
The Impact of Neurotransmitters on the Neurobiology of Neurodegenerative Diseases

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Dopaminergic Neurotransmission
2.1. Regulation of Dopamine Synthesis
2.2. Dopamine Transporter Regulation
2.3. Vesicular Monoamine Transporter Regulation
3. Cholinergic Neurotransmission
3.1. Regulation of Acetylcholine Transmission
3.2. Acetylcholine Signal Transduction
3.3. Vesicular Acetylcholine Transporter Regulation
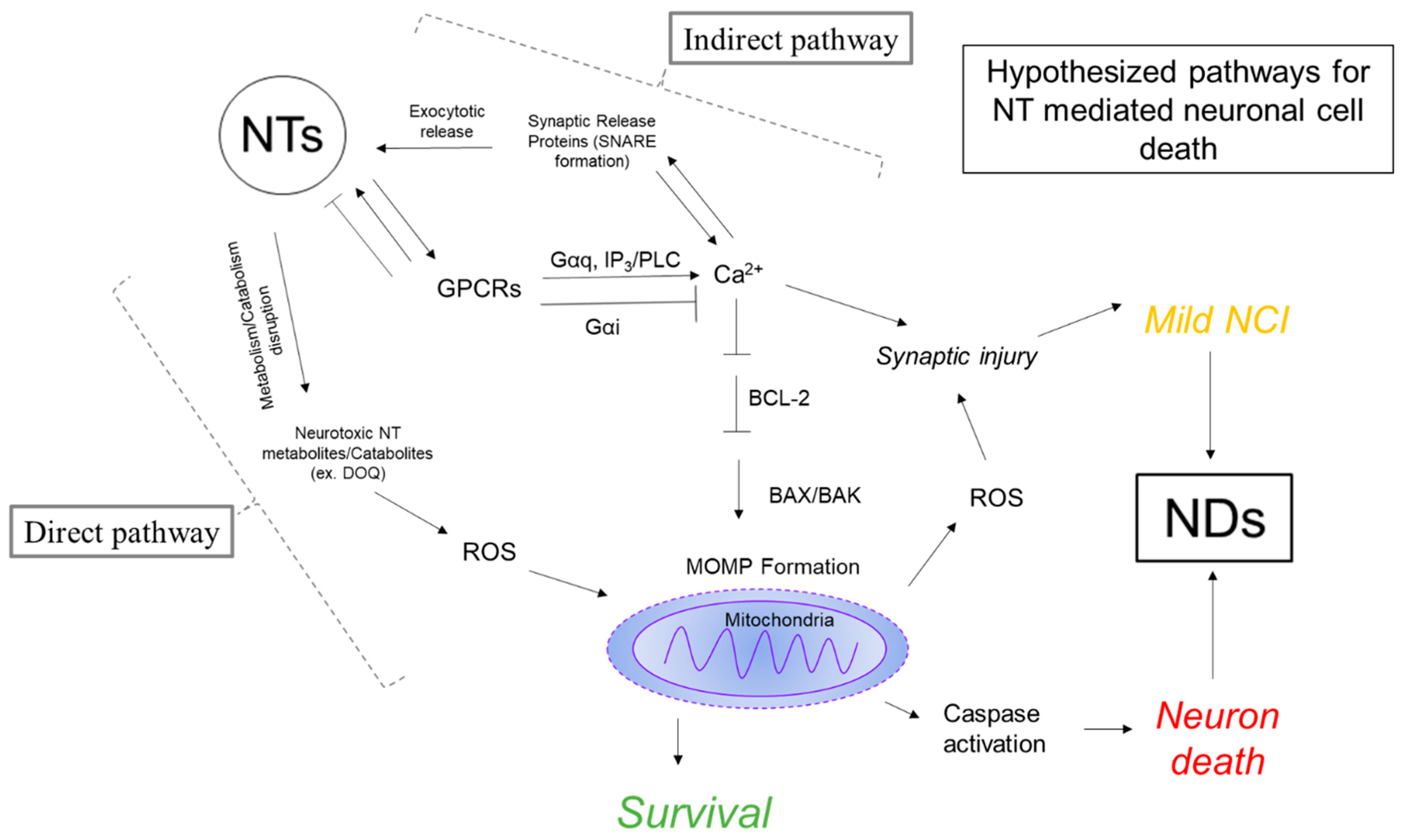
4. Neurotransmitter Hypothesis of Neurodegeneration
4.1. Evidence for Pyroptosis in Neurodegenerative Diseases
4.2. Dysregulation of Neurotransmission in Neurodegenerative Disorders
4.3. Evidence for Neurotransmission as a Mediator of Neurodegeneration
4.3.1. Calcium Function in the Neuron
4.3.2. Mitochondrial Function in the Neuron
4.3.3. Neurotransmitter Mediated Dysregulation of Ca2+
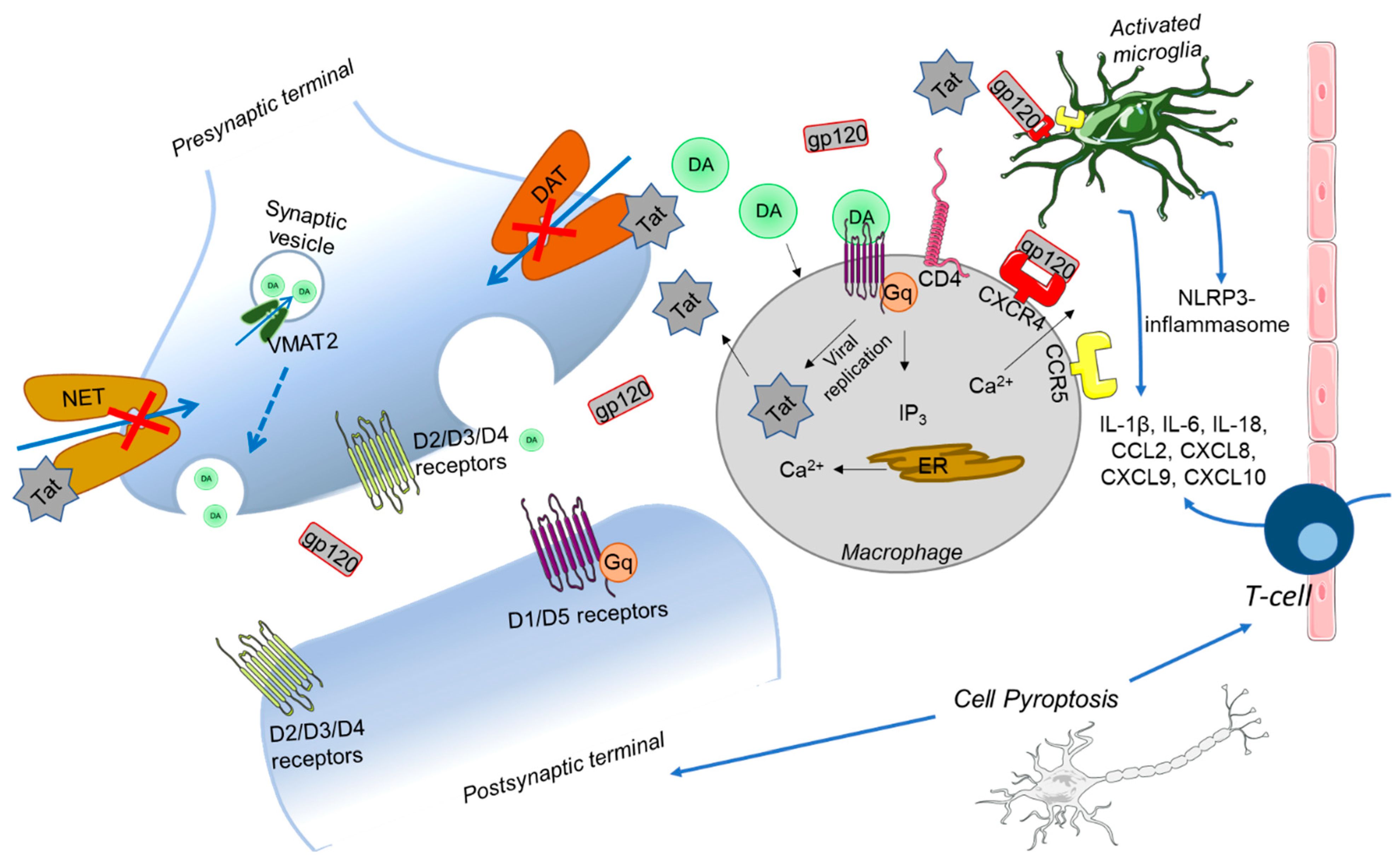
5. Neurodegeneration and HIV-1 Associated Neurocognitive Disorders
5.1. NeuroHIV Background
5.2. Disturbances in Neurotransmitter Systems in HAND
5.3. Evidence for Mitochondria and Ca2+ Disturbances in HAND
5.4. Mechanisms for Synaptic Injury
6. Crosstalk between Inflammation and Mitochondrial Induced Cell Death
7. Conclusions
Funding
Conflicts of Interest
References
- Zhang, X.X.; Tian, Y.; Wang, Z.T.; Ma, Y.H.; Tan, L.; Yu, J.T. The epidemiology of alzheimer’s disease modifiable risk factors and prevention. J. Prev. Alzheimers Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global trends in the incidence, prevalence, and years lived with disability of parkinson’s disease in 204 countries/territories from 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive review on alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatr. 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Beshir, S.A.; Aadithsoorya, A.M.; Parveen, A.; Goh, S.S.L.; Hussain, N.; Menon, V.B. Aducanumab therapy to treat alzheimer’s disease: A narrative review. Int. J. Alzheimers Dis. 2022, 2022, 9343514. [Google Scholar] [CrossRef]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef]
- German, C.L.; Baladi, M.G.; McFadden, L.M.; Hanson, G.R.; Fleckenstein, A.E. Regulation of the dopamine and vesicular monoamine transporters: Pharmacological targets and implications for disease. Pharmacol. Rev. 2015, 67, 1005–1024. [Google Scholar] [CrossRef]
- Liu, Y.; Edwards, R.H. The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu. Rev. Neurosci. 1997, 20, 125–156. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, K.G. Presynaptic regulation of dopamine release: Role of the DAT and VMAT2 transporters. Neurochem. Int. 2019, 122, 94–105. [Google Scholar] [CrossRef]
- Nirenberg, M.J.; Vaughan, R.A.; Uhl, G.R.; Kuhar, M.J.; Pickel, V.M. The dopamine transporter is localized to dendritic and axonal plasma membranes of nigrostriatal dopaminergic neurons. J. Neurosci. 1996, 16, 436–447. [Google Scholar] [CrossRef]
- Predescu, D.-V.; Crețoiu, S.M.; Crețoiu, D.; Pavelescu, L.A.; Suciu, N.; Radu, B.M.; Voinea, S.-C. G Protein-Coupled Receptors (GPCRs)-Mediated Calcium Signaling in Ovarian Cancer: Focus on GPCRs activated by Neurotransmitters and Inflammation-Associated Molecules. Int. J. Mol. Sci. 2019, 20, 5568. [Google Scholar] [CrossRef]
- Vaughan, R.A.; Foster, J.D. Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol. Sci. 2013, 34, 489–496. [Google Scholar] [CrossRef]
- Guptaroy, B.; Fraser, R.; Desai, A.; Zhang, M.; Gnegy, M.E. Site-directed mutations near transmembrane domain 1 alter conformation and function of norepinephrine and dopamine transporters. Mol. Pharmacol. 2011, 79, 520–532. [Google Scholar] [CrossRef]
- Jones, S.R.; Joseph, J.D.; Barak, L.S.; Caron, M.G.; Wightman, R.M. Dopamine neuronal transport kinetics and effects of amphetamine. J. Neurochem. 1999, 73, 2406–2414. [Google Scholar] [CrossRef]
- Carvelli, L.; McDonald, P.W.; Blakely, R.D.; DeFelice, L.J. Dopamine transporters depolarize neurons by a channel mechanism. Proc. Natl. Acad. Sci. USA 2004, 101, 16046–16051. [Google Scholar] [CrossRef]
- Shan, J.; Javitch, J.A.; Shi, L.; Weinstein, H. The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PLoS ONE 2011, 6, e16350. [Google Scholar] [CrossRef]
- Zhu, J.; Quizon, P.M.; Wang, Y.; Adeniran, C.A.; Strauss, M.J.; Jiménez-Torres, A.C.; Patel, P.; Cirino, T.J.; Eans, S.O.; Hammond, H.R.; et al. SRI-32743, a novel allosteric modulator, attenuates HIV-1 Tat protein-induced inhibition of the dopamine transporter and alleviates the potentiation of cocaine reward in HIV-1 Tat transgenic mice. Neuropharmacology 2022, 220, 109239. [Google Scholar] [CrossRef]
- Cao, J.; Slack, R.D.; Bakare, O.M.; Burzynski, C.; Rais, R.; Slusher, B.S.; Kopajtic, T.; Bonifazi, A.; Ellenberger, M.P.; Yano, H.; et al. Novel and High Affinity 2-[(Diphenylmethyl)sulfinyl]acetamide (Modafinil) Analogues as Atypical Dopamine Transporter Inhibitors. J. Med. Chem. 2016, 59, 10676–10691. [Google Scholar] [CrossRef] [PubMed]
- Adkins, E.M.; Samuvel, D.J.; Fog, J.U.; Eriksen, J.; Jayanthi, L.D.; Vaegter, C.B.; Ramamoorthy, S.; Gether, U. Membrane mobility and microdomain association of the dopamine transporter studied with fluorescence correlation spectroscopy and fluorescence recovery after photobleaching. Biochemistry 2007, 46, 10484–10497. [Google Scholar] [CrossRef] [PubMed]
- Gorentla, B.K.; Moritz, A.E.; Foster, J.D.; Vaughan, R.A. Proline-directed phosphorylation of the dopamine transporter N-terminal domain. Biochemistry 2009, 48, 1067–1076. [Google Scholar] [CrossRef]
- Foster, J.D.; Pananusorn, B.; Vaughan, R.A. Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J. Biol. Chem. 2002, 277, 25178–25186. [Google Scholar] [CrossRef] [PubMed]
- Cervinski, M.A.; Foster, J.D.; Vaughan, R.A. Psychoactive substrates stimulate dopamine transporter phosphorylation and down-regulation by cocaine-sensitive and protein kinase C-dependent mechanisms. J. Biol. Chem. 2005, 280, 40442–40449. [Google Scholar] [CrossRef] [PubMed]
- Loder, M.K.; Melikian, H.E. The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J. Biol. Chem. 2003, 278, 22168–22174. [Google Scholar] [CrossRef] [PubMed]
- Bolland, D.E.; Moritz, A.E.; Stanislowski, D.J.; Vaughan, R.A.; Foster, J.D. Palmitoylation by multiple DHHC enzymes enhances dopamine transporter function and stability. ACS Chem. Neurosci. 2019, 10, 2707–2717. [Google Scholar] [CrossRef]
- Lee, F.J.S.; Pei, L.; Moszczynska, A.; Vukusic, B.; Fletcher, P.J.; Liu, F. Dopamine transporter cell surface localization facilitated by a direct interaction with the dopamine D2 receptor. EMBO J. 2007, 26, 2127–2136. [Google Scholar] [CrossRef]
- Su, P.; Liu, F. A peptide disrupting the D2R-DAT interaction protects against dopamine neurotoxicity. Exp. Neurol. 2017, 295, 176–183. [Google Scholar] [CrossRef]
- Nguyen, E.C.; McCracken, K.A.; Liu, Y.; Pouw, B.; Matsumoto, R.R. Involvement of sigma (sigma) receptors in the acute actions of methamphetamine: Receptor binding and behavioral studies. Neuropharmacology 2005, 49, 638–645. [Google Scholar] [CrossRef]
- Sambo, D.O.; Lebowitz, J.J.; Khoshbouei, H. The sigma-1 receptor as a regulator of dopamine neurotransmission: A potential therapeutic target for methamphetamine addiction. Pharmacol. Ther. 2018, 186, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Hedges, D.M.; Obray, J.D.; Yorgason, J.T.; Jang, E.Y.; Weerasekara, V.K.; Uys, J.D.; Bellinger, F.P.; Steffensen, S.C. Methamphetamine Induces Dopamine Release in the Nucleus Accumbens Through a Sigma Receptor-Mediated Pathway. Neuropsychopharmacology 2018, 43, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Tarakad, A.; Jimenez-Shahed, J. VMAT2 inhibitors in neuropsychiatric disorders. CNS Drugs 2018, 32, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Lawal, H.O.; Krantz, D.E. SLC18: Vesicular neurotransmitter transporters for monoamines and acetylcholine. Mol. Aspects Med. 2013, 34, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Lohr, K.M.; Bernstein, A.I.; Stout, K.A.; Dunn, A.R.; Lazo, C.R.; Alter, S.P.; Wang, M.; Li, Y.; Fan, X.; Hess, E.J.; et al. Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 9977–9982. [Google Scholar] [CrossRef]
- Lee, N.-R.; Zheng, G.; Leggas, M.; Janganati, V.; Nickell, J.R.; Crooks, P.A.; Bardo, M.T.; Dwoskin, L.P. GZ-11608, a Vesicular Monoamine Transporter-2 Inhibitor, Decreases the Neurochemical and Behavioral Effects of Methamphetamine. J. Pharmacol. Exp. Ther. 2019, 371, 526–543. [Google Scholar] [CrossRef]
- Yehuda, S.; Rabinovitz, S.; Mostofsky, D.I. Treatment with a polyunsaturated fatty acid prevents deleterious effects of Ro4-1284. Eur. J. Pharmacol. 1999, 365, 27–34. [Google Scholar] [CrossRef]
- Nickell, J.R.; Siripurapu, K.B.; Vartak, A.; Crooks, P.A.; Dwoskin, L.P. The vesicular monoamine transporter-2: An important pharmacological target for the discovery of novel therapeutics to treat methamphetamine abuse. Adv. Pharmacol. 2014, 69, 71–106. [Google Scholar] [CrossRef]
- Yao, J.; Hersh, L.B. The vesicular monoamine transporter 2 contains trafficking signals in both its N-glycosylation and C-terminal domains. J. Neurochem. 2007, 100, 1387–1396. [Google Scholar] [CrossRef]
- Fei, H.; Grygoruk, A.; Brooks, E.S.; Chen, A.; Krantz, D.E. Trafficking of vesicular neurotransmitter transporters. Traffic 2008, 9, 1425–1436. [Google Scholar] [CrossRef]
- Torres, B.; Ruoho, A.E. N-terminus regulation of VMAT2 mediates methamphetamine-stimulated efflux. Neuroscience 2014, 259, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Bye, L.J.; Finol-Urdaneta, R.K.; Tae, H.-S.; Adams, D.J. Nicotinic acetylcholine receptors: Key targets for attenuating neurodegenerative diseases. Int. J. Biochem. Cell Biol. 2023, 157, 106387. [Google Scholar] [CrossRef] [PubMed]
- Warburton, E.C.; Koder, T.; Cho, K.; Massey, P.V.; Duguid, G.; Barker, G.R.I.; Aggleton, J.P.; Bashir, Z.I.; Brown, M.W. Cholinergic neurotransmission is essential for perirhinal cortical plasticity and recognition memory. Neuron 2003, 38, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Sam, C.; Bordoni, B. Physiology, Acetylcholine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Waxenbaum, J.A.; Reddy, V.; Varacallo, M. Anatomy, autonomic nervous system. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Prado, M.A.M.; Reis, R.A.M.; Prado, V.F.; de Mello, M.C.; Gomez, M.V.; de Mello, F.G. Regulation of acetylcholine synthesis and storage. Neurochem. Int. 2002, 41, 291–299. [Google Scholar] [CrossRef]
- Dey, S.; Ray, K. Cholinergic activity is essential for maintaining the anterograde transport of Choline Acetyltransferase in Drosophila. Sci. Rep. 2018, 8, 8028. [Google Scholar] [CrossRef]
- Han, J.; Pluhackova, K.; Böckmann, R.A. The multifaceted role of SNARE proteins in membrane fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef]
- Rodríguez Cruz, P.M.; Cossins, J.; Beeson, D.; Vincent, A. The neuromuscular junction in health and disease: Molecular mechanisms governing synaptic formation and homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef]
- Trang, A.; Khandhar, P.B. Physiology, Acetylcholinesterase. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Carlson, A.B.; Kraus, G.P. Physiology, Cholinergic Receptors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ojiakor, O.A.; Rylett, R.J. Modulation of sodium-coupled choline transporter CHT function in health and disease. Neurochem. Int. 2020, 140, 104810. [Google Scholar] [CrossRef]
- Papke, R.L. Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharmacol. 2014, 89, 1–11. [Google Scholar] [CrossRef]
- Ho, T.N.T.; Abraham, N.; Lewis, R.J. Synthesis of full-length homodimer αD-VxXXB that targets human α7 nicotinic acetylcholine receptors. RSC Med. Chem. 2022, 13, 1410–1419. [Google Scholar] [CrossRef]
- Wittenberg, R.E.; Wolfman, S.L.; De Biasi, M.; Dani, J.A. Nicotinic acetylcholine receptors and nicotine addiction: A brief introduction. Neuropharmacology 2020, 177, 108256. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed]
- Millar, N.S.; Harkness, P.C. Assembly and trafficking of nicotinic acetylcholine receptors (Review). Mol. Membr. Biol. 2008, 25, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.F.; Mazzo, F.; Pistillo, F.; Gotti, C. Biogenesis, trafficking and up-regulation of nicotinic ACh receptors. Biochem. Pharmacol. 2013, 86, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.; Hall, M.; Adler, L.E.; Leonard, S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol. Psychiatry 1995, 38, 22–33. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef]
- D’Andrea, M.R.; Nagele, R.G. Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer’s disease pyramidal neurons. Curr. Pharm. Des. 2006, 12, 677–684. [Google Scholar] [CrossRef]
- Kudlak, M.; Tadi, P. Physiology, Muscarinic Receptor. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Moran, S.P.; Maksymetz, J.; Conn, P.J. Targeting muscarinic acetylcholine receptors for the treatment of psychiatric and neurological disorders. Trends Pharmacol. Sci. 2019, 40, 1006–1020. [Google Scholar] [CrossRef]
- Prado, V.F.; Roy, A.; Kolisnyk, B.; Gros, R.; Prado, M.A.M. Regulation of cholinergic activity by the vesicular acetylcholine transporter. Biochem. J. 2013, 450, 265–274. [Google Scholar] [CrossRef]
- Nguyen, M.L.; Cox, G.D.; Parsons, S.M. Kinetic parameters for the vesicular acetylcholine transporter: Two protons are exchanged for one acetylcholine. Biochemistry 1998, 37, 13400–13410. [Google Scholar] [CrossRef]
- Chen, K.H.; Reese, E.A.; Kim, H.-W.; Rapoport, S.I.; Rao, J.S. Disturbed neurotransmitter transporter expression in Alzheimer’s disease brain. J. Alzheimers Dis. 2011, 26, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Yang, B.; Li, Y.; Qian, A.; Kang, Y.; Shan, X. Focus on the mechanisms and functions of pyroptosis, inflammasomes, and inflammatory caspases in infectious diseases. Oxid. Med. Cell Longev. 2022, 2022, 2501279. [Google Scholar] [CrossRef] [PubMed]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, Y.-H.; Chen, N.-H.; Wang, H.-B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 2019, 67, 458–464. [Google Scholar] [CrossRef]
- Mangalmurti, A.; Lukens, J.R. How neurons die in Alzheimer’s disease: Implications for neuroinflammation. Curr. Opin. Neurobiol. 2022, 75, 102575. [Google Scholar] [CrossRef]
- Milner, M.T.; Maddugoda, M.; Götz, J.; Burgener, S.S.; Schroder, K. The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer’s disease. Curr. Opin. Immunol. 2021, 68, 116–124. [Google Scholar] [CrossRef]
- Moonen, S.; Koper, M.J.; Van Schoor, E.; Schaeverbeke, J.M.; Vandenberghe, R.; von Arnim, C.A.F.; Tousseyn, T.; De Strooper, B.; Thal, D.R. Pyroptosis in Alzheimer’s disease: Cell type-specific activation in microglia, astrocytes and neurons. Acta Neuropathol. 2023, 145, 175–195. [Google Scholar] [CrossRef]
- Vitet, H.; Brandt, V.; Saudou, F. Traffic signaling: New functions of huntingtin and axonal transport in neurological disease. Curr. Opin. Neurobiol. 2020, 63, 122–130. [Google Scholar] [CrossRef]
- Paldino, E.; Fusco, F.R. Emerging role of NLRP3 inflammasome/pyroptosis in huntington’s disease. Int. J. Mol. Sci. 2022, 23, 8363. [Google Scholar] [CrossRef]
- Chen, K.-P.; Hua, K.-F.; Tsai, F.-T.; Lin, T.-Y.; Cheng, C.-Y.; Yang, D.-I.; Hsu, H.-T.; Ju, T.-C. A selective inhibitor of the NLRP3 inflammasome as a potential therapeutic approach for neuroprotection in a transgenic mouse model of Huntington’s disease. J. Neuroinflammation 2022, 19, 56. [Google Scholar] [CrossRef]
- Wong, K.Y.; Roy, J.; Fung, M.L.; Heng, B.C.; Zhang, C.; Lim, L.W. Relationships between Mitochondrial Dysfunction and Neurotransmission Failure in Alzheimer’s Disease. Aging Dis. 2020, 11, 1291–1316. [Google Scholar] [CrossRef] [PubMed]
- Quartey, M.O.; Nyarko, J.N.K.; Pennington, P.R.; Heistad, R.M.; Klassen, P.C.; Baker, G.B.; Mousseau, D.D. Alzheimer Disease and Selected Risk Factors Disrupt a Co-regulation of Monoamine Oxidase-A/B in the Hippocampus, but Not in the Cortex. Front. Neurosci. 2018, 12, 419. [Google Scholar] [CrossRef] [PubMed]
- Rabbito, A.; Dulewicz, M.; Kulczyńska-Przybik, A.; Mroczko, B. Biochemical markers in alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 1989. [Google Scholar] [CrossRef] [PubMed]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The dual role of glutamatergic neurotransmission in alzheimer’s disease: From pathophysiology to pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of gabaergic neurotransmission in alzheimer’s disease. Front. Aging Neurosci. 2016, 8, 31. [Google Scholar] [CrossRef]
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, Signaling, and Association with Neurological Diseases. Cell Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef]
- Latif, S.; Jahangeer, M.; Maknoon Razia, D.; Ashiq, M.; Ghaffar, A.; Akram, M.; El Allam, A.; Bouyahya, A.; Garipova, L.; Ali Shariati, M.; et al. Dopamine in Parkinson’s disease. Clin. Chim. Acta 2021, 522, 114–126. [Google Scholar] [CrossRef]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef]
- Muñoz, A.; Lopez-Lopez, A.; Labandeira, C.M.; Labandeira-Garcia, J.L. Interactions Between the Serotonergic and Other Neurotransmitter Systems in the Basal Ganglia: Role in Parkinson’s Disease and Adverse Effects of L-DOPA. Front. Neuroanat. 2020, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Krebs, J. Why calcium? how calcium became the best communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C.; Lee, A. Presynaptic calcium channels: Specialized control of synaptic neurotransmitter release. Nat. Rev. Neurosci. 2020, 21, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhou, P.; Wang, A.L.; Wu, D.; Zhao, M.; Südhof, T.C.; Brunger, A.T. The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 2017, 548, 420–425. [Google Scholar] [CrossRef]
- Verhage, M.; Sørensen, J.B. Vesicle docking in regulated exocytosis. Traffic 2008, 9, 1414–1424. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Intracellular calcium homeostasis and signaling. Met. Ions Life Sci. 2013, 12, 119–168. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, G. Gq-Coupled Receptors in Autoimmunity. J. Immunol. Res. 2016, 2016, 3969023. [Google Scholar] [CrossRef]
- Mizuno, N.; Itoh, H. Functions and regulatory mechanisms of Gq-signaling pathways. Neurosignals 2009, 17, 42–54. [Google Scholar] [CrossRef]
- Wang, Q.J. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol. Sci. 2006, 27, 317–323. [Google Scholar] [CrossRef]
- Woll, K.A.; Van Petegem, F. Calcium-release channels: Structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 2022, 102, 209–268. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 463–470. [Google Scholar] [CrossRef]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. Biophys. Acta 2006, 1757, 639–647. [Google Scholar] [CrossRef]
- Betke, K.M.; Wells, C.A.; Hamm, H.E. GPCR mediated regulation of synaptic transmission. Prog. Neurobiol. 2012, 96, 304–321. [Google Scholar] [CrossRef]
- Spangler, S.M.; Bruchas, M.R. Optogenetic approaches for dissecting neuromodulation and GPCR signaling in neural circuits. Curr. Opin. Pharmacol. 2017, 32, 56–70. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Fuxe, K. Oligomeric Receptor Complexes and Their Allosteric Receptor-Receptor Interactions in the Plasma Membrane Represent a New Biological Principle for Integration of Signals in the CNS. Front. Mol. Neurosci. 2019, 12, 230. [Google Scholar] [CrossRef]
- Akam, E.C.; Challiss, R.A.; Nahorski, S.R. G(q/11) and G(i/o) activation profiles in CHO cells expressing human muscarinic acetylcholine receptors: Dependence on agonist as well as receptor-subtype. Br. J. Pharmacol. 2001, 132, 950–958. [Google Scholar] [CrossRef]
- Hasbi, A.; O’Dowd, B.F.; George, S.R. Heteromerization of dopamine D2 receptors with dopamine D1 or D5 receptors generates intracellular calcium signaling by different mechanisms. Curr. Opin. Pharmacol. 2010, 10, 93–99. [Google Scholar] [CrossRef]
- Spoida, K.; Masseck, O.A.; Deneris, E.S.; Herlitze, S. Gq/5-HT2c receptor signals activate a local GABAergic inhibitory feedback circuit to modulate serotonergic firing and anxiety in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 6479–6484. [Google Scholar] [CrossRef] [PubMed]
- McElligott, Z.A.; Klug, J.R.; Nobis, W.P.; Patel, S.; Grueter, B.A.; Kash, T.L.; Winder, D.G. Distinct forms of Gq-receptor-dependent plasticity of excitatory transmission in the BNST are differentially affected by stress. Proc. Natl. Acad. Sci. USA 2010, 107, 2271–2276. [Google Scholar] [CrossRef] [PubMed]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABA(B) receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Mackiewicz, J.; Sobolczyk, M.; Wawrzyniak, J.; Lisek, M.; Ferenc, B.; Guo, F.; Zylinska, L. The Role of G Protein-Coupled Receptors (GPCRs) and Calcium Signaling in Schizophrenia. Focus on GPCRs Activated by Neurotransmitters and Chemokines. Cells 2021, 10, 1228. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.C. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 2007, 6, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Sushma; Mondal, A.C. Role of GPCR signaling and calcium dysregulation in Alzheimer’s disease. Mol. Cell Neurosci. 2019, 101, 103414. [Google Scholar] [CrossRef]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochim. Biophys. Acta 2009, 1787, 335–344. [Google Scholar] [CrossRef]
- Zampese, E.; Surmeier, D.J. Calcium, bioenergetics, and parkinson’s disease. Cells 2020, 9, 2045. [Google Scholar] [CrossRef]
- Antinori, A.; Arendt, G.; Becker, J.T.; Brew, B.J.; Byrd, D.A.; Cherner, M.; Clifford, D.B.; Cinque, P.; Epstein, L.G.; Goodkin, K.; et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef]
- Berger, J.R.; Arendt, G. HIV dementia: The role of the basal ganglia and dopaminergic systems. J. Psychopharmacol. 2000, 14, 214–221. [Google Scholar] [CrossRef]
- Kannan, M.; Singh, S.; Chemparathy, D.T.; Oladapo, A.A.; Gawande, D.Y.; Dravid, S.M.; Buch, S.; Sil, S. HIV-1 Tat induced microglial EVs leads to neuronal synaptodendritic injury: Microglia-neuron cross-talk in NeuroHIV. Extracell. Vesicles Circ. Nucl. Acids 2022, 3, 133–149. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yang, W.; Zeng, Z.; Wei, Y.; Gao, J.; Zhang, B.; Li, L.; Liu, L.; Wan, Y.; Zeng, Q.; et al. NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology. Cell Mol. Immunol. 2020, 17, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Aylward, E.H.; Henderer, J.D.; McArthur, J.C.; Brettschneider, P.D.; Harris, G.J.; Barta, P.E.; Pearlson, G.D. Reduced basal ganglia volume in HIV-1-associated dementia: Results from quantitative neuroimaging. Neurology 1993, 43, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Scheller, C.; Arendt, G.; Nolting, T.; Antke, C.; Sopper, S.; Maschke, M.; Obermann, M.; Angerer, A.; Husstedt, I.W.; Meisner, F.; et al. Increased dopaminergic neurotransmission in therapy-naïve asymptomatic HIV patients is not associated with adaptive changes at the dopaminergic synapses. J. Neural Transm. 2010, 117, 699–705. [Google Scholar] [CrossRef]
- Horn, A.; Scheller, C.; du Plessis, S.; Arendt, G.; Nolting, T.; Joska, J.; Sopper, S.; Maschke, M.; Obermann, M.; Husstedt, I.W.; et al. Increases in CSF dopamine in HIV patients are due to the dopamine transporter 10/10-repeat allele which is more frequent in HIV-infected individuals. J. Neural Transm. 2013, 120, 1411–1419. [Google Scholar] [CrossRef]
- Berger, J.R.; Kumar, M.; Kumar, A.; Fernandez, J.B.; Levin, B. Cerebrospinal fluid dopamine in HIV-1 infection. AIDS 1994, 8, 67–71. [Google Scholar] [CrossRef]
- Sardar, A.M.; Czudek, C.; Reynolds, G.P. Dopamine deficits in the brain: The neurochemical basis of parkinsonian symptoms in AIDS. Neuroreport 1996, 7, 910–912. [Google Scholar] [CrossRef]
- Larsson, M.; Hagberg, L.; Forsman, A.; Norkrans, G. Cerebrospinal fluid catecholamine metabolites in HIV-infected patients. J. Neurosci. Res. 1991, 28, 406–409. [Google Scholar] [CrossRef]
- Kumar, A.M.; Fernandez, J.B.; Singer, E.J.; Commins, D.; Waldrop-Valverde, D.; Ownby, R.L.; Kumar, M. Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J. Neurovirol. 2009, 15, 257–274. [Google Scholar] [CrossRef]
- Kumar, A.M.; Ownby, R.L.; Waldrop-Valverde, D.; Fernandez, B.; Kumar, M. Human immunodeficiency virus infection in the CNS and decreased dopamine availability: Relationship with neuropsychological performance. J. Neurovirol. 2011, 17, 26–40. [Google Scholar] [CrossRef]
- Zhu, J.; Mactutus, C.F.; Wallace, D.R.; Booze, R.M. HIV-1 Tat protein-induced rapid and reversible decrease in [3H]dopamine uptake: Dissociation of [3H]dopamine uptake and [3H]2beta-carbomethoxy-3-beta-(4-fluorophenyl)tropane (WIN 35,428) binding in rat striatal synaptosomes. J. Pharmacol. Exp. Ther. 2009, 329, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ananthan, S.; Zhan, C.-G. The role of human dopamine transporter in NeuroAIDS. Pharmacol. Ther. 2018, 183, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.J.; Porter, K.D.; Quizon, P.M.; Davis, S.E.; Lin, S.; Yuan, Y.; Martinez-Muniz, G.A.; Sun, W.-L.; Zhan, C.-G.; Zhu, J. Mutations of tyrosine 467 in the human norepinephrine transporter attenuate HIV-1 Tat-induced inhibition of dopamine transport while retaining physiological function. PLoS ONE 2022, 17, e0275182. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.; O’Donovan, B.; Ma, Y.; Xiao, Z.; Lin, S.; Bardo, M.T.; Ortinski, P.I.; McLaughlin, J.P.; Zhu, J. [3H]Dopamine Uptake through the Dopamine and Norepinephrine Transporters is Decreased in the Prefrontal Cortex of Transgenic Mice Expressing HIV-1 Transactivator of Transcription Protein. J. Pharmacol. Exp. Ther. 2020, 374, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Adeniran, C.; Yuan, Y.; Davis, S.E.; Lin, C.; Xu, J.; Zhu, J.; Zhan, C.-G. Binding Mode of Human Norepinephrine Transporter Interacting with HIV-1 Tat. ACS Chem. Neurosci. 2021, 12, 1519–1527. [Google Scholar] [CrossRef]
- Kim, B.O.; Liu, Y.; Ruan, Y.; Xu, Z.C.; Schantz, L.; He, J.J. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am. J. Pathol. 2003, 162, 1693–1707. [Google Scholar] [CrossRef]
- Henderson, L.J.; Johnson, T.P.; Smith, B.R.; Reoma, L.B.; Santamaria, U.A.; Bachani, M.; Demarino, C.; Barclay, R.A.; Snow, J.; Sacktor, N.; et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019, 33 (Suppl. 2), S145–S157. [Google Scholar] [CrossRef]
- Gerena, Y.; Menéndez-Delmestre, R.; Delgado-Nieves, A.; Vélez, J.; Méndez-Álvarez, J.; Sierra-Pagan, J.E.; Skolasky, R.L.; Henderson, L.; Nath, A.; Wojna, V. Release of soluble insulin receptor from neurons by cerebrospinal fluid from patients with neurocognitive dysfunction and HIV infection. Front. Neurol. 2019, 10, 285. [Google Scholar] [CrossRef]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef]
- Davis, S.E.; Ferris, M.J.; Ananthan, S.; Augelli-Szafran, C.E.; Zhu, J. Novel Allosteric Modulator Southern Research Institute-32743 Reverses HIV-1 Transactivator of Transcription-Induced Increase in Dopamine Release in the Caudate Putamen of Inducible Transactivator of Transcription Transgenic Mice. J. Pharmacol. Exp. Ther. 2023, 384, 306–314. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Lokensgard, J.R.; Peterson, P.K.; Rock, R.B. Preferential sensitivity of human dopaminergic neurons to gp120-induced oxidative damage. J. Neurovirol. 2009, 15, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.A.; Rusyniak, D.E.; Hollingsworth, C.K. HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res. 1995, 705, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.R.; Dodson, S.; Nath, A.; Booze, R.M. Estrogen attenuates gp120- and tat1-72-induced oxidative stress and prevents loss of dopamine transporter function. Synapse 2006, 59, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-J.; Chang, L.; Volkow, N.D.; Telang, F.; Logan, J.; Ernst, T.; Fowler, J.S. Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain 2004, 127, 2452–2458. [Google Scholar] [CrossRef]
- Silverstein, P.S.; Shah, A.; Weemhoff, J.; Kumar, S.; Singh, D.P.; Kumar, A. HIV-1 gp120 and drugs of abuse: Interactions in the central nervous system. Curr. HIV Res. 2012, 10, 369–383. [Google Scholar] [CrossRef]
- Rappaport, J.; Joseph, J.; Croul, S.; Alexander, G.; Del Valle, L.; Amini, S.; Khalili, K. Molecular pathway involved in HIV-1-induced CNS pathology: Role of viral regulatory protein, Tat. J. Leukoc. Biol. 1999, 65, 458–465. [Google Scholar] [CrossRef]
- Fitting, S.; Booze, R.M.; Mactutus, C.F. HIV-1 proteins, Tat and gp120, target the developing dopamine system. Curr. HIV Res. 2015, 13, 21–42. [Google Scholar] [CrossRef]
- Bansal, A.K.; Mactutus, C.F.; Nath, A.; Maragos, W.; Hauser, K.F.; Booze, R.M. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 2000, 879, 42–49. [Google Scholar] [CrossRef]
- Agrawal, L.; Louboutin, J.-P.; Marusich, E.; Reyes, B.A.S.; Van Bockstaele, E.J.; Strayer, D.S. Dopaminergic neurotoxicity of HIV-1 gp120: Reactive oxygen species as signaling intermediates. Brain Res. 2010, 1306, 116–130. [Google Scholar] [CrossRef]
- Haughey, N.J.; Mattson, M.P. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune Defic. Syndr. 2002, 31 (Suppl. 2), S55–S61. [Google Scholar] [CrossRef]
- Haughey, N.J.; Holden, C.P.; Nath, A.; Geiger, J.D. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J. Neurochem. 1999, 73, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Nath, A.; Mattson, M.P. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 1998, 154, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Andhavarapu, S.; Katuri, A.; Bryant, J.; Patel, V.; Gupta, U.; Asemu, G.; Makar, T.K. Intersecting roles of ER stress, mitochondrial dysfunction, autophagy, and calcium homeostasis in HIV-associated neurocognitive disorder. J. NeuroVirology 2020, 26, 664–675. [Google Scholar] [CrossRef]
- Strijbos, P.J.; Zamani, M.R.; Rothwell, N.J.; Arbuthnott, G.; Harkiss, G. Neurotoxic mechanisms of transactivating protein Tat of Maedi-Visna virus. Neurosci. Lett. 1995, 197, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Nath, A.; Mattson, M.P.; Slevin, J.T.; Geiger, J.D. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem. 2001, 78, 457–467. [Google Scholar] [CrossRef]
- Norman, J.P.; Perry, S.W.; Reynolds, H.M.; Kiebala, M.; De Mesy Bentley, K.L.; Trejo, M.; Volsky, D.J.; Maggirwar, S.B.; Dewhurst, S.; Masliah, E.; et al. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS ONE 2008, 3, e3731. [Google Scholar] [CrossRef]
- Hu, X.-T. HIV-1 Tat-Mediated Calcium Dysregulation and Neuronal Dysfunction in Vulnerable Brain Regions. Curr. Drug Targets 2016, 17, 4–14. [Google Scholar] [CrossRef]
- Musante, V.; Summa, M.; Neri, E.; Puliti, A.; Godowicz, T.T.; Severi, P.; Battaglia, G.; Raiteri, M.; Pittaluga, A. The HIV-1 viral protein Tat increases glutamate and decreases GABA exocytosis from human and mouse neocortical nerve endings. Cereb. Cortex 2010, 20, 1974–1984. [Google Scholar] [CrossRef]
- Feligioni, M.; Raiteri, L.; Pattarini, R.; Grilli, M.; Bruzzone, S.; Cavazzani, P.; Raiteri, M.; Pittaluga, A. The human immunodeficiency virus-1 protein Tat and its discrete fragments evoke selective release of acetylcholine from human and rat cerebrocortical terminals through species-specific mechanisms. J. Neurosci. 2003, 23, 6810–6818. [Google Scholar] [CrossRef]
- Yuan, Y.; Huang, X.; Midde, N.M.; Quizon, P.M.; Sun, W.-L.; Zhu, J.; Zhan, C.-G. Molecular mechanism of HIV-1 Tat interacting with human dopamine transporter. ACS Chem. Neurosci. 2015, 6, 658–665. [Google Scholar] [CrossRef]
- Neri, E.; Musante, V.; Pittaluga, A. Effects of the HIV-1 viral protein TAT on central neurotransmission: Role of group I metabotropic glutamate receptors. In Neuroinflammation in Neuronal Death and Repair; Elsevier: Amsterdam, The Netherlands, 2007; Volume 82, pp. 339–356. [Google Scholar]
- Kesby, J.P.; Najera, J.A.; Romoli, B.; Fang, Y.; Basova, L.; Birmingham, A.; Marcondes, M.C.G.; Dulcis, D.; Semenova, S. HIV-1 TAT protein enhances sensitization to methamphetamine by affecting dopaminergic function. Brain Behav. Immun. 2017, 65, 210–221. [Google Scholar] [CrossRef]
- Brailoiu, G.C.; Deliu, E.; Barr, J.L.; Console-Bram, L.M.; Ciuciu, A.M.; Abood, M.E.; Unterwald, E.M.; Brailoiu, E. HIV Tat excites D1 receptor-like expressing neurons from rat nucleus accumbens. Drug Alcohol. Depend. 2017, 178, 7–14. [Google Scholar] [CrossRef]
- Nickoloff-Bybel, E.A.; Mackie, P.; Runner, K.; Matt, S.M.; Khoshbouei, H.; Gaskill, P.J. Dopamine increases HIV entry into macrophages by increasing calcium release via an alternative signaling pathway. Brain Behav. Immun. 2019, 82, 239–252. [Google Scholar] [CrossRef]
- Nolan, R.A.; Muir, R.; Runner, K.; Haddad, E.K.; Gaskill, P.J. Role of Macrophage Dopamine Receptors in Mediating Cytokine Production: Implications for Neuroinflammation in the Context of HIV-Associated Neurocognitive Disorders. J. Neuroimmune Pharmacol. 2019, 14, 134–156. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder-pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Irollo, E.; Luchetta, J.; Ho, C.; Nash, B.; Meucci, O. Mechanisms of neuronal dysfunction in HIV-associated neurocognitive disorders. Cell Mol. Life Sci. 2021, 78, 4283–4303. [Google Scholar] [CrossRef] [PubMed]
- Fitting, S.; Knapp, P.E.; Zou, S.; Marks, W.D.; Bowers, M.S.; Akbarali, H.I.; Hauser, K.F. Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na⁺ influx, mitochondrial instability, and Ca²⁺ overload. J. Neurosci. 2014, 34, 12850–12864. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Babcock, I.W.; Minamide, L.S.; Shaw, A.E.; Bamburg, J.R.; Kuhn, T.B. Direct interaction of HIV gp120 with neuronal CXCR4 and CCR5 receptors induces cofilin-actin rod pathology via a cellular prion protein- and NOX-dependent mechanism. PLoS ONE 2021, 16, e0248309. [Google Scholar] [CrossRef]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef]
- Chen, X.; Guo, X.; Ge, Q.; Zhao, Y.; Mu, H.; Zhang, J. ER stress activates the NLRP3 inflammasome: A novel mechanism of atherosclerosis. Oxid. Med. Cell Longev. 2019, 2019, 3462530. [Google Scholar] [CrossRef]
- Ni, H.; Ou, Z.; Wang, Y.; Liu, Y.; Sun, K.; Zhang, J.; Zhang, J.; Deng, W.; Zeng, W.; Xia, R.; et al. XBP1 modulates endoplasmic reticulum and mitochondria crosstalk via regulating NLRP3 in renal ischemia/reperfusion injury. Cell Death Discov. 2023, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Ureña-Peralta, J.R.; Morillo-Bargues, M.J.; Oliver-De La Cruz, J.; Guerri, C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front. Cell Neurosci. 2014, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, S.E.; Cirincione, A.B.; Jimenez-Torres, A.C.; Zhu, J. The Impact of Neurotransmitters on the Neurobiology of Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 15340. https://doi.org/10.3390/ijms242015340
Davis SE, Cirincione AB, Jimenez-Torres AC, Zhu J. The Impact of Neurotransmitters on the Neurobiology of Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(20):15340. https://doi.org/10.3390/ijms242015340
Chicago/Turabian StyleDavis, Sarah E., Abagail B. Cirincione, Ana Catya Jimenez-Torres, and Jun Zhu. 2023. "The Impact of Neurotransmitters on the Neurobiology of Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 20: 15340. https://doi.org/10.3390/ijms242015340