
Integrated Transcriptomic and Metabolomics Analysis of the Root Responses of Orchardgrass to Submergence Stress
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Root Appearance after Submergence Stress
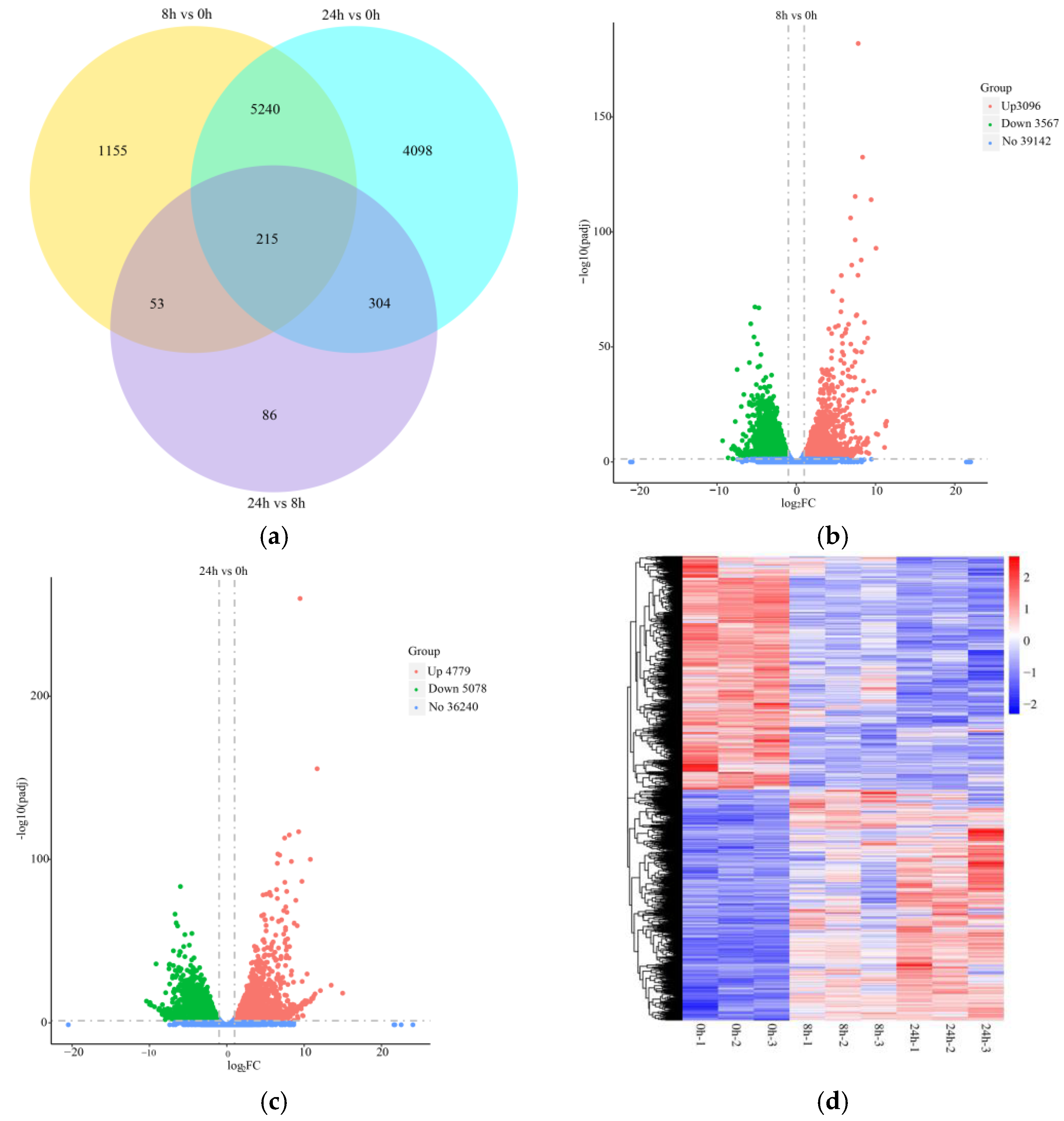
2.2. Transcriptome Sequencing Analysis and Identification of DEGs
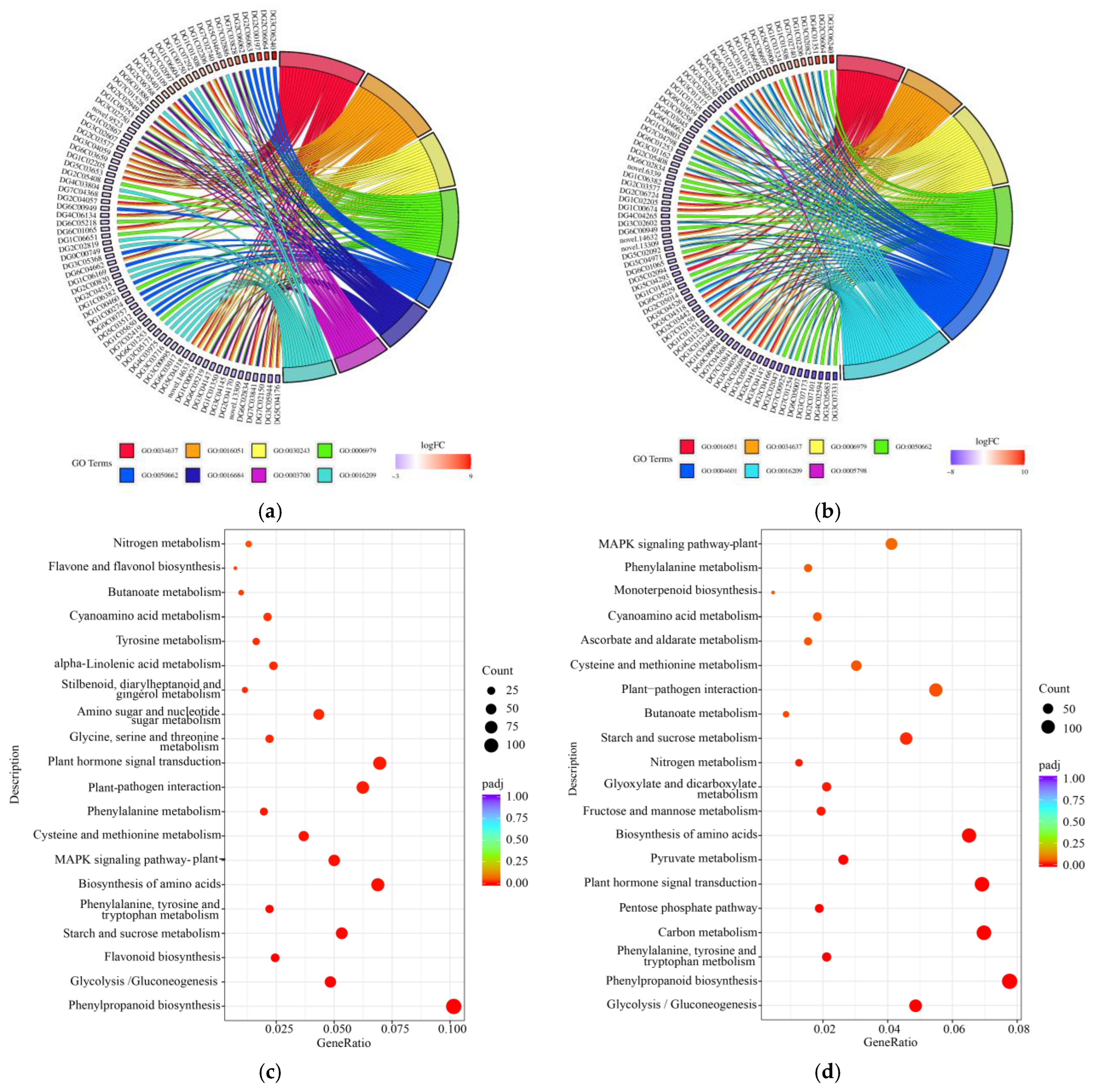
2.3. GO Analysis and KEGG Pathway Analysis
2.4. qRT-PCR Validation
2.5. Quality Control of Metabolomic Data
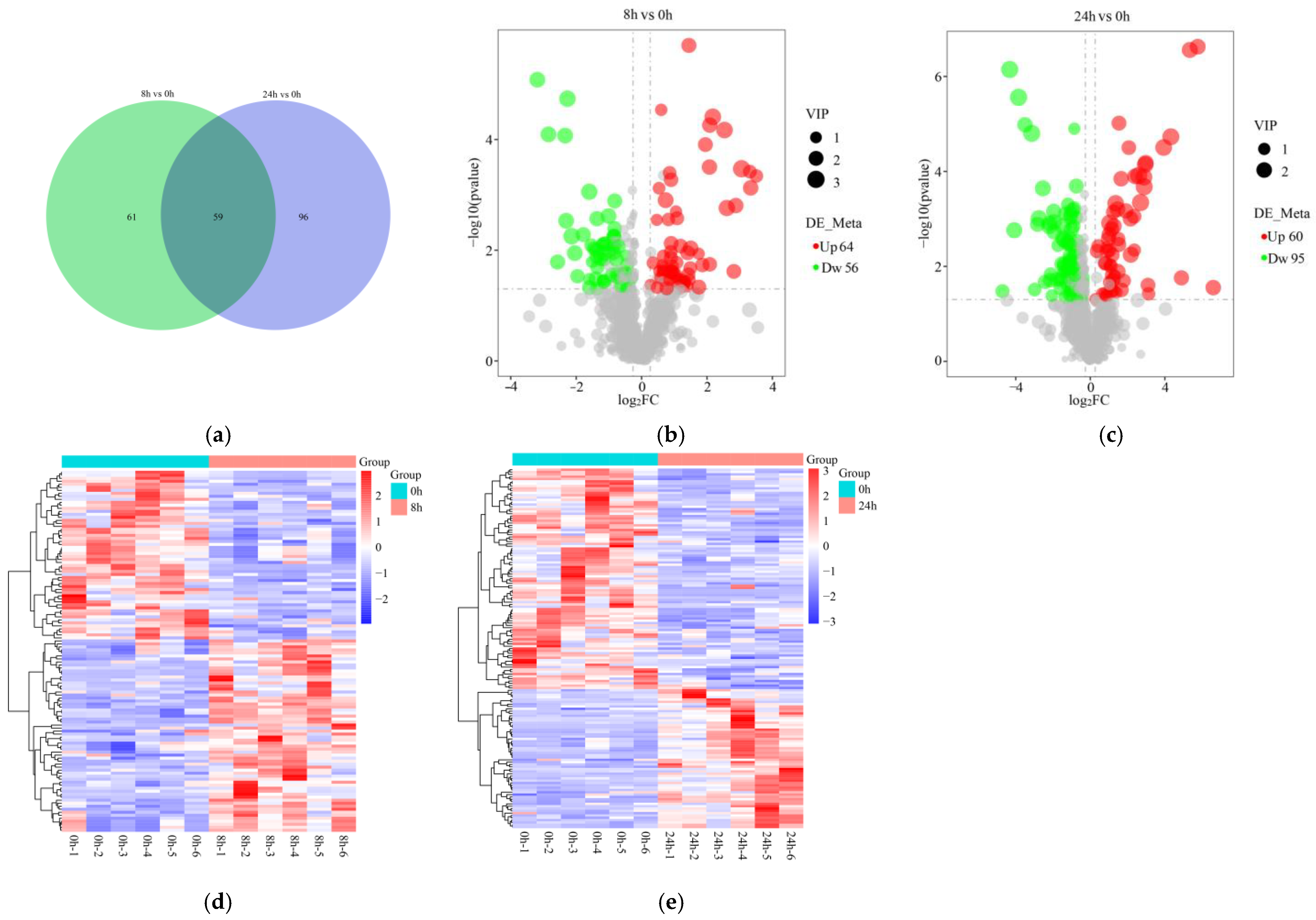
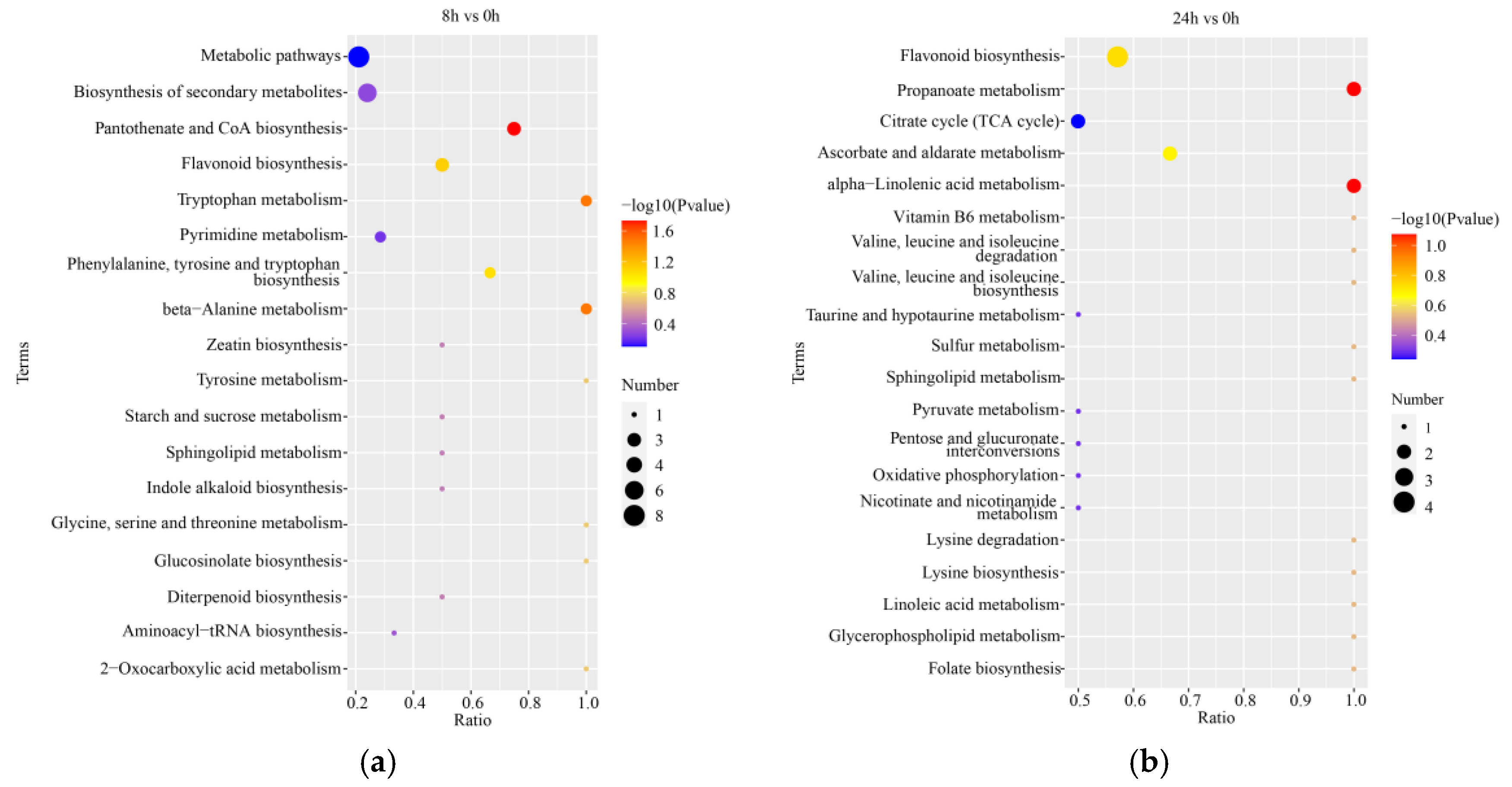
2.6. Differentially Expressed Metabolites (DEMs) and Functional Enrichment
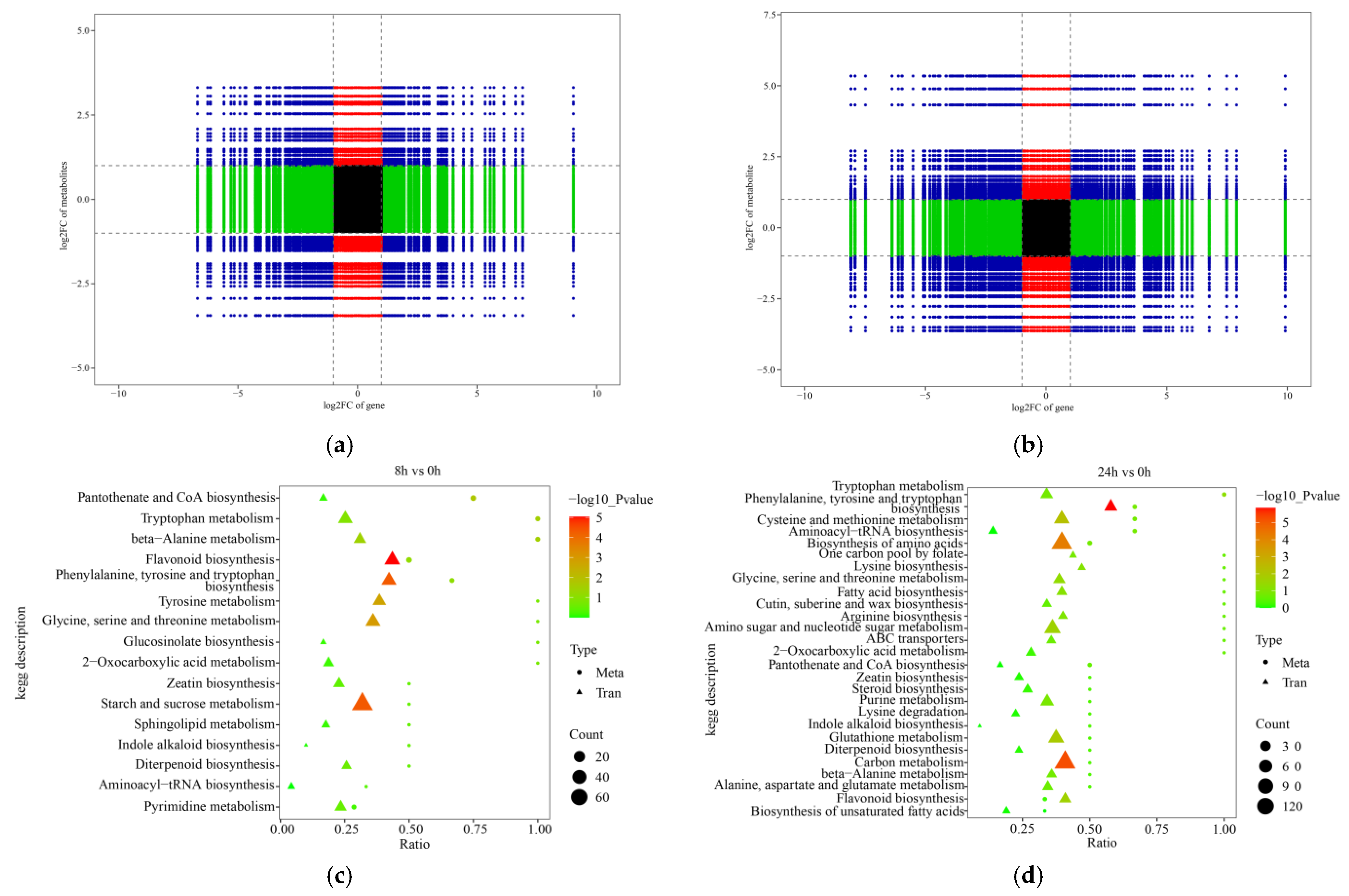
2.7. C orrelation and Enrichment Analysis between DEGs and DEMs
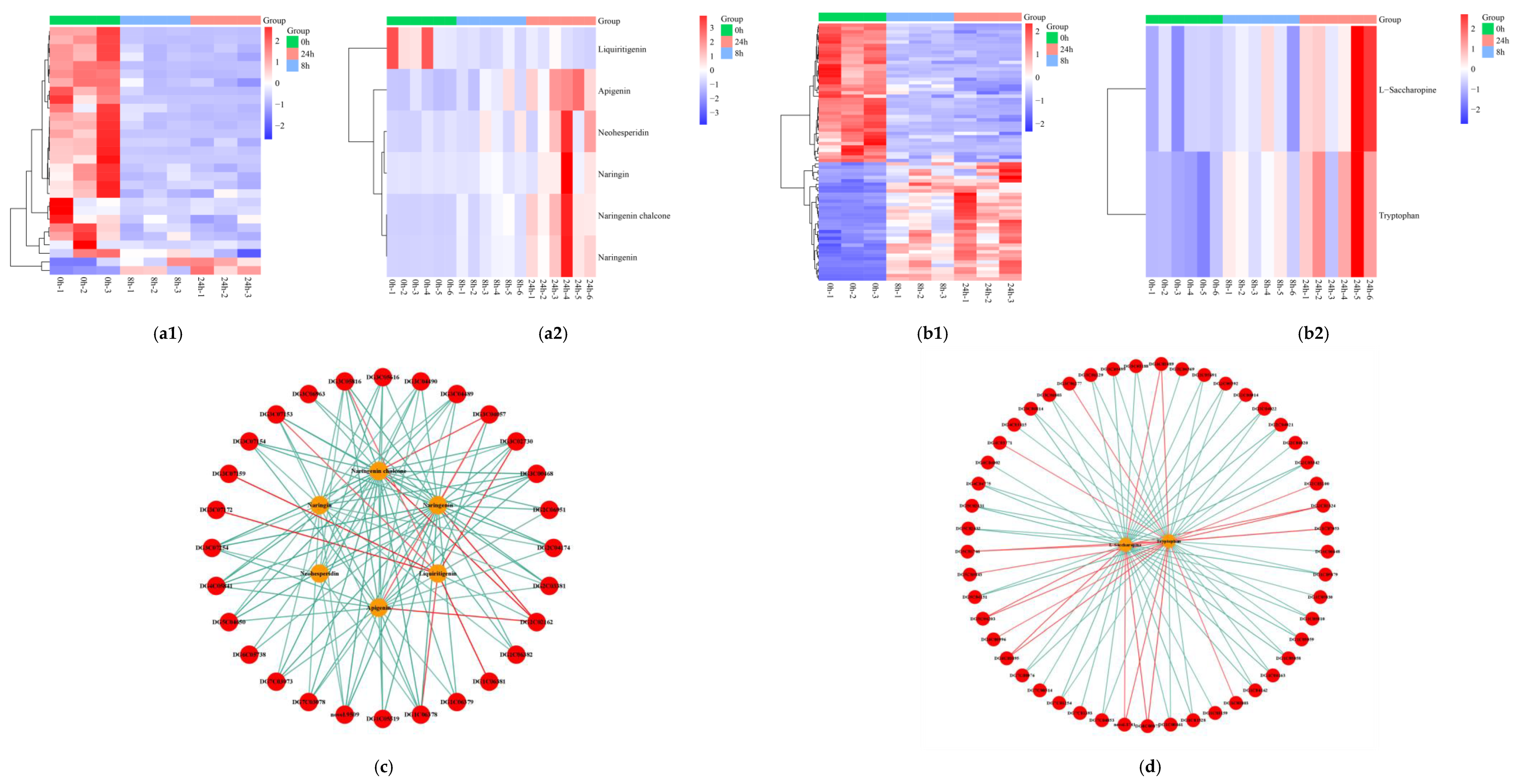
2.8. Biosynthesis of Flavonoid and Amino Acids
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Submergence Treatment and Harvesting
4.2. Transcriptome Sequencing and Data Analysis
4.3. qRT-PCR Analysis
4.4. Metabolomics Data Analysis
4.5. Integration of Transcriptomic and Metabolomic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, X.Q.; Luo, N.; Yan, J.P.; Tang, J.C.; Liu, S.W.; Jiang, Y.W. Differential growth response and carbohydrate metabolism of global collection of perennial ryegrass accessions to submergence and recovery following de-submergence. J. Plant Physiol. 2012, 169, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Mackill, D.J.; Ismail, A.M. Physiological basis of tolerance to complete submergence in rice involves genetic factors in addition to the SUB1 gene. AoB Plants 2014, 6, plu060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamang, B.G.; Magliozzi, J.O.; Maroof, M.; Fukao, T. Physiological and transcriptomic characterization of submergence and reoxygenation responses in soybean seedlings. Plant Cell Environ. 2014, 37, 2350–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.X.; Jiang, Y.W. Genotypic variation in growth and metabolic responses of perennial ryegrass exposed to short-term waterlogging and submergence stress. Plant Physiol. Bioch. 2015, 95, 57–64. [Google Scholar] [CrossRef]
- Samanta, P.; Chakrabarti, A.; Dey, N. Study on physiological responses with allelic diversity of Sub1A and SK loci in rice seedlings under complete submergence. Plant Physiol. 2022, 27, 275–281. [Google Scholar] [CrossRef]
- Gan, L.; Han, L.B.; Yin, S.X.; Jiang, Y.W. Chlorophyll metabolism and gene expression in response to submergence stress and subsequent recovery in perennial ryegrass accessions differing in growth habits. J. Plant Physiol. 2020, 251, 153195. [Google Scholar] [CrossRef]
- Basu, S.; Kumari, S.; Kumar, P.; Kumar, G.; Rajwanshi, R. Redox imbalance impedes photosynthetic activity in rice by disrupting cellular membrane integrity and induces programmed cell death under submergence. Physiol Plant. 2021, 172, 1764–1778. [Google Scholar] [CrossRef]
- Kumar, A.; Nayak, A.K.; Hanjagi, P.S.; Kumari, K.; Vijayakumar, S.; Mohanty, S.; Tripathi, R.; Panneerselvam, P. Submergence stress in rice: Adaptive mechanisms, coping strategies and future research needs. Environ. Exp. Bot. 2021, 186, 104448. [Google Scholar] [CrossRef]
- Simova-Stoilova, L.; Demirevska, K.; Kingston-Smith, A.; Feller, U. Involvement of the leaf antioxidant system in the response to soil flooding in two Trifolium genotypes differing in their tolerance to waterlogging. Plant Sci. 2012, 183, 43–49. [Google Scholar] [CrossRef]
- Bui, L.T.; Ella, E.S.; Dionisio-Sese, M.L.; Ismail, A.M. Morpho-physiological changes in roots of rice seedling upon submergence. Rice Sci. 2019, 26, 167–177. [Google Scholar] [CrossRef]
- Sasidharan, R.; Mustroph, A.; Boonman, A.; Akman, M.; Ammerlaan, A.; Breit, T.; Schranz, M.E.; Voesenek, L.; van Tienderen, P.H. Root transcript profiling of two rorippa species reveals gene clusters associated with extreme submergence tolerance. Plant Physiol. 2013, 163, 1277–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, A.; Yano, K.; Gamuyao, R.; Nagai, K.; Kuroha, T.; Ayano, M.; Nakamori, M.; Koike, M.; Kondo, Y.; Niimi, Y.; et al. Time-Course transcriptomics analysis reveals key responses of submerged deepwater rice to flooding. Plant Physiol. 2018, 176, 3081–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaas, M.; Haiminen, N.; Grant, J.; Cormican, P.; Finnan, J.; Arojju, S.K.; Utro, F.; Vellani, T.; Parida, L.; Barth, S. Transcriptome characterization and differentially expressed genes under flooding and drought stress in the biomass grasses Phalaris arundinacea and Dactylis glomerata. Ann. Bot. 2019, 124, 717–730. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira-Busatto, L.A.; Giordano, C.; Da Silva, M.F.; Uhry, D.J.; Guzman, F.; Wiebke-Strohm, B.; Li, Z.L.; Bredemeier, C.; Bodanese-Zanettini, M.H. Identification of functional genetic variations underlying flooding tolerance in Brazilian soybean genotypes. Int. J. Mol. Sci. 2022, 23, 10611. [Google Scholar] [CrossRef] [PubMed]
- Keska, K.; Szczesniak, M.W.; Makalowska, I.; Czernicka, M. Long-term waterlogging as factor contributing to hypoxia stress tolerance enhancement in cucumber: Comparative transcriptome analysis of waterlogging sensitive and tolerant accessions. Gene 2021, 12, 189. [Google Scholar] [CrossRef]
- Taylor-Kearney, L.J.; Flashman, E. Targeting plant cysteine oxidase activity for improved submergence tolerance. Plant J. 2022, 109, 779–788. [Google Scholar] [CrossRef]
- John, D.; Shylaraj, K.S. Mechanism of anoxic tolerance in backcross lines developed through Jyothi x Swarna-Sub 1 under submergence stress. Indian J. Genet. Plant Breed. 2020, 80, 375–383. [Google Scholar] [CrossRef]
- Li, Z.; Bai, D.F.; Zhong, Y.P.; Lin, M.M.; Sun, L.M.; Qi, X.J.; Hu, C.G.; Fang, J.B. Full-length transcriptome and RNA-Seq analyses reveal the mechanisms underlying waterlogging tolerance in Kiwifruit (Actinidia valvata). Int. J. Mol. Sci. 2022, 23, 3237. [Google Scholar] [CrossRef]
- Barding, G.A.; Beni, S.; Fukao, T.; Bailey-Serres, J.; Larive, C.K. Comparison of GC-MS and NMR for metabolite profiling of rice subjected to submergence stress. J. Proteome Res. 2013, 12, 898–909. [Google Scholar] [CrossRef]
- Pompeiano, A.; Guglielminetti, L. Carbohydrate metabolism in germinating caryopses of Oryza sativa L. exposed to prolonged anoxia. J. Plant Res. 2016, 129, 833–840. [Google Scholar] [CrossRef]
- Coutinho, I.D.; Henning, L.; Dopp, S.A.; Nepomuceno, A.; Moraes, L.; Marcolino-Gomes, J.; Richter, C.; Schwalbe, H.; Colnago, L.A. Flooded soybean metabolomic analysis reveals important primary and secondary metabolites involved in the hypoxia stress response and tolerance. Environ. Exp. Bot. 2018, 153, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zou, Q.J.; Guo, Q.S.; Yang, F.; Wu, L.W.; Zhang, W.Y. Widely targeted metabolomics analysis reveals the effect of flooding stress on the synthesis of flavonoids in Chrysanthemum morifolium. Molecules 2019, 24, 3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, A.; Kuroha, T.; Nagai, K.; Hattori, Y.; Kobayashi, M.; Nishizawa, T.; Kojima, M.; Utsumi, Y.; Oikawa, A.; Seki, M.; et al. Metabolite and phytohormone profiling illustrates metabolic reprogramming as an escape strategy of deepwater rice during partially submerged stress. Metabolites 2020, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Herzog, M.; Fukao, T.; Winkel, A.; Konnerup, D.; Lamichhane, S.; Alpuerto, J.B.; Hasler-Sheetal, H.; Pedersen, O. Physiology, gene expression, and metabolome of two wheat cultivars with contrasting submergence tolerance. Plant Cell Environ. 2018, 41, 1632–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.W.; Tang, S.H.; Li, J.Q.; Fan, C.F.; Xing, L.B.; Luo, K.M. Integrative transcriptomic and metabolomic analyses provide insight into the long-term submergence response mechanisms of young Salix variegata stems. Planta 2021, 253, 88. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Lu, W.X.; Ran, L.Y.; Dou, L.W.; Yao, S.; Hu, J.; Fan, D.; Li, C.F.; Luo, K.M. R2R3-MYB transcription factor MYB6 promotes anthocyanin and proanthocyanidin biosynthesis but inhibits secondary cell wall formation in Populus tomentosa. Plant J. 2019, 99, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.A.; Clark, S.G.; Reed, K.; Nie, Z.N.; Smith, K.F. Novel Festuca arundinacea Shreb. and Dactylis glomerata L. germplasm to improve adaptation for marginal environments. Aust. J. Exp. Agr. 2008, 48, 436–448. [Google Scholar] [CrossRef]
- Sanada, Y.; Tamura, K.; Yamada, T. Relationship between water-soluble carbohydrates in fall and spring and vigor of spring regrowth in orchardgrass. Crop Sci. 2010, 50, 380–390. [Google Scholar] [CrossRef]
- Bushman, B.S.; Singh, D.; Lamp, R.; Young, C.A.; Charlton, N.D.; Robins, J.G.; Anderson, N. Variation among orchardgrass (Dactylis glomerata) Germplasm for choke prevalence caused by Epichloe typhina. Plant Dis. 2019, 103, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Volaire, F.; Norton, M.R.; Norton, G.M.; Lelievre, F. Seasonal patterns of growth, dehydrins and water-soluble carbohydrates in genotypes of Dactylis glomerata varying in summer dormancy. Ann. Bot. 2005, 95, 981–990. [Google Scholar] [CrossRef]
- Mandrone, M.; Marincich, L.; Chiocchio, I.; Zannini, P.; Guarino, R.; Poli, F. Metabolomic study of Dactylis glomerata growing on Aeolian Archipelago (Italy). Metabolites 2022, 12, 533. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.D.; Zhang, Y.J.; Xiong, X.M.; Li, M.Y.; Cai, K.; Luo, H.; Zeng, B. Transcriptome analysis on responses of orchardgrass (Dactylis glomerata L.) leaves to a short term flooding. Hereditas 2020, 157, 20. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Zhang, Y.J.; Zhang, A.L.; Qiao, D.D.; Ren, J.C.; Li, M.Y.; Cai, K.; Zhang, J.H.; Huang, L.K. Transcriptome profiling of two Dactylis glomerata L. cultivars with different tolerance in response to submergence stress. Phytochemistry 2020, 175, 112378. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Rivera, G.; Sanz, A.; Cifuentes, A.; Ibanez, E.; Paape, T.; Lucas, M.M.; Pueyo, J.J. Flavonoid accumulation varies in medicago truncatula in response to mercury stress. Front. Plant Sci. 2022, 13, 933209. [Google Scholar] [CrossRef]
- Meng, L.D.; Yang, Y.P.; Ma, Z.W.; Jiang, J.W.; Zhang, X.M.; Chen, Z.R.; Cui, G.W.; Yin, X.J. Integrated physiological, transcriptomic and metabolomic analysis of the response of Trifolium pratense L. to Pb toxicity. J. Hazard. Mater. 2022, 436, 129128. [Google Scholar] [CrossRef]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as important molecules of plant interactions with the environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.H.; Huang, K.X.; Yang, X.L.; Xu, H.B. Free radical scavenging and antioxidant activities of flavonoids extracted from the radix of Scutellaria baicalensis Georgi. Biochim. Biophys. Acta Gen. Subj. 1999, 1472, 643–650. [Google Scholar] [CrossRef]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Ravaglia, D.; Espley, R.V.; Henry-Kirk, R.A.; Andreotti, C.; Ziosi, V.; Hellens, R.P.; Costa, G.; Allan, A.C. Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors. BMC Plant Biol. 2013, 13, 68. [Google Scholar] [CrossRef] [Green Version]
- Chacon, D.S.; Santos, M.; Bonilauri, B.; Vilasboa, J.; Da Costa, C.T.; Da Silva, I.B.; Torres, T.D.; de Araujo, T.F.; Roque, A.D.; Pilon, A.C.; et al. Non-target molecular network and putative genes of flavonoid biosynthesis in Erythrina velutina Willd, a Brazilian semiarid native woody plant. Front. Plant Sci. 2022, 13, 947558. [Google Scholar] [CrossRef]
- Nemesio-Gorriz, M.; Blair, P.B.; Dalman, K.; Hammerbacher, A.; Arnerup, J.; Stenlid, J.; Mukhtar, S.M.; Elfstrand, M. Identification of norway spruce MYB-bHLH-WDR transcription factor complex members linked to regulation of the flavonoid pathway. Front. Plant Sci. 2017, 8, 00305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.W.; Reichelt, M.; Yoshida, K.; Gershenzon, J.; Constabel, C.P. Two R2R3-MYB proteins are broad repressors of flavonoid and phenylpropanoid metabolism in poplar. Plant J. 2018, 96, 949–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shingaki-Wells, R.; Millar, A.H.; Whelan, J.; Narsai, R. What happens to plant mitochondria under low oxygen? An omics review of the responses to low oxygen and reoxygenation. Plant Cell Environ. 2014, 37, 2260–2277. [Google Scholar] [CrossRef]
- Rohini, G.; Mohit, V.; Shashank, A.; Rama, S.; Manoj, M.; Mukesh, J. Deep transcriptome sequencing of wild halophyte rice, Porteresia coarctata, provides novel insights into the salinity and submergence tolerance factors. DNA Res. 2014, 21, 69–84. [Google Scholar] [CrossRef]
- Ren, C.G.; Kong, C.C.; Yan, K.; Zhang, H.; Luo, Y.M.; Xie, Z.H. Elucidation of the molecular responses to waterlogging in Sesbania cannabina roots by transcriptome profiling. Sci. Rep. 2017, 7, 9256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M.; Cortleven, A.; Novak, O.; Schmulling, T. Root-derived trans-zeatin cytokinin protects Arabidopsis plants against photoperiod stress. Plant Cell Environ. 2020, 43, 2637–2649. [Google Scholar] [CrossRef]
- Antoniadi, I.; Mateo-Bonmati, E.; Pernisova, M.; Brunoni, F.; Antoniadi, M.; Villalonga, M.; Ament, A.; Karady, M.; Turnbull, C.; Dolezal, K.; et al. IPT9, a cis-zeatin cytokinin biosynthesis gene, promotes root growth. Front. Plant Sci. 2022, 13, 932008. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, H.Z.; Jin, J.J.; Ma, X.Y.; Li, K.L. Physiological and Transcriptome analysis on diploid and polyploid Populus ussuriensis Kom. under salt stress. Int. J. Mol. Sci. 2022, 23, 7529. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, X.; Zhao, C.F.; Fang, Z.M.; Jiao, P.P. Transcriptomic and metabolomic analysis reveals the role of CoA in the salt tolerance of Zygophyllum spp. BMC Plant Biol. 2020, 20, 9. [Google Scholar] [CrossRef]
- Huang, L.K.; Feng, G.Y.; Yan, H.D.; Zhang, Z.R.; Bushman, B.S.; Wang, J.P.; Bombarely, A.; Li, M.Z.; Yang, Z.F.; Nie, G.; et al. Genome assembly provides insights into the genome evolution and flowering regulation of orchardgrass. Plant Biotechnol. J. 2020, 18, 373–388. [Google Scholar] [CrossRef]
- Shuai, Y.; Feng, G.Y.; Yang, Z.F.; Liu, Q.X.; Han, J.T.; Xu, X.H.; Nie, G.; Huang, L.K.; Zhang, X.Q. Genome-wide identification of C2H2-type zinc finger gene family members and their expression during abiotic stress responses in orchardgrass (Dactylis glomerata). Genome 2022, 65, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topcu, H. Reference gene selection for RT-qPCR normalization of strawberry at various organs, different growing stages of fruits, and salt-stress treatment. Turk. J. Agric. For. 2022, 46, 509–524. [Google Scholar] [CrossRef]
- Huang, L.K.; Yan, H.D.; Jiang, X.M.; Zhang, Y.; Zhang, X.Q.; Ji, Y.; Zeng, B.; Xu, B.; Yin, G.H.; Lee, S.; et al. Reference gene selection for quantitative real-time reverse-transcriptase PCR in orchardgrass subjected to various abiotic stresses. Gene 2014, 553, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Arocho, A.; Chen, B.; Ladanyi, M.; Pan, Q. Validation of the 2-DeltaDeltaCt calculation as an alternate method of data analysis for quantitative PCR of BCR-ABL P210 transcripts. Diagn. Mol. Pathol. 2006, 15, 56–61. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, P.; Shen, B.; Zeng, B.; Bi, L.; Qu, M.; Zheng, Y.; Ye, Y.; Li, W.; Zhou, X.; Yang, X.; et al. Integrated Transcriptomic and Metabolomics Analysis of the Root Responses of Orchardgrass to Submergence Stress. Int. J. Mol. Sci. 2023, 24, 2089. https://doi.org/10.3390/ijms24032089
Shang P, Shen B, Zeng B, Bi L, Qu M, Zheng Y, Ye Y, Li W, Zhou X, Yang X, et al. Integrated Transcriptomic and Metabolomics Analysis of the Root Responses of Orchardgrass to Submergence Stress. International Journal of Molecular Sciences. 2023; 24(3):2089. https://doi.org/10.3390/ijms24032089
Chicago/Turabian StyleShang, Panpan, Bingna Shen, Bing Zeng, Lei Bi, Minghao Qu, Yuqian Zheng, Yujing Ye, Wenwen Li, Xiaoli Zhou, Xingyun Yang, and et al. 2023. "Integrated Transcriptomic and Metabolomics Analysis of the Root Responses of Orchardgrass to Submergence Stress" International Journal of Molecular Sciences 24, no. 3: 2089. https://doi.org/10.3390/ijms24032089