
Ketogenic Diet Regulates Cardiac Remodeling and Calcium Homeostasis in Diabetic Rat Cardiomyopathy
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
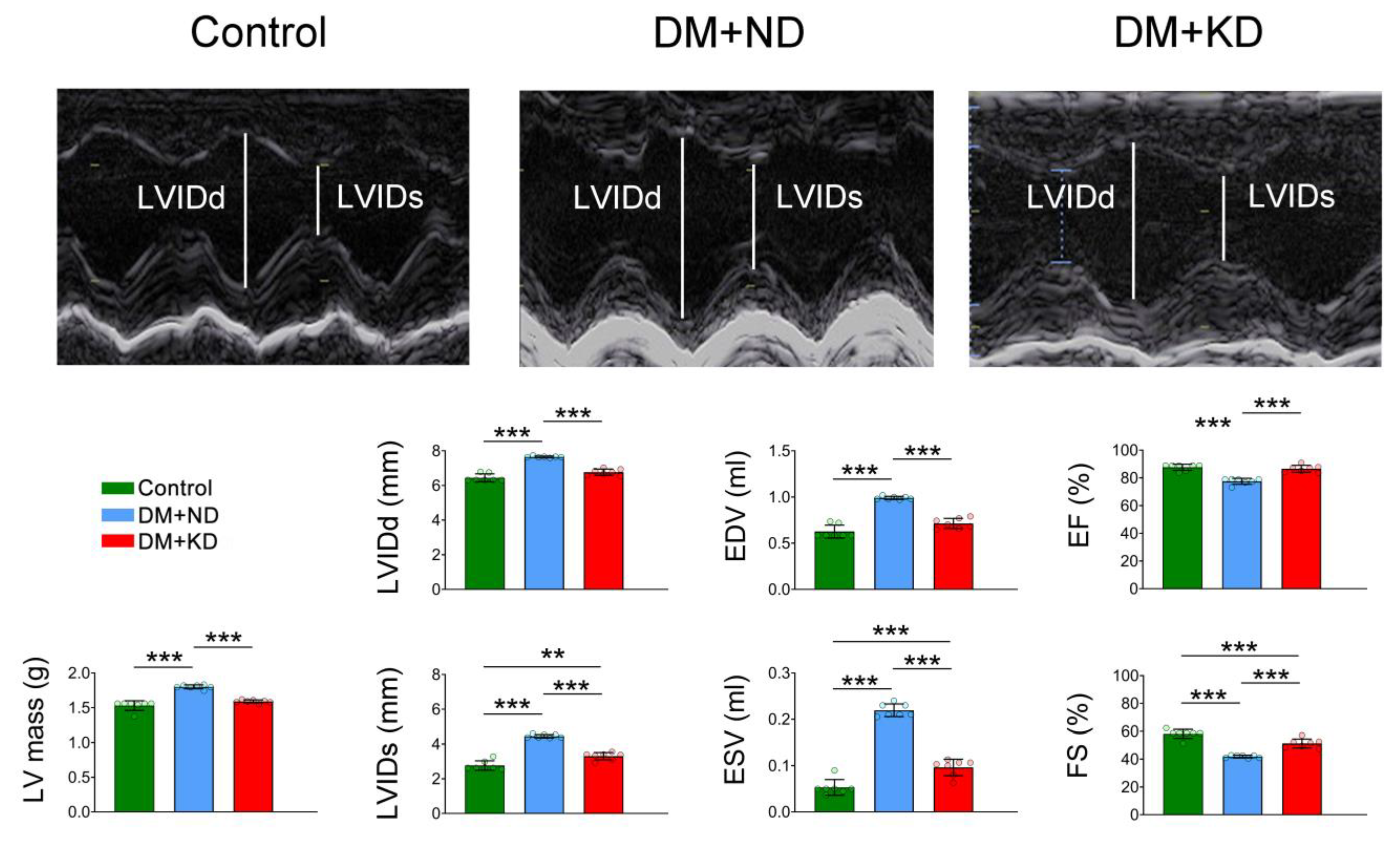
2.1. Myocardial Function and Electrophysiology of the Ventricular Myocytes of Control Rats, DM Rats on a Normal Diet (ND), and DM Rats on a KD
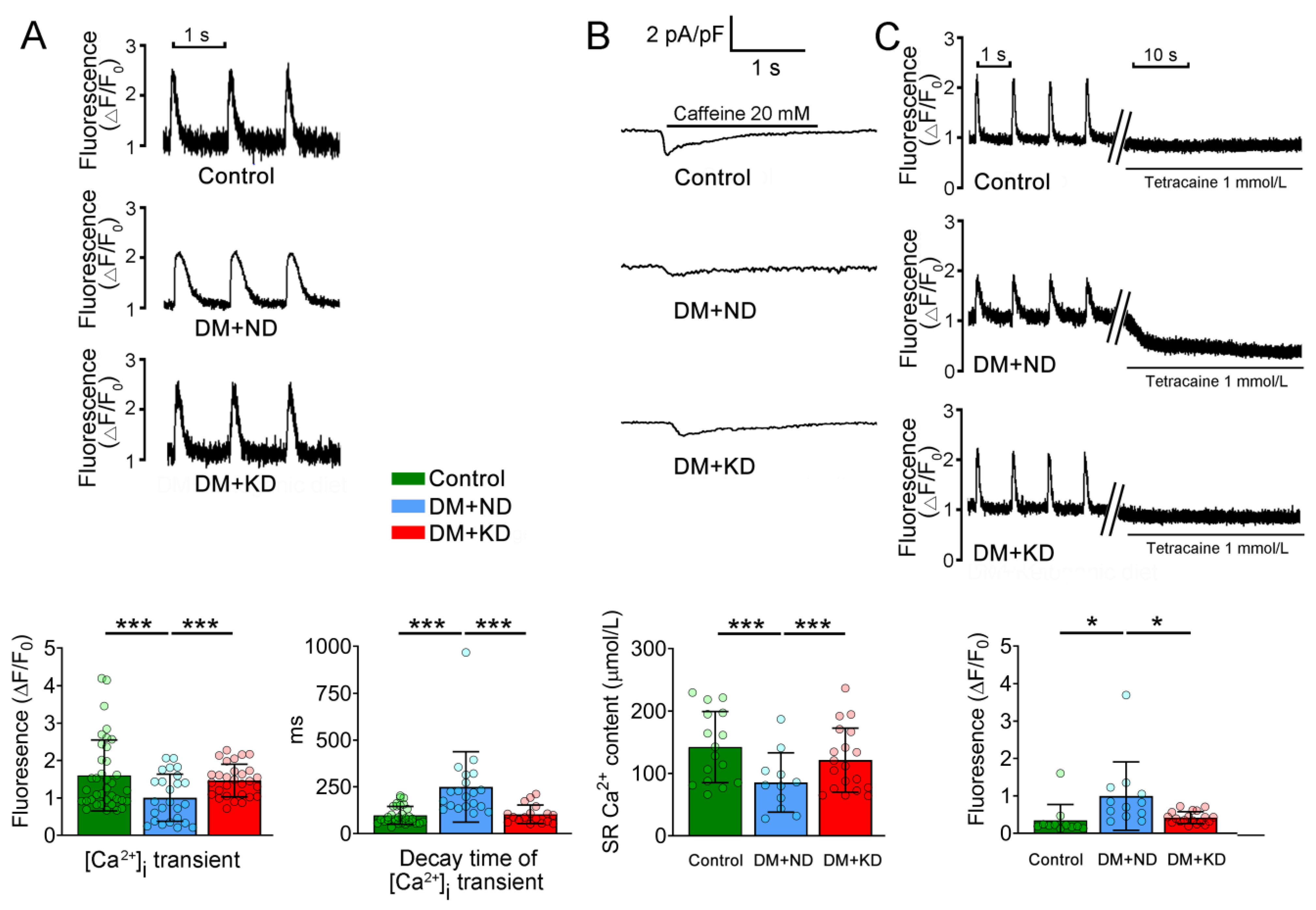
2.2. Ca2+ Stores in the Ventricular Myocytes of DM with or without a KD
2.3. Effects of KD L-Type Ca2+ Current, (ICa-L) Current, and Na+/Ca2+ Exchanger (NCX) Current in the Ventricular Myocytes of DM Rats with and without KD
2.4. Effects of KD on Late Sodium Current (INa-Late) and Sodium/Hydrogen (Na+/H+) Exchanger Current on the Ventricular Myocytes DM Rats with or without a KD
2.5. Oxidative Stress of the Ventricular Myocytes of the Control Rats and DM with or without a KD
2.6. The Effects of Ca2+ Regulatory Proteins in DM Hearts with or without a KD
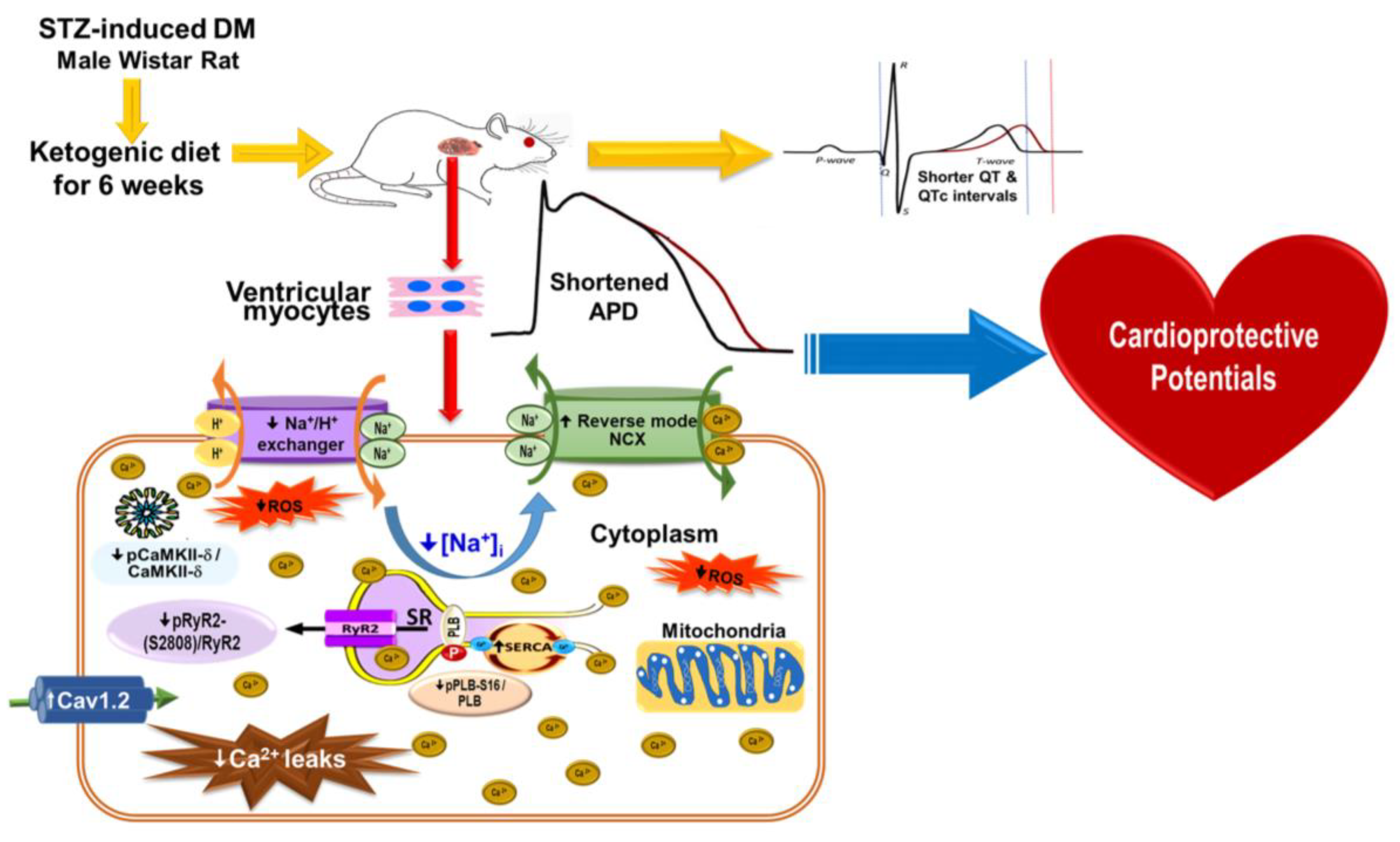
3. Discussion
Study Limitation
4. Material and Methods
4.1. Animal Experiments
4.2. Cardiomyocyte Isolation
4.3. Measurement of Intracellular Ca2+ ([Ca2+]i), SR Ca2+ Transient, and Ca2+ Store
4.4. AP and Ionic Currents
4.5. Intracellular Na+ and Reactive Oxygen Species (ROS) Measurements
4.6. Western Blot
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baranano, K.W.; Hartman, A.L. The ketogenic diet: Uses in epilepsy and other neurologic illnesses. Curr. Treat. Options Neurol. 2008, 10, 410–419. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Branco, A.F.; Ferreira, A.; Simoes, R.F.; Magalhaes-Novais, S.; Zehowski, C.; Cope, E.; Silva, A.M.; Pereira, D.; Sardao, V.A.; Cunha-Oliveira, T. Ketogenic diets: From cancer to mitochondrial diseases and beyond. Eur. J. Clin. Investig. 2016, 46, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, C.; Shang, F.F.; Luo, M.; You, Y.; Zhai, Q.; Xia, Y.; Suxin, L. Ketogenic Diet Ameliorates Cardiac Dysfunction via Balancing Mitochondrial Dynamics and Inhibiting Apoptosis in Type 2 Diabetic Mice. Aging Dis. 2020, 11, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional Ketosis and Mitohormesis: Potential Implications for Mitochondrial Function and Human Health. J. Nutr. Metab. 2018, 2018, 5157645. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B. Ketone metabolism in the failing heart. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158813. [Google Scholar] [CrossRef]
- Yurista, S.R.; Matsuura, T.R.; Sillje, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Heart Fail. 2021, 14, e007684. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Westermeier, F.; Riquelme, J.A.; Pavez, M.; Garrido, V.; Diaz, A.; Verdejo, H.E.; Castro, P.F.; Garcia, L.; Lavandero, S. New Molecular Insights of Insulin in Diabetic Cardiomyopathy. Front. Physiol. 2016, 7, 125. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Chen, Y.C.; Lin, Y.K.; Chung, C.C.; Lu, Y.Y.; Kao, Y.H.; Chen, Y.J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. [Google Scholar] [CrossRef] [PubMed]
- Yurista, S.R.; Chong, C.R.; Badimon, J.J.; Kelly, D.P.; de Boer, R.A.; Westenbrink, B.D. Therapeutic Potential of Ketone Bodies for Patients with Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Trang, N.N.; Chung, C.C.; Lee, T.W.; Cheng, W.L.; Kao, Y.H.; Huang, S.Y.; Lee, T.I.; Chen, Y.J. Empagliflozin and Liraglutide Differentially Modulate Cardiac Metabolism in Diabetic Cardiomyopathy in Rats. Int. J. Mol. Sci. 2021, 22, 1177. [Google Scholar] [CrossRef]
- Trang, N.N.; Lee, T.W.; Kao, Y.H.; Chao, T.F.; Lee, T.I.; Chen, Y.J. Ketogenic diet modulates cardiac metabolic dysregulation in streptozocin-induced diabetic rats. J. Nutr. Biochem. 2023, 111, 109161. [Google Scholar] [CrossRef] [PubMed]
- Garbow, J.R.; Doherty, J.M.; Schugar, R.C.; Travers, S.; Weber, M.L.; Wentz, A.E.; Ezenwajiaku, N.; Cotter, D.G.; Brunt, E.M.; Crawford, P.A. Hepatic steatosis, inflammation, and ER stress in mice maintained long term on a very low-carbohydrate ketogenic diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G956–G967. [Google Scholar] [CrossRef]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary carbohydrate restriction as the first approach in diabetes management: Critical review and evidence base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef]
- Westman, E.C.; Feinman, R.D.; Mavropoulos, J.C.; Vernon, M.C.; Volek, J.S.; Wortman, J.A.; Yancy, W.S.; Phinney, S.D. Low-carbohydrate nutrition and metabolism. Am. J. Clin. Nutr. 2007, 86, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, I.; Lepore, J.J.; Epstein, J.A.; Parmacek, M.S.; Gruber, P.J. MRL mice fail to heal the heart in response to ischemia-reperfusion injury. Wound Repair Regen. 2005, 13, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Vetter, R.; Rehfeld, U.; Reissfelder, C.; Weiss, W.; Wagner, K.D.; Gunther, J.; Hammes, A.; Tschope, C.; Dillmann, W.; Paul, M. Transgenic overexpression of the sarcoplasmic reticulum Ca2+ ATPase improves reticular Ca2+ handling in normal and diabetic rat hearts. FASEB J. 2002, 16, 1657–1659. [Google Scholar] [CrossRef]
- Trost, S.U.; Belke, D.D.; Bluhm, W.F.; Meyer, M.; Swanson, E.; Dillmann, W.H. Overexpression of the sarcoplasmic reticulum Ca(2+)-ATPase improves myocardial contractility in diabetic cardiomyopathy. Diabetes 2002, 51, 1166–1171. [Google Scholar] [CrossRef]
- Teshima, Y.; Takahashi, N.; Saikawa, T.; Hara, M.; Yasunaga, S.; Hidaka, S.; Sakata, T. Diminished expression of sarcoplasmic reticulum Ca(2+)-ATPase and ryanodine sensitive Ca(2+)Channel mRNA in streptozotocin-induced diabetic rat heart. J. Mol. Cell Cardiol. 2000, 32, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Matsuda, N.; Kimura, J.; Ishitani, T.; Tamada, A.; Gando, S.; Kemmotsu, O.; Kanno, M. Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: Implication in Ca2+ overload. J. Physiol. 2000, 527 Pt 1, 85–94. [Google Scholar] [CrossRef]
- Kahn, J.K.; Sisson, J.C.; Vinik, A.I. QT interval prolongation and sudden cardiac death in diabetic autonomic neuropathy. J. Clin. Endocrinol. Metab. 1987, 64, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Magyar, J.; Rusznak, Z.; Szentesi, P.; Szucs, G.; Kovacs, L. Action potentials and potassium currents in rat ventricular muscle during experimental diabetes. J. Mol. Cell Cardiol. 1992, 24, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Movahed, M.R.; Bahrami, A.; Manrique, C.; Hashemzadeh, M. Strong independent association between third-degree AV-block and diabetes mellitus using a large database. Diabetes Res. Clin. Pract. 2023, 205, 110948. [Google Scholar] [CrossRef] [PubMed]
- Pescariu, S.A.; Tudoran, C.; Pop, G.N.; Pescariu, S.; Timar, R.Z.; Tudoran, M. Impact of COVID-19 Pandemic on the Implantation of Intra-Cardiac Devices in Diabetic and Non-Diabetic Patients in the Western of Romania. Medicina 2021, 57, 441. [Google Scholar] [CrossRef]
- Lee, T.I.; Chen, Y.C.; Kao, Y.H.; Hsiao, F.C.; Lin, Y.K.; Chen, Y.J. Rosiglitazone induces arrhythmogenesis in diabetic hypertensive rats with calcium handling alteration. Int. J. Cardiol. 2013, 165, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Ungvari, Z.; Nanasi, P.P.; Kecskemeti, V. Electrophysiological changes in rat ventricular and atrial myocardium at different stages of experimental diabetes. Acta Physiol. Scand. 1999, 166, 7–13. [Google Scholar] [CrossRef]
- Lichtenstein, A.H.; Appel, L.J.; Vadiveloo, M.; Hu, F.B.; Kris-Etherton, P.M.; Rebholz, C.M.; Sacks, F.M.; Thorndike, A.N.; Van Horn, L.; Wylie-Rosett, J. 2021 Dietary Guidance to Improve Cardiovascular Health: A Scientific Statement From the American Heart Association. Circulation 2021, 144, e472–e487. [Google Scholar] [CrossRef]
- D’Souza, M.S.; Dong, T.A.; Ragazzo, G.; Dhindsa, D.S.; Mehta, A.; Sandesara, P.B.; Freeman, A.M.; Taub, P.; Sperling, L.S. From Fad to Fact: Evaluating the Impact of Emerging Diets on the Prevention of Cardiovascular Disease. Am. J. Med. 2020, 133, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Kuchkuntla, A.R.; Limketkai, B.; Nanda, S.; Hurt, R.T.; Mundi, M.S. Fad Diets: Hype or Hope? Curr. Nutr. Rep. 2018, 7, 310–323. [Google Scholar] [CrossRef]
- Asghar, O.; Al-Sunni, A.; Khavandi, K.; Khavandi, A.; Withers, S.; Greenstein, A.; Heagerty, A.M.; Malik, R.A. Diabetic cardiomyopathy. Clin. Sci. 2009, 116, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.K.; Pierce, G.N.; Dhalla, K.S.; Dhalla, N.S. Defective sarcoplasmic reticular calcium transport in diabetic cardiomyopathy. Am. J. Physiol. 1983, 244, E528–E535. [Google Scholar] [CrossRef]
- Lacombe, V.A.; Viatchenko-Karpinski, S.; Terentyev, D.; Sridhar, A.; Emani, S.; Bonagura, J.D.; Feldman, D.S.; Gyorke, S.; Carnes, C.A. Mechanisms of impaired calcium handling underlying subclinical diastolic dysfunction in diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1787–R1797. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Zhong, Y.; Hoit, B.D.; Grupp, I.L.; Hahn, H.; Dilly, K.W.; Guatimosim, S.; Lederer, W.J.; Matlib, M.A. Defective intracellular Ca(2+) signaling contributes to cardiomyopathy in Type 1 diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1398–H1408. [Google Scholar] [CrossRef]
- Lu, Z.; Jiang, Y.P.; Xu, X.H.; Ballou, L.M.; Cohen, I.S.; Lin, R.Z. Decreased L-type Ca2+ current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes 2007, 56, 2780–2789. [Google Scholar] [CrossRef]
- Bracken, N.; Howarth, F.C.; Singh, J. Effects of streptozotocin-induced diabetes on contraction and calcium transport in rat ventricular cardiomyocytes. Ann. N. Y. Acad. Sci. 2006, 1084, 208–222. [Google Scholar] [CrossRef]
- Al Kury, L.T. Calcium Homeostasis in Ventricular Myocytes of Diabetic Cardiomyopathy. J. Diabetes Res. 2020, 2020, 1942086. [Google Scholar] [CrossRef]
- Pereira, L.; Matthes, J.; Schuster, I.; Valdivia, H.H.; Herzig, S.; Richard, S.; Gomez, A.M. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes 2006, 55, 608–615. [Google Scholar] [CrossRef]
- Wang, D.W.; Kiyosue, T.; Shigematsu, S.; Arita, M. Abnormalities of K+ and Ca2+ currents in ventricular myocytes from rats with chronic diabetes. Am. J. Physiol. 1995, 269, H1288–H1296. [Google Scholar] [CrossRef] [PubMed]
- Chattou, S.; Diacono, J.; Feuvray, D. Decrease in sodium-calcium exchange and calcium currents in diabetic rat ventricular myocytes. Acta Physiol. Scand. 1999, 166, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Despa, S. Myocyte [Na(+)](i) Dysregulation in Heart Failure and Diabetic Cardiomyopathy. Front. Physiol. 2018, 9, 1303. [Google Scholar] [CrossRef]
- Gonano, L.A.; Jones, P.P. FK506-binding proteins 12 and 12.6 (FKBPs) as regulators of cardiac Ryanodine Receptors: Insights from new functional and structural knowledge. Channels 2017, 11, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.M.; Wang, Y.L.; Guo, C.Y.; Chen, J.L.; Wu, Y.Q. Progressive decay of Ca2+ homeostasis in the development of diabetic cardiomyopathy. Cardiovasc. Diabetol. 2014, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Yaras, N.; Ugur, M.; Ozdemir, S.; Gurdal, H.; Purali, N.; Lacampagne, A.; Vassort, G.; Turan, B. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes 2005, 54, 3082–3088. [Google Scholar] [CrossRef]
- Hamilton, S.; Terentyev, D. Proarrhythmic Remodeling of Calcium Homeostasis in Cardiac Disease; Implications for Diabetes and Obesity. Front. Physiol. 2018, 9, 1517. [Google Scholar] [CrossRef]
- Tang, C.; Koulajian, K.; Schuiki, I.; Zhang, L.; Desai, T.; Ivovic, A.; Wang, P.; Robson-Doucette, C.; Wheeler, M.B.; Minassian, B.; et al. Glucose-induced beta cell dysfunction in vivo in rats: Link between oxidative stress and endoplasmic reticulum stress. Diabetologia 2012, 55, 1366–1379. [Google Scholar] [CrossRef]
- Anderson, M.E.; Brown, J.H.; Bers, D.M. CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell Cardiol. 2011, 51, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Daniels, L.; Bell, J.R.; Delbridge, L.M.; McDonald, F.J.; Lamberts, R.R.; Erickson, J.R. The role of CaMKII in diabetic heart dysfunction. Heart Fail. Rev. 2015, 20, 589–600. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef]
- Zhang, T.; Kohlhaas, M.; Backs, J.; Mishra, S.; Phillips, W.; Dybkova, N.; Chang, S.; Ling, H.; Bers, D.M.; Maier, L.S.; et al. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 2007, 282, 35078–35087. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Maurer, U.; Schotola, H.; Hartmann, N.; Didie, M.; Zimmermann, W.H.; Jacobshagen, C.; Wagner, S.; Maier, L.S. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res. Cardiol. 2011, 106, 263–272. [Google Scholar] [CrossRef]
- Bueno, N.B.; de Melo, I.S.; de Oliveira, S.L.; da Rocha Ataide, T. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2013, 110, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Jeon, S.M.; Shin, S. Impact of a Ketogenic Diet on Metabolic Parameters in Patients with Obesity or Overweight and with or without Type 2 Diabetes: A Meta-Analysis of Randomized Controlled Trials. Nutrients 2020, 12, 2005. [Google Scholar] [CrossRef] [PubMed]
- Barrea, L.; Verde, L.; Santangeli, P.; Luca, S.; Docimo, A.; Savastano, S.; Colao, A.; Muscogiuri, G. Very low-calorie ketogenic diet (VLCKD): An antihypertensive nutritional approach. J. Transl. Med. 2023, 21, 128. [Google Scholar] [CrossRef] [PubMed]
- Dynka, D.; Kowalcze, K.; Charuta, A.; Paziewska, A. The Ketogenic Diet and Cardiovascular Diseases. Nutrients 2023, 15, 3368. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lin, G.; Chen, J.; Chen, Z.; Xu, F.; Zhu, F.; Zhang, J.; Yuan, S. The effect of periodic ketogenic diet on newly diagnosed overweight or obese patients with type 2 diabetes. BMC Endocr. Disord. 2022, 22, 34. [Google Scholar] [CrossRef]
- Westman, E.C.; Tondt, J.; Maguire, E.; Yancy, W.S., Jr. Implementing a low-carbohydrate, ketogenic diet to manage type 2 diabetes mellitus. Expert. Rev. Endocrinol. Metab. 2018, 13, 263–272. [Google Scholar] [CrossRef]
- American Diabetes, A. 5. Lifestyle Management: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42, S46–S60. [Google Scholar] [CrossRef]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; DeFronzo, R.A.; Einhorn, D.; Fonseca, V.A.; et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm—2019 Executive Summary. Endocr. Pract. 2019, 25, 69–100. [Google Scholar] [CrossRef] [PubMed]
- McGaugh, E.; Barthel, B. A Review of Ketogenic Diet and Lifestyle. Mo. Med. 2022, 119, 84–88. [Google Scholar] [PubMed]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rodriguez, E.; Anderson, C.A.; Thayne, K.; Chitwood, W.R.; Kypson, A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H118–H124. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, P.; Yeo, J.L.; Brady, E.M.; McCann, G.P. Role of inflammation in diabetic cardiomyopathy. Ther. Adv. Endocrinol. Metab. 2022, 13, 20420188221083530. [Google Scholar] [CrossRef] [PubMed]
- Elia, E.; Ministrini, S.; Carbone, F.; Montecucco, F. Diabetic cardiomyopathy and inflammation: Development of hostile microenvironment resulting in cardiac damage. Minerva Cardiol. Angiol. 2022, 70, 357–369. [Google Scholar] [CrossRef]
- Srivastava, S.; Pawar, V.A.; Tyagi, A.; Sharma, K.P.; Kumar, V.; Shukla, S.K. Immune Modulatory Effects of Ketogenic Diet in Different Disease Conditions. Immuno 2023, 3, 1–15. [Google Scholar] [CrossRef]
- Lee, T.I.; Kao, Y.H.; Chen, Y.C.; Pan, N.H.; Chen, Y.J. Oxidative stress and inflammation modulate peroxisome proliferator-activated receptors with regional discrepancy in diabetic heart. Eur. J. Clin. Investig. 2010, 40, 692–699. [Google Scholar] [CrossRef]
- Lee, T.I.; Kao, Y.H.; Chen, Y.C.; Pan, N.H.; Lin, Y.K.; Chen, Y.J. Cardiac peroxisome-proliferator-activated receptor expression in hypertension co-existing with diabetes. Clin. Sci. 2011, 121, 305–312. [Google Scholar] [CrossRef]
- Chan, C.S.; Lin, F.J.; Liu, C.M.; Lin, Y.K.; Chen, Y.C.; Hsu, C.C.; Higa, S.; Chen, S.A.; Chen, Y.J. Mirabegron, a beta3-adrenoreceptor agonist, regulates right and left atrial arrhythmogenesis differently. Exp. Ther. Med. 2022, 24, 720. [Google Scholar] [CrossRef]
- Chang, S.H.; Chen, Y.C.; Chiang, S.J.; Higa, S.; Cheng, C.C.; Chen, Y.J.; Chen, S.A. Increased Ca(2+) sparks and sarcoplasmic reticulum Ca(2+) stores potentially determine the spontaneous activity of pulmonary vein cardiomyocytes. Life Sci. 2008, 83, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Shannon, T.R.; Ginsburg, K.S.; Bers, D.M. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ. Res. 2002, 91, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Hove-Madsen, L.; Llach, A.; Bayes-Genis, A.; Roura, S.; Rodriguez Font, E.; Aris, A.; Cinca, J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 2004, 110, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Chen, Y.C.; Kao, Y.H.; Lin, Y.K.; Yeh, Y.H.; Chen, S.A.; Chen, Y.J. Colchicine modulates calcium homeostasis and electrical property of HL-1 cells. J. Cell Mol. Med. 2016, 20, 1182–1190. [Google Scholar] [CrossRef]
- Suenari, K.; Chen, Y.C.; Kao, Y.H.; Cheng, C.C.; Lin, Y.K.; Chen, Y.J.; Chen, S.A. Discrepant electrophysiological characteristics and calcium homeostasis of left atrial anterior and posterior myocytes. Basic Res. Cardiol. 2011, 106, 65–74. [Google Scholar] [CrossRef]
- Huang, S.Y.; Chen, Y.C.; Kao, Y.H.; Hsieh, M.H.; Lin, Y.K.; Chung, C.C.; Lee, T.I.; Tsai, W.C.; Chen, S.A.; Chen, Y.J. Fibroblast growth factor 23 dysregulates late sodium current and calcium homeostasis with enhanced arrhythmogenesis in pulmonary vein cardiomyocytes. Oncotarget 2016, 7, 69231–69242. [Google Scholar] [CrossRef]
- Kondratev, D.; Christ, A.; Gallitelli, M.F. Inhibition of the Na+-H+ exchanger with cariporide abolishes stretch-induced calcium but not sodium accumulation in mouse ventricular myocytes. Cell Calcium 2005, 37, 69–80. [Google Scholar] [CrossRef]
- Viatchenko-Karpinski, S.; Kornyeyev, D.; El-Bizri, N.; Budas, G.; Fan, P.; Jiang, Z.; Yang, J.; Anderson, M.E.; Shryock, J.C.; Chang, C.P.; et al. Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J. Mol. Cell Cardiol. 2014, 76, 247–256. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, X.; Zhang, Q.; Li, L.; Shang, Y.; Wang, Z.; Zhong, M.; Chen, Y.; Zhang, W.; Tang, M. Activin receptor-like kinase 7 silencing alleviates cardiomyocyte apoptosis, cardiac fibrosis, and dysfunction in diabetic rats. Exp. Biol. Med. 2022, 247, 1397–1409. [Google Scholar] [CrossRef]
- Lei, S.; Li, H.; Xu, J.; Liu, Y.; Gao, X.; Wang, J.; Ng, K.F.; Lau, W.B.; Ma, X.L.; Rodrigues, B.; et al. Hyperglycemia-induced protein kinase C beta2 activation induces diastolic cardiac dysfunction in diabetic rats by impairing caveolin-3 expression and Akt/eNOS signaling. Diabetes 2013, 62, 2318–2328. [Google Scholar] [CrossRef]
- Liu, X.; Guo, B.; Zhang, W.; Ma, B.; Li, Y. MiR-20a-5p overexpression prevented diabetic cardiomyopathy via inhibition of cardiomyocyte apoptosis, hypertrophy, fibrosis and JNK/NF-kappaB signalling pathway. J. Biochem. 2021, 170, 349–362. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.-I.; Trang, N.N.; Lee, T.-W.; Higa, S.; Kao, Y.-H.; Chen, Y.-C.; Chen, Y.-J. Ketogenic Diet Regulates Cardiac Remodeling and Calcium Homeostasis in Diabetic Rat Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 16142. https://doi.org/10.3390/ijms242216142
Lee T-I, Trang NN, Lee T-W, Higa S, Kao Y-H, Chen Y-C, Chen Y-J. Ketogenic Diet Regulates Cardiac Remodeling and Calcium Homeostasis in Diabetic Rat Cardiomyopathy. International Journal of Molecular Sciences. 2023; 24(22):16142. https://doi.org/10.3390/ijms242216142
Chicago/Turabian StyleLee, Ting-I, Nguyen Ngoc Trang, Ting-Wei Lee, Satoshi Higa, Yu-Hsun Kao, Yao-Chang Chen, and Yi-Jen Chen. 2023. "Ketogenic Diet Regulates Cardiac Remodeling and Calcium Homeostasis in Diabetic Rat Cardiomyopathy" International Journal of Molecular Sciences 24, no. 22: 16142. https://doi.org/10.3390/ijms242216142