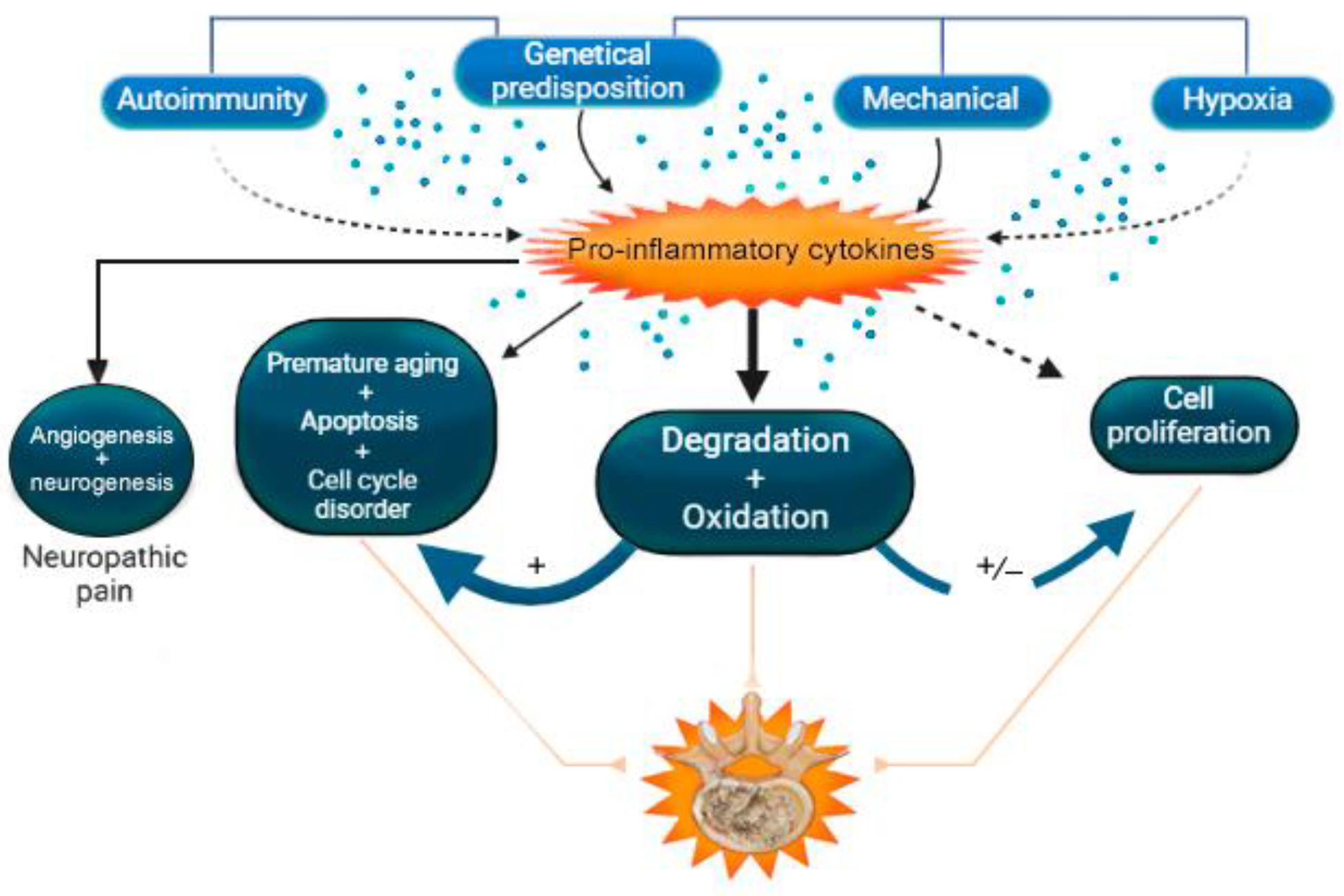
| Theory of degradation of structures of the extracellular matrix of intervertebral disc | IL-1β, IL-6, IL-8, IL-17, IL-18, IL-21, IL-23, TNF-α, IFN-γ | [2,20,67,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100] |
| Theory of oxidation | IL-1β, IL-6, IL-8, IL-17, IL-18, TNF-α, IFN-γ | [67,69,70,71,76,77,78,90,91,92,101,102,103,104,105,106,107,108] |
| Mechanical load theory | IL-1β, IL-6, IL-8, IL-17, TNF-α | [76,109,110,111,112,113,114,115] |
| Theory of programmed cell death | IL-1β, IL-6, IL-17, IL-18, TNF-α | [2,69,72,75,76,116,117,118,119,120,121,122,123,124] |
| Theory of cell proliferation | IL-1β, IL-17, TNF-α | [76,77,90,117,125,126,127,128,129,130,131] |
| Theory of premature aging | IL-1β, IL-6, IL-18, TNF-α | [39,73,74,76,77,124,132,133,134,135] |
| Autophagy theory | IL-1β, IL-17, TNF-α, IFN-β1 | [71,72,90,133,136,137,138] |
| Theory of angiogenesis and neoinnervation | IL-1β, IL-17, TNF-α | [76,90,139,140,141,142] |
| Theory of hypoxia | IL-1β, TNF-α | [76] |
| Cell cycle disorder theory | IL-1β, IL-17, TNF-α | [77,143] |
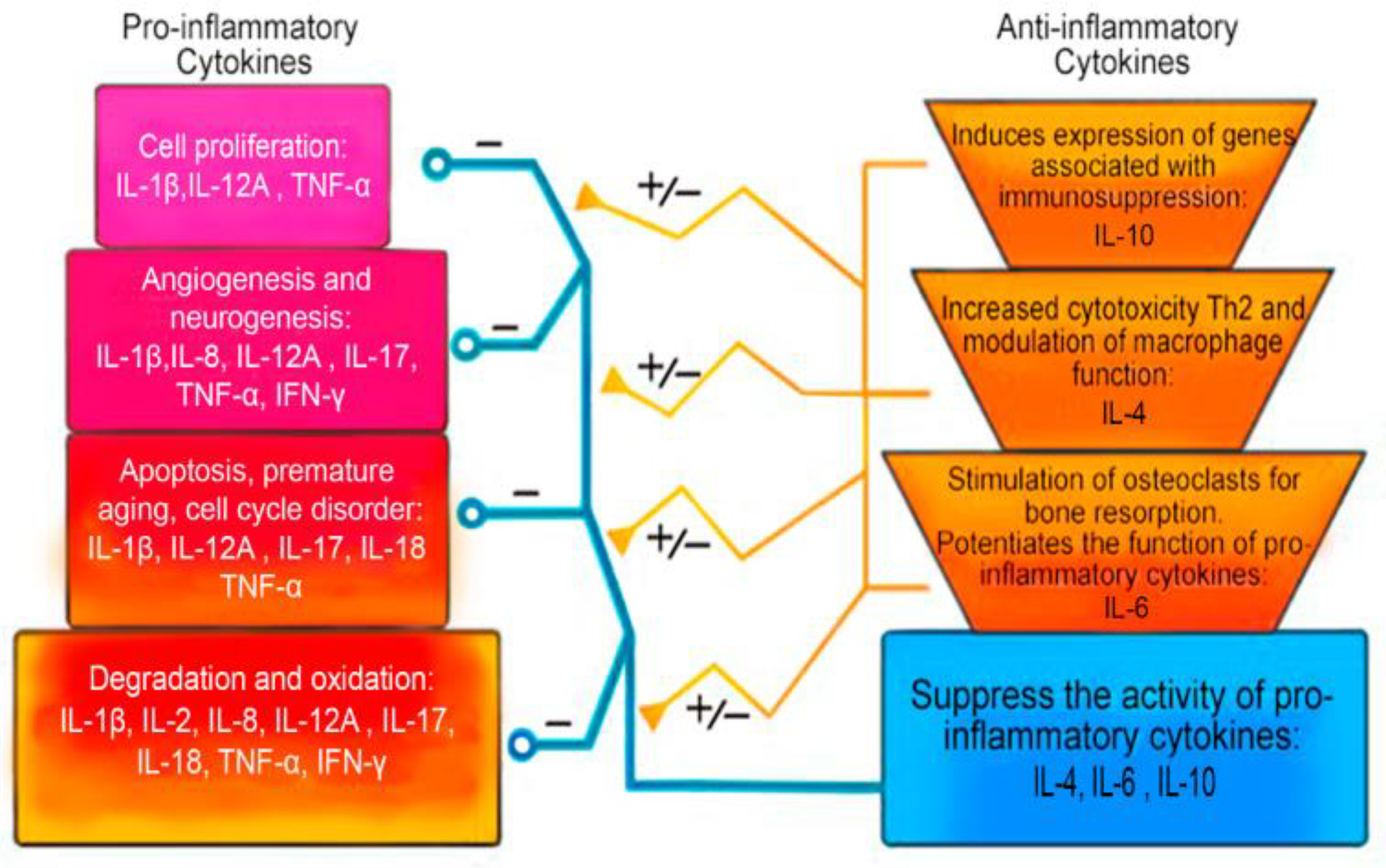
| Pro-Inflammatory Cytokines | Anti-Inflammatory Cytokines |
|---|---|
| Interleukin 1 alpha (IL-1α) Interleukin 1 beta (IL1-β) Interleukin 6 (IL-6) Interleukin 8 (IL-8) Interleukin 11 (IL-11) Interleukin 12 (IL-12) Interleukin 17 (IL-17) Interleukin 18 (IL-18) Interleukin 20 (IL-20) Interleukin 33 (IL-33) Interferon gamma (IFN-γ) Tumor necrosis factor alpha (TNF-α) Transforming growth factor beta (TGF-β) Ciliary neurotrophic factor (CNTF) Granulocytic-macrophage colony-stimulating factor (GM-CSF) Leukemia inhibitory factor (LIF) Oncostatin M (OSM) | Interleukin 1 receptor antagonist (IL-1Ra) Interleukin 4 (IL-4) Interleukin 6 (IL-6) Interleukin 10 (IL-10) Interleukin 11 (IL-11) Interleukin 13 (IL-13) Interleukin-18-binding protein (IL1-8BP) Transforming growth factor beta (TGF-β) |
| Cytokine | Gene: OMIM Number | Role in Intervertebral Disk | Clinical Role in IDD | References |
|---|---|---|---|---|
| IL-1β | IL1B: 147720 | Initiation of inflammatory, oxidative, degenerative, apoptotic cascades. Association with premature aging and cell growth arrest. Over-expression of vascular endothelial growth factor, NGF, and BDNF. | +++ | [67,69,73,75,76,77,102,118,132,141,151] |
| IL-2 | IL2: 147680 | Growth factor. Initiation of the inflammatory and degenerative cascade. | + | [155,156] |
| IL-8 | CXCL8: 146930 | Increased migration (potent chemokine) of neutrophils, T cells, and monocytes, whose enzymes produce free oxygen radicals. Indirect increase in oxidative stress, which can lead to IVD cell death. Involvement in the pathogenesis of acute neuropathic pain. | + | [85,86,87,88,89,106,107,109,110,111,158] |
| IL-12A | IL12A: 161560 | Stimulation of proliferation. Activation and increase in the cytotoxicity of NK cells and T cells. Stimulation of differentiation in Th1. Induction of IFN-γ and TNF-α secretion, synergism with pro-inflammatory cytokines with IL-18. | ++ | [141,144] |
| IL-17 | IL17A: 603149 | Initiation of the inflammatory and degenerative cascade. Association with cell growth arrest. Stimulation of angiogenesis. | +++ | [68,72,90,91,92,93,94,96,113,115,165,167,172] |
| IL-18 | IL18: 600953 | Initiation of the inflammatory and degenerative cascade (IFN-γ activation). Initiation of the apoptotic and oxidative cascade. Association with premature aging of IVD cells. | +++ | [70,83,105,121,122,123] |
| TNF-α | TNF: 191160 | Initiation of inflammatory, apoptotic, oxidation, and degenerative cascades. Association with premature aging and cell growth arrest. Autophagy promoter While TNF-α signaling via TNFR2 is anti-inflammatory and cytoprotective, resulting in the induction of proliferation, differentiation, angiogenesis, and tissue repair. | +++ | [71,74,76,77,79,80,104,116,117,126,127,130,133,135,144] |
| IFN-γ | IFNG: 147570 | Initiation of the inflammatory and degenerative cascade in IVD cells. Involvement in the pathogenesis of acute neuropathic pain in IDD. | +++ | [2,156,164,191,192] |
| Cytokine | Gene: OMIM Number | Role in Intervertebral Disk | Clinical Role in IDD | References |
|---|---|---|---|---|
| IL-4 | IL4: 147780 | Initiation of Th differentiation into Th2 lymphocytes. Increased Th2 cytotoxicity. Modulation of the function of macrophage cells. Decreased cytotoxicity. Inhibition of LPS, IFN gamma, and induction of TNF-α, IL-1α pathways of degeneration. Induction of production of IL-1β and TNF-α. Stimulation of IL-6 activation and participation together with it in anti-inflammatory, antioxidant activity. Decreased LPS-induced expression of IL-8, IL-12. | +++ | [157,164,194,195,196,199,200,202,203,205,207] |
| IL-6 | IL6: 147620 | A key role in the processes associated with immunity and inflammation. Potentiates the inflammatory, degenerative and oxidative cascade. May act as an anti-inflammatory cytokine. Able to stimulate osteoclasts and bone resorption. Suppresses H2O2-associated premature aging and apoptosis. | +++ | [2,32,97,98,99,100,104,108,114,124,157,210,212,213,215] |
| IL-10 | IL10: 124092 | Initiation of cellular effects through canonical JAK/STAT, which includes JAK1 and STAT3. Induction of expression of genes associated with immunosuppression. Providing antigens and enhancing immune tolerance. Anti-inflammatory, anti-catabolic, and anti-apoptotic action. Decreased production of IL-1α, IL-1β, IL-6, IL-8, IL-12, TNF-α, IFN-γ, GM-GSF and GCSF. | +++ | [104,224,225,227,228,229,230,231,235,236] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shnayder, N.A.; Ashhotov, A.V.; Trefilova, V.V.; Nurgaliev, Z.A.; Novitsky, M.A.; Vaiman, E.E.; Petrova, M.M.; Nasyrova, R.F. Cytokine Imbalance as a Biomarker of Intervertebral Disk Degeneration. Int. J. Mol. Sci. 2023, 24, 2360. https://doi.org/10.3390/ijms24032360
Shnayder NA, Ashhotov AV, Trefilova VV, Nurgaliev ZA, Novitsky MA, Vaiman EE, Petrova MM, Nasyrova RF. Cytokine Imbalance as a Biomarker of Intervertebral Disk Degeneration. International Journal of Molecular Sciences. 2023; 24(3):2360. https://doi.org/10.3390/ijms24032360
Chicago/Turabian StyleShnayder, Natalia A., Azamat V. Ashhotov, Vera V. Trefilova, Zaitun A. Nurgaliev, Maxim A. Novitsky, Elena E. Vaiman, Marina M. Petrova, and Regina F. Nasyrova. 2023. "Cytokine Imbalance as a Biomarker of Intervertebral Disk Degeneration" International Journal of Molecular Sciences 24, no. 3: 2360. https://doi.org/10.3390/ijms24032360