
Molecular Mechanisms of Noncoding RNA in the Occurrence of Castration-Resistant Prostate Cancer

Abstract
:1. Introduction
2. CRPC and Its Mechanisms
3. Noncoding RNAs and Metabolic Remodeling in CRPC
4. Noncoding RNAs and Epigenetic Dysregulation in CRPC
4.1. Noncoding RNAs and Epigenetics Modifications
4.2. The Interaction between ncRNAs and Epigenetics in CRPC
4.3. Noncoding RNAs Related Signaling Pathway in CRPC
5. Noncoding RNAs and AR-Related Signaling
5.1. AR Structure and Functions
5.2. AR-Related Signaling in CRPC
5.3. AR-Related ncRNAs in CRPC
6. ncRNAs and Drug Resistance
7. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohler, J.L.; Antonarakis, E.S.; Armstrong, A.J.; D’Amico, A.V.; Davis, B.J.; Dorff, T.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, W.C.; Hartman, H.E.; Dess, R.T.; Birer, S.R.; Soni, P.D.; Hearn, J.W.D.; Reichert, Z.R.; Kishan, A.U.; Mahal, B.A.; Zumsteg, Z.S. Addition of Androgen-Deprivation Therapy or Brachytherapy Boost to External Beam Radiotherapy for Localized Prostate Cancer: A Network Meta-Analysis of Randomized Trials. J. Clin. Oncol. 2020, 38, 3024–3031. [Google Scholar] [CrossRef]
- Connor, M.J.; Shah, T.T.; Horan, G.; Bevan, C.L.; Winkler, M.; Ahmed, H.U. Cytoreductive treatment strategies for de novo metastatic prostate cancer. Nat. Rev. Clin. Oncol. 2020, 17, 168–182. [Google Scholar] [CrossRef]
- Libertini, S.J.; Tepper, C.G.; Rodriguez, V.; Asmuth, D.M.; Kung, H.J.; Mudryj, M. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res. 2007, 67, 9001–9005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logothetis, C.J.; Gallick, G.E.; Maity, S.N.; Kim, J.; Aparicio, A.; Efstathiou, E.; Lin, S.-H. Molecular classification of prostate cancer progression: Foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013, 3, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.F.; Li, W.; Uo, T.; et al. ARv7 Represses Tumor-Suppressor Genes in Castration-Resistant Prostate Cancer. Cancer Cell 2019, 35, 401–413.e6. [Google Scholar] [CrossRef] [Green Version]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.Z.; Chen, Y.D.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berruti, A.; Pia, A.; Terzolo, M. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 365, 766. [Google Scholar] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; Wit, R.D.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Halabi, S.; Kelly, W.K.; Ma, H.; Zhou, H.; Solomon, N.C.; Fizazi, K.; Tangen, C.M.; Rosenthal, M.; Petrylak, D.P.; Hussain, M.; et al. Meta-Analysis Evaluating the Impact of Site of Metastasis on Overall Survival in Men with Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2016, 34, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Cha, H.R.; Lee, J.H.; Ponnazhagan, S. Revisiting Immunotherapy: A Focus on Prostate Cancer. Cancer Res. 2020, 80, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Subudhi, S.K.; Aparicio, A.; Ge, Z.; Guan, B.; Miura, Y.; Sharma, P. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell 2019, 179, 1177–1190.e13. [Google Scholar] [CrossRef]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Flechon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e3. [Google Scholar] [CrossRef]
- Wei, L.; Sun, J.; Zhang, N.; Zheng, Y.; Wang, X.; Lv, L.; Liu, J.D.; Xu, Y.Y.; Shen, Y.; Yang, M. Noncoding RNAs in gastric cancer: Implications for drug resistance. Mol. Cancer 2020, 19, 62. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xu, J.; Wang, Y.; Cao, X. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science 2017, 358, 1051–1055. [Google Scholar] [CrossRef]
- Keller, C.; Buhler, M. Chromatin-associated ncRNA activities. Chromosome Res. 2013, 21, 627–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Weng, H.; Chen, J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Gebetsberger, J.; Polacek, N. Ribosome-associated ncRNAs: An emerging class of translation regulators. RNA. Biol. 2014, 11, 1335–1339. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Yuan, T.; Liang, M.; Du, M.; Xia, S.; Dittmar, R.; Wang, D.; See, W.; Costello, B.A.; Quevedo, F.; et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur. Urol. 2015, 67, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug. Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.; Girnita, L.; Varani, G.; Calin, G.A. Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 2019, 29, 1377–1388. [Google Scholar] [CrossRef] [Green Version]
- Zamore, P.D.; Haley, B. Ribo-gnome: The big world of small RNAs. Science 2005, 309, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Zhao, Y.; Srivastava, D. A developmental view of microRNA function. Trends. Biochem. Sci. 2007, 32, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Deng, X.; Ma, W.; Berletch, J.B.; Rabaia, N.; Wei, G.; Moore, J.M.; Filippova, G.N.; Xu, J.; Liu, Y.J.; et al. The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 2015, 16, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornford, P.; Bellmunt, J.; Bolla, M.; Briers, E.; De Santis, M.; Gross, T.; Henry, A.M.; Joniau, S.; Lam, T.B.; Mason, M.D.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 630–642. [Google Scholar] [CrossRef]
- Hu, C.Y.; Wu, K.Y.; Lin, T.Y.; Chen, C.C. The Crosstalk of Long Non-Coding RNA and MicroRNA in Castration-Resistant and Neuroendocrine Prostate Cancer: Their Interaction and Clinical Importance. Int. J. Mol. Sci. 2021, 23, 392. [Google Scholar] [CrossRef]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Knudsen, K.E.; Penning, T.M. Partners in crime: Deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol. Metab. 2010, 21, 315–324. [Google Scholar] [CrossRef]
- Conteduca, V.; Mosca, A.; Brighi, N.; de Giorgi, U.; Rescigno, P. New Prognostic Biomarkers in Metastatic Castration-Resistant Prostate Cancer. Cells 2021, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Titus, M.A.; Schell, M.J.; Lih, F.B.; Tomer, K.B.; Mohler, J.L. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin. Cancer Res. 2005, 11, 4653–4657. [Google Scholar] [CrossRef] [Green Version]
- Mansinho, A.; Macedo, D.; Fernandes, I.; Costa, L. Castration-Resistant Prostate Cancer: Mechanisms, Targets and Treatment. Adv. Exp. Med. Biol. 2018, 1096, 117–133. [Google Scholar] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.J.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreira, S.; Romanel, A.; Goodall, J.; Grist, E.; Ferraldeschi, R.; Miranda, S.; Prandi, D.; Lorente, D.; Frenel, J.S.; Pezaro, C.; et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014, 6, 254ra125. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Naznus, D.M.; et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Sung, Y.Y.; Cheung, E. Androgen receptor co-regulatory networks in castration-resistant prostate cancer. Endocr. Relat. Cancer 2014, 21, R1–R11. [Google Scholar] [CrossRef]
- Taplin, M.E.; Bubley, G.J.; Ko, Y.J.; Small, E.J.; Upton, M.; Rajeshkumar, B.; Balk, S.P. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999, 59, 2511–2515. [Google Scholar]
- Steinkamp, M.P.; O’Mahony, O.A.; Brogley, M.; Rehman, H.; Lapensee, E.W.; Dhanasekaran, S.; Hofer, M.D.; Kuefer, R.; Chinnaiyan, A.; Rubin, M.A.; et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009, 69, 4434–4442. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprenger, C.C.; Plymate, S.R. The link between androgen receptor splice variants and castration-resistant prostate cancer. Horm. Cancer 2014, 5, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014, 5, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalava, S.E.; Urbanucci, A.; Latonen, L.; Waltering, K.K.; Sahu, B.; Janne, O.A.; Seppala, J.; Lahdesmaki, H.; Tammela, T.L.J.; Visakorpi, T.; et al. Androgen-regulated miR-32 targets BTG2 and is overexpressed in castration-resistant prostate cancer. Oncogene 2012, 31, 4460–4471. [Google Scholar] [CrossRef] [Green Version]
- Stuopelyte, K.; Daniunaite, K.; Bakavicius, A.; Lazutka, J.R.; Jankevicius, F.; Jarmalaite, S. The utility of urine-circulating miRNAs for detection of prostate cancer. Br. J. Cancer 2016, 115, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Layer, R.; Mueller, A.C.; Cichewicz, M.A.; Negishi, M.; Paschal, B.M.; Dutta, A. Regulation of several androgen-induced genes through the repression of the miR-99a/let-7c/miR-125b-2 miRNA cluster in prostate cancer cells. Oncogene 2014, 33, 1448–1457. [Google Scholar] [CrossRef] [Green Version]
- Sabahi, A.; Salahandish, R.; Ghaffarinejad, A.; Omidinia, E. Electrochemical nano-genosensor for highly sensitive detection of miR-21 biomarker based on SWCNT-grafted dendritic Au nanostructure for early detection of prostate cancer. Talanta 2020, 209, 120595. [Google Scholar] [CrossRef]
- Kurul, N.O.; Ates, F.; Yilmaz, I.; Narli, G.; Yesildal, C.; Senkul, T. The association of let-7c, miR-21, miR-145, miR-182, and miR-221 with clinicopathologic parameters of prostate cancer in patients diagnosed with low-risk disease. Prostate 2019, 79, 1125–1132. [Google Scholar] [CrossRef]
- Folini, M.; Gandellini, P.; Longoni, N.; Profumo, V.; Callari, M.; Pennati, M.; Colecchia, M.; Supino, R.; Veneroni, S.; Salvioni, R.; et al. miR-21: An oncomir on strike in prostate cancer. Mol. Cancer 2010, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Zhang, L.; Wang, S.; Long, L.; Zhou, H.; Qian, S.; Ma, M.; Bai, F.; Meng, Q.H.; Lyu, J. Upregulation of MicroRNA-21 promotes tumorigenesis of prostate cancer cells by targeting KLF5. Cancer Biol. Ther. 2019, 20, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wang, X.; He, H.H.; Sweeney, C.J.; Liu, S.X.; Brown, M.; Balk, S.; Lee, G.-S.; Kantoff, P.W. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene 2014, 33, 2790–2800. [Google Scholar] [CrossRef] [Green Version]
- Kneitz, B.; Krebs, M.; Kalogirou, C.; Schubert, M.; Joniau, S.; van Poppel, H.; Lerut, E.; Kneitz, S.; Scholz, C.J.; Strobel, P.; et al. Survival in patients with high-risk prostate cancer is predicted by miR-221, which regulates proliferation, apoptosis, and invasion of prostate cancer cells by inhibiting IRF2 and SOCS3. Cancer Res. 2014, 74, 2591–2603. [Google Scholar] [CrossRef] [Green Version]
- Hagman, Z.; Haflidadottir, B.S.; Ceder, J.A.; Larne, O.; Bjartell, A.; Lilja, H.; Edsjo, A.; Ceder, Y. miR-205 negatively regulates the androgen receptor and is associated with adverse outcome of prostate cancer patients. Br. J. Cancer 2013, 108, 1668–1676. [Google Scholar] [CrossRef] [Green Version]
- Tucci, P.; Agostini, M.; Grespi, F.; Markert, E.K.; Terrinoni, A.; Vousden, K.H.; Muller, P.A.J.; Dotsch, V.; Kehrloesser, S.; Sayan, B.S.; et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 15312–15317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagirath, D.; Yang, T.L.; Bucay, N.; Sekhon, K.; Majid, S.; Shahryari, V.; Dahiya, R.; Tanaka, Y.; Saini, S. microRNA-1246 Is an Exosomal Biomarker for Aggressive Prostate Cancer. Cancer Res. 2018, 78, 1833–1844. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Liu, Z.; Ning, H.; Zhang, K.; Pan, D.; Ding, K.; Huang, W.; Kang, X.L.; Wang, Y.; Chen, X. MicroRNA-21 in peripheral blood mononuclear cells as a novel biomarker in the diagnosis and prognosis of prostate cancer. Cancer Biomark. 2016, 17, 223–230. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Reon, B.J.; Anaya, J.; Dutta, A. The lncRNA DRAIC/PCAT29 Locus Constitutes a Tumor-Suppressive Nexus. Mol. Cancer Res. 2015, 13, 828–838. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Zhao, J.C.; Kim, J.; Fong, K.W.; Yang, Y.A.; Chakravarti, D.; Mo, Y.-Y.; Yu, J. LncRNA HOTAIR Enhances the Androgen-Receptor-Mediated Transcriptional Program and Drives Castration-Resistant Prostate Cancer. Cell Rep. 2015, 13, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; He, C.; Shi, S.; Wang, M.; Ma, J.; Yang, P.; Dong, Y.; Mou, X.; Han, S. Linc00963 Promote Cell Proliferation and Tumor Growth in Castration-Resistant Prostate Cancer by Modulating miR-655/TRIM24 Axis. Front. Oncol. 2021, 11, 636965. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Han, S.; Jin, G.; Zhou, X.; Li, M.; Ying, X.; Wang, L.; Wu, H.; Zhu, Q. Linc00963: A novel, long non-coding RNA involved in the transition of prostate cancer from androgen-dependence to androgen-independence. Int. J. Oncol. 2014, 44, 2041–2049. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Liu, Y.; Xu, W.; Sun, Y.; Lu, J.; Wang, F.; Wei, M.; Shen, J.; Hou, J.; Gao, X. Long noncoding RNA MALAT-1 is a new potential therapeutic target for castration resistant prostate cancer. J. Urol. 2013, 190, 2278–2287. [Google Scholar] [CrossRef]
- Malik, R.; Patel, L.; Prensner, J.R.; Shi, Y.; Iyer, M.K.; Subramaniyan, S.; Carley, A.; Niknafs, Y.S.; Sahu, A.; Han, S. The lncRNA PCAT29 inhibits oncogenic phenotypes in prostate cancer. Mol. Cancer Res. 2014, 12, 1081–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovics, G.; Zhang, W.; Makarem, M.; Street, J.P.; Connelly, R.; Sun, L.; Sesterhenn, I.A.; Srikantan, V.; Moul, J.W.; Srivastava, S. Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene 2004, 23, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, M.; Shi, X.; Li, Y.; Xiao, Y.; Butler, W.; Huang, Y.; Du, L.; Wu, T.; Bian, X.; Shi, G. LINC00675 activates androgen receptor axis signaling pathway to promote castration-resistant prostate cancer progression. Cell Death. Dis. 2020, 11, 638. [Google Scholar] [CrossRef]
- Parolia, A.; Venalainen, E.; Xue, H.; Mather, R.; Lin, D.; Wu, R.; Pucci, P.; Rogalski, J.; Evans, J.R.; Feng, F.; et al. The long noncoding RNA HORAS5 mediates castration-resistant prostate cancer survival by activating the androgen receptor transcriptional program. Mol. Oncol. 2019, 13, 1121–1136. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Zhang, X.; Niknafs, Y.S.; Xiao, L.; Mehra, R.; Cieslik, M.; Ross, A.; Schaeffer, E.; Malik, B.; Guo, S.; et al. Identification and Validation of PCAT14 as Prognostic Biomarker in Prostate Cancer. Neoplasia 2016, 18, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Calve, B.; Rynkowski, M.; Le Mercier, M.; Bruyere, C.; Lonez, C.; Gras, T.; Haibe-Kains, B.; Bontempi, G.; Decaestecker, C.; Ruysschaert, J.M.; et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 2010, 12, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Saylor, P.J.; Karoly, E.D.; Smith, M.R. Prospective study of changes in the metabolomic profiles of men during their first three months of androgen deprivation therapy for prostate cancer. Clin. Cancer Res. 2012, 18, 3677–3685. [Google Scholar] [CrossRef] [Green Version]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Shankaraiah, R.C.; Veronese, A.; Sabbioni, S.; Negrini, M. Non-coding RNAs in the reprogramming of glucose metabolism in cancer. Cancer Lett. 2018, 419, 167–174. [Google Scholar] [CrossRef]
- Redis, R.S.; Vela, L.E.; Lu, W.; Ferreira de Oliveira, J.; Ivan, C.; Rodriguez-Aguayo, C.; Adamoski, D.; Pasculli, B.; Taguchi, A.; Chen, Y.; et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol. Cell 2016, 61, 640. [Google Scholar] [CrossRef]
- Hung, C.L.; Wang, L.Y.; Yu, Y.L.; Chen, H.W.; Srivastava, S.; Petrovics, G.; Kung, H.-J. A long noncoding RNA connects c-Myc to tumor metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 18697–18702. [Google Scholar] [CrossRef] [Green Version]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: Connecting the dots. Mol. Cancer 2006, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebron, J.S.; Shankar, E.; Singh, J.; Sikand, K.; Weyman, C.M.; Gupta, S.; Lindner, D.J.; Liu, X.; Campbell, M.J.; Shukla, G.C. MiR-644a Disrupts Oncogenic Transformation and Warburg Effect by Direct Modulation of Multiple Genes of Tumor-Promoting Pathways. Cancer Res. 2019, 79, 1844–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Archer, M.C. Sp1 coordinately regulates de novo lipogenesis and proliferation in cancer cells. Int. J. Cancer 2010, 126, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Eisermann, K.; Broderick, C.J.; Bazarov, A.; Moazam, M.M.; Fraizer, G.C. Androgen up-regulates vascular endothelial growth factor expression in prostate cancer cells via an Sp1 binding site. Mol. Cancer 2013, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Ling, Z.; Liu, D.; Zhang, G.; Liang, Q.; Xiang, P.; Xu, Y.; Han, C.; Tao, T. miR-361–5p modulates metabolism and autophagy via the Sp1-mediated regulation of PKM2 in prostate cancer. Oncol. Rep. 2017, 38, 1621–1628. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Shen, Z.; Lin, Y.; Xiao, J.; Xia, K.; Xu, C.; Chen, B.; Shi, R.; Zhu, A.; Sun, X.; et al. LncRNA-MALAT1 Regulates Cancer Glucose Metabolism in Prostate Cancer via MYBL2/mTOR Axis. Oxid. Med. Cell Longev. 2022, 2022, 8693259. [Google Scholar] [CrossRef]
- Kanagasabai, T.; Li, G.; Shen, T.H.; Gladoun, N.; Castillo-Martin, M.; Celada, S.I.; Xie, Y.; Brown, L.K.; Mark, Z.A.; Ochieng, J.; et al. MicroRNA-21 deficiency suppresses prostate cancer progression through downregulation of the IRS1-SREBP-1 signaling pathway. Cancer Lett. 2022, 525, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Kang, H.; Xiao, Y.; Du, Y.; Xiao, Y.; Song, G.; Zhan, Y.; Guo, Y.; Yang, F.; He, F.; et al. LncRNA GAL promotes colorectal cancer liver metastasis through stabilizing GLUT1. Oncogene 2022, 41, 1882–1894. [Google Scholar] [CrossRef] [PubMed]
- Cannistraci, A.; Hascoet, P.; Ali, A.; Mundra, P.; Clarke, N.W.; Pavet, V.; Marais, R. MiR-378a inhibits glucose metabolism by suppressing GLUT1 in prostate cancer. Oncogene 2022, 41, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.T.; Josson, S.; Mukhopadhyay, N.K.; Kim, J.; Freeman, M.R.; Huang, W.-C. MicroRNA-185 and 342 inhibit tumorigenicity and induce apoptosis through blockade of the SREBP metabolic pathway in prostate cancer cells. PLoS ONE 2013, 8, e70987. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, Y.; Shu, Y.; He, J.; Gao, W. Interaction between N(6)-methyladenosine (m(6)A) modification and noncoding RNAs in cancer. Mol. Cancer 2020, 19, 94. [Google Scholar] [CrossRef]
- Frye, M.; Jaffrey, S.R.; Pan, T.; Rechavi, G.; Suzuki, T. RNA modifications: What have we learned and where are we headed? Nat. Rev. Genet. 2016, 17, 365–372. [Google Scholar] [CrossRef]
- Lee, J.T. Lessons from X-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes. Dev. 2009, 23, 1831–1842. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Hou, P.; Fan, D.; Dong, M.; Ma, M.; Li, H.; Yao, R.; Li, Y.; Wang, G.; Geng, P.; et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. CellDeath. Differ. 2017, 24, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, L.; Zhong, T.; Mueller, M.; Men, Y.; Zhang, N.; Xie, J.; Giang, K.; Chung, H.; Sun, X.; et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat. Commun. 2015, 6, 10221. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Xing, Y.; Wen, X.; Jia, R.; Ni, H.; He, J.; Ding, X.; Pan, H.; Qian, G.; Ge, S.; et al. Long non-coding RNA ROR decoys gene-specific histone methylation to promote tumorigenesis. Genome Biol. 2015, 16, 139. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Chen, C.; Ji, X.; Liu, J.; Zhou, Q.; Wang, G.; Yuan, W.; Kan, Q.; Sun, Z. The interplay between m6A RNA methylation and noncoding RNA in cancer. J. Hematol. Oncol. 2019, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Jiao, Z.; Rong, W.; Qu, S.; Liao, Z.; Sun, X.; Wei, Y.; Zhao, Q.; Wang, J.; Liu, Y.; et al. 3’-Terminal 2’-O-methylation of lung cancer miR-21–5p enhances its stability and association with Argonaute 2. Nucleic Acids Res. 2020, 48, 7027–7040. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; Liguori, L.; De Antonellis, P.; Cusanelli, E.; Marinaro, F.; Pistollato, F.; Garzia, L.; Vita, G.D.; Petrosino, G.; Accordi, B.; et al. The micro-RNA 199b-5p regulatory circuit involves Hes1, CD15, and epigenetic modifications in medulloblastoma. Neuro. Oncol. 2012, 14, 596–612. [Google Scholar] [CrossRef] [Green Version]
- Ghildiyal, R.; Sawant, M.; Renganathan, A.; Mahajan, K.; Kim, E.H.; Luo, J.; Dang, H.X.; Maher, C.A.; Feng, F.Y.; Mahajan, N.P.; et al. Loss of Long Noncoding RNA NXTAR in Prostate Cancer Augments Androgen Receptor Expression and Enzalutamide Resistance. Cancer Res. 2022, 82, 155–168. [Google Scholar] [CrossRef]
- Sachamitr, P.; Ho, J.C.; Ciamponi, F.E.; Ba-Alawi, W.; Coutinho, F.J.; Guilhamon, P.; Kushida, M.M.; Cavalli, F.M.; Lee, L.; Rastegar, N.; et al. PRMT5 inhibition disrupts splicing and stemness in glioblastoma. Nat. Commun. 2021, 12, 979. [Google Scholar] [CrossRef]
- Yao, B.; Zhu, S.; Wei, X.; Chen, M.K.; Feng, Y.; Li, Z.; Xu, X.; Zhang, Y.; Wang, Y.; Zhou, J.; et al. The circSPON2/miR-331–3p axis regulates PRMT5, an epigenetic regulator of CAMK2N1 transcription and prostate cancer progression. Mol. Cancer 2022, 21, 119. [Google Scholar] [CrossRef]
- Barros-Silva, D.; Costa-Pinheiro, P.; Duarte, H.; Sousa, E.J.; Evangelista, A.F.; Graca, I.; Carneiro, I.; Martins, A.T.; Oliveira, J.; Carvalho, A.L.; et al. MicroRNA-27a-5p regulation by promoter methylation and MYC signaling in prostate carcinogenesis. Cell Death. Dis. 2018, 9, 167. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gao, H.; Ren, L.; Gu, J.; Zhang, Y.; Zhang, Y. Demethylation of the miR-146a promoter by 5-Aza-2’-deoxycytidine correlates with delayed progression of castration-resistant prostate cancer. BMC Cancer 2014, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Wilting, S.M.; van Boerdonk, R.A.; Henken, F.E.; Meijer, C.J.; Diosdado, B.; Meijer, G.A.; Sage, C.L.; Agami, R.; Snijders, P.J.F.; Steenbergen, R.D.M. Methylation-mediated silencing and tumour suppressive function of hsa-miR-124 in cervical cancer. Mol. Cancer 2010, 9, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Chen, L.; Zhang, J.; Chen, H.; Fan, J.; Wang, K.; Luo, J.; Chen, Z.; Meng, Z.; Liu, L.; et al. Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene 2014, 33, 514–524. [Google Scholar] [CrossRef]
- Chu, M.; Chang, Y.; Guo, Y.; Wang, N.; Cui, J.; Gao, W.Q. Regulation and methylation of tumor suppressor miR-124 by androgen receptor in prostate cancer cells. PLoS ONE 2015, 10, e0116197. [Google Scholar] [CrossRef] [PubMed]
- Raynal, N.J.; Si, J.; Taby, R.F.; Gharibyan, V.; Ahmed, S.; Jelinek, J. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 2012, 72, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Pak, S.; Park, S.; Kim, Y.; Park, J.H.; Park, C.H.; Lee, K.J.; Kim, C.; Ahn, H. The small molecule WNT/beta-catenin inhibitor CWP232291 blocks the growth of castration-resistant prostate cancer by activating the endoplasmic reticulum stress pathway. J. Exp. Clin. Cancer Res. 2019, 38, 342. [Google Scholar] [CrossRef]
- Lee, G.T.; Kwon, S.J.; Kim, J.; Kwon, Y.S.; Lee, N.; Hong, J.H.; Jamieson, C.; Kim, W.; Kim, I.Y. WNT5A induces castration-resistant prostate cancer via CCL2 and tumour-infiltrating macrophages. Br. J. Cancer 2018, 118, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Banuelos, C.A.; Imamura, Y.; Leung, J.K.; Caley, D.P.; Wang, J.; Mawji, N.R.; Sadar, M.D. Cotargeting Androgen Receptor Splice Variants and mTOR Signaling Pathway for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 2744–2754. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Yang, L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends. Cell Biol. 2018, 28, 287–301. [Google Scholar] [CrossRef]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Shang, Z.; Yu, J.; Sun, L.; Tian, J.; Zhu, S.; Zhang, B.; Dong, Q.; Jiang, N.; Flores-Morales, A.; Chang, C.; et al. LncRNA PCAT1 activates AKT and NF-kappaB signaling in castration-resistant prostate cancer by regulating the PHLPP/FKBP51/IKKalpha complex. Nucleic Acids. Res. 2019, 47, 4211–4225. [Google Scholar] [CrossRef] [PubMed]
- Gasparian, A.V.; Yao, Y.J.; Kowalczyk, D.; Lyakh, L.A.; Karseladze, A.; Slaga, T.J.; Budunova, I.V. The role of IKK in constitutive activation of NF-kappaB transcription factor in prostate carcinoma cells. J. Cell Sci. 2002, 115 Pt 1, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, L.; Wu, X.; Li, R.; Wen, J.; Sha, J.; Wen, X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front. Biosci. 2016, 21, 1084–1091. [Google Scholar] [PubMed] [Green Version]
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.D.; Deng, S.; Hoover, E.; Chen, C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 2020, 37, 584–598.e11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Ando, T.; Izumi, H.; Feng, X.; Arang, N.; Gilardi, M.; Wang, Z.; Ando, K.; Gutkind, J.S. Muscarinic receptors promote castration-resistant growth of prostate cancer through a FAK-YAP signaling axis. Oncogene 2020, 39, 4014–4027. [Google Scholar] [CrossRef]
- Guo, Y.; Cui, J.; Ji, Z.; Cheng, C.; Zhang, K.; Zhang, C.; Chu, M.; Zhao, Q.; Yu, Z.; Zhang, Y.; et al. miR-302/367/LATS2/YAP pathway is essential for prostate tumor-propagating cells and promotes the development of castration resistance. Oncogene 2017, 36, 6336–6347. [Google Scholar] [CrossRef]
- Mu, H.; Xiang, L.; Li, S.; Rao, D.; Wang, S.; Yu, K. MiR-10a functions as a tumor suppressor in prostate cancer via targeting KDM4A. J. Cell Biochem. 2019, 120, 4987–4997. [Google Scholar] [CrossRef]
- Kono, M.; Fujii, T.; Lim, B.; Karuturi, M.S.; Tripathy, D.; Ueno, N.T. Androgen Receptor Function and Androgen Receptor-Targeted Therapies in Breast Cancer: A Review. JAMA Oncol. 2017, 3, 1266–1273. [Google Scholar] [CrossRef]
- McCrea, E.; Sissung, T.M.; Price, D.K.; Chau, C.H.; Figg, W.D. Androgen receptor variation affects prostate cancer progression and drug resistance. Pharmacol. Res. 2016, 114, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; He, S.; Wang, D.; Patel, H.K.; Miller, C.P.; Brown, J.L.; Hattersley, G.; Saeh, J.C. Selective Androgen Receptor Modulator RAD140 Inhibits the Growth of Androgen/Estrogen Receptor-Positive Breast Cancer Models with a Distinct Mechanism of Action. Clin. Cancer Res. 2017, 23, 7608–7620. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chanbon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luisi, B.F.; Xu, W.X.; Otwinowski, Z.; Freedman, L.P.; Yamamoto, K.R.; Sigler, P.B. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 1991, 352, 497–505. [Google Scholar] [CrossRef]
- Gelmann, E.P. Molecular biology of the androgen receptor. J. Clin. Oncol. 2002, 20, 3001–3015. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Armstrong, A.J.; Dehm, S.M.; Luo, J. Androgen receptor variant-driven prostate cancer: Clinical implications and therapeutic targeting. Prostate. Cancer Prostatic. Dis. 2016, 19, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Panizo, A.; Perez, P.; Rojas, A.M.; Fuentes-Prior, P.; Estebanez-Perpina, E. Non-canonical dimerization of the androgen receptor and other nuclear receptors: Implications for human disease. Endocr. Relat. Cancer 2019, 26, R479–R497. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, R.C.; Hickey, T.E.; Tilley, W.D.; Selth, L.A. Interplay between the androgen receptor signaling axis and microRNAs in prostate cancer. Endocr. Relat. Cancer 2019, 26, R237–R257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.G.; McKinnon, R.A.; Hulin, J.A.; Mackenzie, P.I.; Meech, R. Novel Nine-Exon AR Transcripts (Exon 1/Exon 1b/Exons 2–8) in Normal and Cancerous Breast and Prostate Cells. Int. J. Mol. Sci. 2016, 18, 40. [Google Scholar] [CrossRef] [Green Version]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J. 2005, 272, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Hillebrand, A.C.; Pizzolato, L.S.; Neto, B.S.; Branchini, G.; Brum, I.S. Androgen receptor isoforms expression in benign prostatic hyperplasia and primary prostate cancer. PLoS ONE 2018, 13, e0200613. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wu, D.; Thomas-Ahner, J.M.; Lu, C.; Zhao, P.; Zhang, Q.; Geraghty, C.; Yan, P.S.; Hankey, W.; Sunkel, B.; et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. USA 2018, 115, 6810–6815. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Paschalis, A.; Sharp, A.; Welti, J.C.; Neeb, A.; Raj, G.V.; Luo, J.; Plymate, S.R.; Bono, J.S.D. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Kohli, M.; Ho, Y.; Hillman, D.W.; Van Etten, J.L.; Henzler, C.; Yang, R.; Sperger, J.M.; Li, Y.; Tseng, E.; Hon, T.; et al. Androgen Receptor Variant AR-V9 Is Coexpressed with AR-V7 in Prostate Cancer Metastases and Predicts Abiraterone Resistance. Clin. Cancer Res. 2017, 23, 4704–4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Zhang, G.; Wang, X.; Qi, Y.; Bai, S.; Li, D.; Ma, T.; Sartor, O.; Flemington, E.K.; Zhang, H.; et al. Interplay between Cytoplasmic and Nuclear Androgen Receptor Splice Variants Mediates Castration Resistance. Mol. Cancer Res. 2017, 15, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhan, Y.; Liu, X.; Qi, Y.; Zhang, G.; Sartor, O.; Dong, Y. Splicing variants of androgen receptor in prostate cancer. Am. J. Clin. Exp. Urol. 2013, 1, 18–24. [Google Scholar]
- Cioni, B.; Zaalberg, A.; van Beijnum, J.R.; Melis, M.H.M.; van Burgsteden, J.; Muraro, M.J.; Hooijberg, E.; Peters, D.; Hofland, I.; Lubeck, Y.; et al. Androgen receptor signalling in macrophages promotes TREM-1-mediated prostate cancer cell line migration and invasion. Nat. Commun. 2020, 11, 4498. [Google Scholar] [CrossRef]
- Lee, Y.G.; Nam, Y.; Shin, K.J.; Yoon, S.; Park, W.S.; Joung, J.Y.; Seo, J.K.; Jang, J.; Lee, S.; Nam, D.; et al. Androgen-induced expression of DRP1 regulates mitochondrial metabolic reprogramming in prostate cancer. Cancer Lett. 2020, 471, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Kishan, A.U.; Wang, X.; Seiferheld, W.; Collette, L.; Sandler, K.A.; Sandler, H.M.; Bolla, M.; Maingon, P.; Reijke, T.D.; Hanks, G.E.; et al. Association of Gleason Grade With Androgen Deprivation Therapy Duration and Survival Outcomes: A Systematic Review and Patient-Level Meta-analysis. JAMA. Oncol. 2019, 5, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, M.T.; Yu, E.Y. Persistent androgen receptor addiction in castration-resistant prostate cancer. J. Hematol. Oncol. 2015, 8, 128. [Google Scholar] [CrossRef] [Green Version]
- Vasaitis, T.; Belosay, A.; Schayowitz, A.; Khandelwal, A.; Chopra, P.; Gediya, L.K.; Guo, Z.; Fang, H.; Njar, V.C.O.; Brodie, A.M.H. Androgen receptor inactivation contributes to antitumor efficacy of 17alpha-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Mol. Cancer Ther. 2008, 7, 2348–2357. [Google Scholar] [CrossRef] [Green Version]
- Vasaitis, T.S.; Bruno, R.D.; Njar, V.C. CYP17 inhibitors for prostate cancer therapy. J. Steroid. Biochem. Mol. Biol. 2011, 125, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, M.G.; Barrie, S.E.; Chan, F.; Houghton, J.; Jarman, M.; McCague, R.; Pottor, G.A. Esters of 3-pyridylacetic acid that combine potent inhibition of 17 alpha-hydroxylase/C17,20-lyase (cytochrome P45017 alpha) with resistance to esterase hydrolysis. J. Med. Chem. 1995, 38, 4191–4197. [Google Scholar] [CrossRef]
- Attard, G.; Reid, A.H.; Yap, T.A.; Raynaud, F.; Dowsett, M.; Settatree, S.; Barrett, M.; Parker, C.; Martins, V.; Folkerd, E.; et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef]
- Mostaghel, E.A. Abiraterone in the treatment of metastatic castration-resistant prostate cancer. Cancer Manag. Res. 2014, 6, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, W.H.; Kabbara, W.K.; Al Basiouni Al Masri, H.S. Enzalutamide for patients with metastatic castration-resistant prostate cancer. Onco. Targets. Ther. 2015, 8, 871–876. [Google Scholar] [CrossRef] [Green Version]
- Rittmaster, R.S. Finasteride. N. Engl. J. Med. 1994, 330, 120–125. [Google Scholar] [PubMed]
- Canby-Hagino, E.D.; Brand, T.C.; Hernandez, J.; Thompson, I.M. Chemoprevention of prostate cancer with finasteride. Expert. Opin. Pharmacother. 2006, 7, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Gormley, G.J.; Stoner, E.; Bruskewitz, R.C.; Imperato-McGinley, J.; Walsh, P.C.; McConnell, J.D.; Andriole, G.L.; Geller, J.; Bracken, B.R.; Tenover, J.S.; et al. The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N. Engl. J. Med. 1992, 327, 1185–1191. [Google Scholar] [CrossRef]
- Hulin-Curtis, S.L.; Petit, D.; Figg, W.D.; Hsing, A.W.; Reichardt, J.K. Finasteride metabolism and pharmacogenetics: New approaches to personalized prevention of prostate cancer. Future. Oncol. 2010, 6, 1897–1913. [Google Scholar] [CrossRef] [PubMed]
- Slater, S.; Dumas, C.; Bubley, G. Dutasteride for the treatment of prostate-related conditions. Expert. Opin. Drug. Saf. 2012, 11, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Dalrymple, S.L.; Coleman, I.; Zheng, S.L.; Xu, J.; Hooper, J.E.; Antonarakis, E.S.; Marzo, A.M.D.; Meeker, A.K.; Nelson, P.S.; et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene 2020, 39, 6935–6949. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danila, D.C.; Healy, P.; Anand, M.; Rothwell, C.J.; et al. Prospective Multicenter Validation of Androgen Receptor Splice Variant 7 and Hormone Therapy Resistance in High-Risk Castration-Resistant Prostate Cancer: The PROPHECY Study. J. Clin. Oncol. 2019, 37, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Armstrong, C.M.; Lou, W.; Lombard, A.P.; Cucchiara, V.; Gu, X.; Yang, J.C.; Nadiminty, N.; Pan, C.; Evans, C.P.; et al. Niclosamide and Bicalutamide Combination Treatment Overcomes Enzalutamide- and Bicalutamide-Resistant Prostate Cancer. Mol. Cancer Ther. 2017, 16, 1521–1530. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.; Horie-Inoue, K.; Katayama, S.; Suzuki, T.; Tsutsumi, S.; Ikeda, K.; Urano, T.; Fujimura, T.; Takagi, K.; Takahashi, S.; et al. Androgen-responsive long noncoding RNA CTBP1-AS promotes prostate cancer. EMBO J. 2013, 32, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, S.; Xie, X.; Xiao, Y.F.; Tang, B.; Hu, C.J.; Wang, S.M.; Wu, Y.; Dong, H.; Li, B.; Yang, S. Long noncoding RNA LINC00675 enhances phosphorylation of vimentin on Ser83 to suppress gastric cancer progression. Cancer Lett. 2018, 412, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; An, N.; Qin, J.; Yang, J.; Sun, H.; Yang, W. Long non-coding RNA Linc00675 suppresses cell proliferation and metastasis in colorectal cancer via acting on miR-942 and Wnt/beta-catenin signaling. Biomed. Pharmacother. 2018, 101, 769–776. [Google Scholar] [CrossRef]
- Ma, S.; Deng, X.; Yang, Y.; Zhang, Q.; Zhou, T.; Liu, Z. The lncRNA LINC00675 regulates cell proliferation, migration, and invasion by affecting Wnt/beta-catenin signaling in cervical cancer. Biomed. Pharmacother. 2018, 108, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Chen, X.; Xie, R.; Xie, W.; Huang, L.; Dong, W.; Han, J.; Liu, X.; Shen, J.; Huang, J. A novel AR translational regulator lncRNA LBCS inhibits castration resistance of prostate cancer. Mol. Cancer 2019, 18, 109. [Google Scholar] [CrossRef]
- Mishra, S.; Deng, J.J.; Gowda, P.S.; Rao, M.K.; Lin, C.L.; Chen, C.L.; Huang, T.; Sun, L.-Z. Androgen receptor and microRNA-21 axis downregulates transforming growth factor beta receptor II (TGFBR2) expression in prostate cancer. Oncogene 2014, 33, 4097–4106. [Google Scholar] [CrossRef] [Green Version]
- Ribas, J.; Ni, X.; Haffner, M.; Wentzel, E.A.; Salmasi, A.H.; Chowdhury, W.H.; Kudrolli, T.A.; Yegnasubramanian, S.; Luo, J.; Rodriguez, R.; et al. miR-21: An androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res. 2009, 69, 7165–7169. [Google Scholar] [CrossRef] [Green Version]
- Aakula, A.; Leivonen, S.K.; Hintsanen, P.; Aittokallio, T.; Ceder, Y.; Borresen-Dale, A.L.; Perala, M.; Ostling, P.; Kallioniemi, O. MicroRNA-135b regulates ERalpha, AR and HIF1AN and affects breast and prostate cancer cell growth. Mol. Oncol. 2015, 9, 1287–1300. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.Y.; Ruan, Y.; Wang, X.H.; Zhao, W.; Jiang, Q.; Jing, Y.F.; Han, B.; Xia, S.; Zhao, F. MiR-185 attenuates androgen receptor function in prostate cancer indirectly by targeting bromodomain containing 8 isoform 2, an androgen receptor co-activator. Mol Cell Endocrinol. 2016, 427, 13–20. [Google Scholar] [CrossRef]
- Kashat, M.; Azzouz, L.; Sarkar, S.H.; Kong, D.; Li, Y.; Sarkar, F.H. Inactivation of AR and Notch-1 signaling by miR-34a attenuates prostate cancer aggressiveness. Am. J. Transl. Res. 2012, 4, 432–442. [Google Scholar]
- Yang, L.; Lin, C.; Jin, C.; Yang, J.C.; Tanasa, B.; Li, W.; Merkurjev, D.; Ohgi, K.A.; Meng, D.; Zhang, J.; et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 2013, 500, 598–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parolia, A.; Crea, F.; Xue, H.; Wang, Y.; Mo, F.; Ramnarine, V.R.; Liu, H.H.; Lin, D.; Saidy, N.R.N.; Clermont, P.; et al. The long non-coding RNA PCGEM1 is regulated by androgen receptor activity in vivo. Mol. Cancer 2015, 14, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Xu, C.; Li, Y.; Cai, X.; Ren, S.; Liu, H.; Wang, Y.; Wang, F.; Chen, R.; Qu, M.; et al. A feed-forward regulatory loop between androgen receptor and PlncRNA-1 promotes prostate cancer progression. Cancer Lett. 2016, 374, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pitchiaya, S.; Cieslik, M.; Niknafs, Y.S.; Tien, J.C.; Hosono, Y.; Iyer, M.K.; Yazdani, S.; Subramaniam, S.; Shukla, S.K.; et al. Analysis of the androgen receptor-regulated lncRNA landscape identifies a role for ARLNC1 in prostate cancer progression. Nat. Genet. 2018, 50, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Lingadahalli, S.; Jadhao, S.; Sung, Y.Y.; Chen, M.; Hu, L.; Chen, X.; Gheung, E. Novel lncRNA LINC00844 Regulates Prostate Cancer Cell Migration and Invasion through AR Signaling. Mol. Cancer Res. 2018, 16, 1865–1878. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Jia, D.; Kim, H.; Abd Elmageed, Z.Y.; Datta, A.; Davis, R.; Srivastav, S.; Moroz, K.; Crawford, B.E.; Moparty, K.; et al. Dysregulation of miR-212 Promotes Castration Resistance through hnRNPH1-Mediated Regulation of AR and AR-V7: Implications for Racial Disparity of Prostate Cancer. Clin. Cancer Res. 2016, 22, 1744–1756. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, C.E.; Sulpice, E.; Combe, S.; Shibakawa, A.; Leach, D.A.; Hamilton, M.P.; Chrysostomou, S.L.; Sharp, A.; Welti, J.; Yuan, W.; et al. Androgen receptor-modulatory microRNAs provide insight into therapy resistance and therapeutic targets in advanced prostate cancer. Oncogene 2019, 38, 5700–5724. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Ma, T.; Ungerleider, N.; Roberts, C.; Kobelski, M.; Jin, L.; Concha, M.; Wang, X.; Baddoo, M.; Nguyen, H.M.; et al. Circular RNAs add diversity to androgen receptor isoform repertoire in castration-resistant prostate cancer. Oncogene 2019, 38, 7060–7072. [Google Scholar] [CrossRef]
- Rizzo, M. Mechanisms of docetaxel resistance in prostate cancer: The key role played by miRNAs. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188481. [Google Scholar] [CrossRef]
- Chen, L.; Cao, H.; Feng, Y. MiR-199a suppresses prostate cancer paclitaxel resistance by targeting YES1. World J. Urol. 2018, 36, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Kojima, K.; Ohhashi, R.; Hamada, N.; Nozawa, Y.; Kitamoto, A.; Sato, A.; Kondo, S.; Kojima, T.; Deguchi, T.; et al. MiR-148a attenuates paclitaxel resistance of hormone-refractory, drug-resistant prostate cancer PC3 cells by regulating MSK1 expression. J. Biol. Chem. 2010, 285, 19076–19084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zou, L.; Li, X.; Chen, Z.; Lin, Q.; Wu, X. MicroRNA-195 regulates docetaxel resistance by targeting clusterin in prostate cancer. Biomed. Pharmacother. 2018, 99, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Wang, J.; Chen, G.; Zhao, X. microRNA-204 modulates chemosensitivity and apoptosis of prostate cancer cells by targeting zinc-finger E-box-binding homeobox 1 (ZEB1). Am J Transl Res. 2017, 9, 3599–3610. [Google Scholar]
- Gao, W.; Lin, S.; Cheng, C.; Zhu, A.; Hu, Y.; Shi, Z.; Zhang, X.; Hong, Z. Long non-coding RNA CASC2 regulates Sprouty2 via functioning as a competing endogenous RNA for miR-183 to modulate the sensitivity of prostate cancer cells to docetaxel. Arch. Biochem. Biophys. 2019, 665, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Tang, J.; Wang, G.; Zhu, Y.S. Long Non-Coding RNA as Potential Biomarker for Prostate Cancer: Is It Making a Difference? Int. J. Environ. Res. Public. Health 2017, 14, 270. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
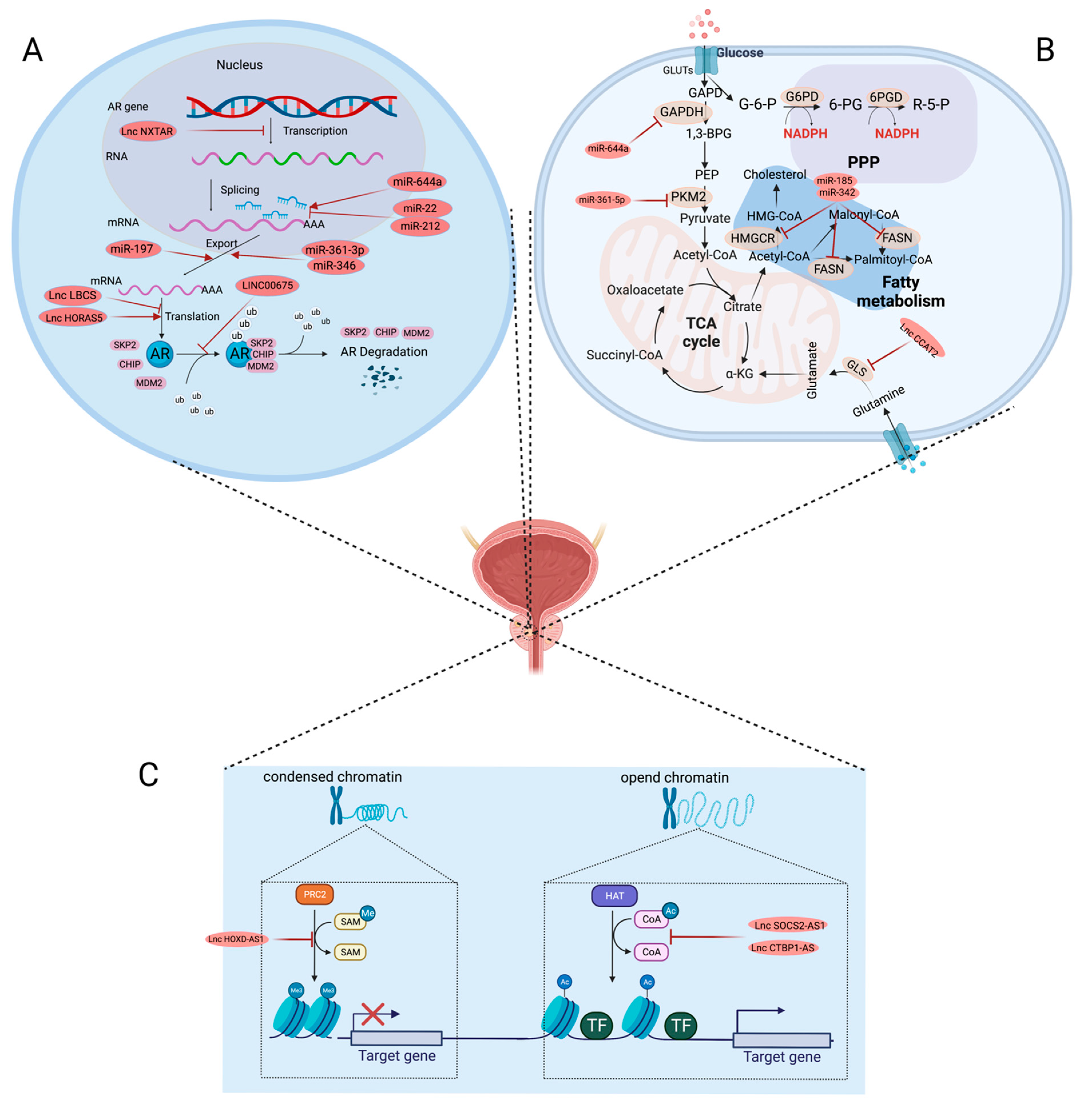{kind=link}
| Noncoding RNAs | Expression | Functions | Clinical Significance | Reference |
|---|---|---|---|---|
| miR-32 | Up | Proliferation (+); Apoptosis (−) | Potential marker for aggressive disease | [37,38] |
| miR-148a | Up | Proliferation (+); Cell cycle (+) | Urine-circulating mir-148a used for detection of PCa | [37] |
| miR-99a | Down | Proliferation (−) | N/A | [39] |
| miR-21 | Up | Migration (+); Invasion (+); Proliferation (+); Apoptosis (+); Chemo- and radiosensitivity (−) | Associated with biochemical recurrence in low-risk PCa | [40,41,42,43] |
| miR-221 | Up | Proliferation (+); Metastasis (+); EMT (+) | Prognostic marker in high-risk prostate cancer | [44,45] |
| miR-205 | Down | Metastasis (−); Migration (−) | Attenuates progression of prostate cancer | [46,47] |
| miR-1246 | Down | Proliferation (−); Migration (−); Invasion(−); Apoptosis (+) | Correlated with increasing pathologic grade, positive metastasis and poor prognosis | [48] |
| miR-34a | Down | Metastasis (−) | Potential invasive biomarker; Influences response to docetaxel | [49,50] |
| LncRNA DRAIC | Down | Migration(−); Invasion (−); EMT (−) | Predicts poor patient outcomes | [51] |
| LncRNA HOTAIR | Up | Proliferation(+); Invasion (+); Enzalutamide resistance (+) | May be a biomarker of enzalutamide resistance | [52] |
| LINC00963 | Up | Proliferation(+); Apoptosis (−) | May be a novel diagnosis biomarker | [53,54] |
| LncRNA MALAT-1 | Up | Proliferation (+); Migration(+); invasion (+) Arrest in the G0/G1 phases (+) | Correlated with high Gleason score, prostate specific antigen and tumor stage | [55] |
| LncRNA PCAT29 | Down | Proliferation (−); Migration (−); Apoptosis (+) | Associated with higher rates of biochemical recurrence | [56] |
| LncRNA PCGEM1 | Up | Proliferation (−) | Associated with high-risk prostate cancer patients | [57] |
| LINC00675 | Up | Proliferation (+); Migration (+); EMT (+) | Associated with Gleason score | [58] |
| LncRNA HORAS | Up | Proliferation (+) | Predicts poorer clinical outcomes | [59] |
| LncRNA PCAT14 | Down | Invasion (−) | Associated with Gleason score; predicts disease aggressiveness and recurrence | [60] |
| Noncoding RNAs | Expression | Regulation | Mechanisms | Reference |
|---|---|---|---|---|
| miR-32 | Up | Upregulated by AR | BTG2 is targeted by miR-32 | [37] |
| miR-148a | Up | Upregulated by AR | PIK3IP1 is targeted by miR-148a | [37] |
| miR-99a | Down | Repressed by AR | EZH2 promotes the repression of miR-99a by AR | [39] |
| miR-21 | Up | Repressed by AR | Drives the downregulation of TGFBR2 | [187,188] |
| miR-221 | Up | Abolishes AR-mediated transcription | Mediated by targeting HECTD2 | [44] |
| miR-135 | Down | Reduces AR protein and mRNA levels | Interacts with the 3′UTR of AR mRNA | [189] |
| miR-185 | Down | Reduces AR protein and mRNA levels | Interacts with the 3′UTR of AR mRNA; Suppresses BRD8 ISO2 protein | [190] |
| miR-34a | Down | Reduces AR protein and mRNA levels | N/A | [191] |
| miR-205 | Down | Reduces AR protein and mRNA levels | Interacts with the 3′UTR of AR mRNA | [46] |
| miR-124 | Down | Reduces AR protein and mRNA levels | Interacts with the 3′UTR of AR mRNA | |
| miR-644a | Down | Reduces both AR-FL and AR-V7 protein levels | Interacts with the 3′UTR of AR mRNA; Suppresses the expression of AR coregulators | [96] |
| LncRNA DRAIC | Down | Repressed by AR | The occupation of FOXA1 and NKX3-1 at DRAIC promoter was abolished | [51] |
| LncRNA HOTAIR | Up | Upregulates AR protein | Prevents AR ubiquitination and protein degradation by blocking its interaction with MDM2 | [52] |
| LncRNA PCAT29 | Down | Repressed by AR | AR binds to the PCAT29 promoter | [56] |
| LncRNA PCGEM1 | Up | Upregulates both AR-FL and AR-V7 proteins; Repressed by AR | Binds to the DOT1L-mediated methylated AR N-terminus; Functions as a transcriptional coregulator of the AR | [192,193] |
| PlncRNA-1 | Up | Upregulated by AR; Upregulates AR | Protects AR from miR-34c- and miR-297-mediated suppression | [194] |
| LncRNA ARLNC1 | Up | Upregulated by AR | Directly regulated by AR and modestly regulated by FOXA1; Interacts with 3′UTR of AR mRNA | [195] |
| LINC00844 | Down | Repressed by AR; Enhances AR activity | AR binds to the TSS of LINC00844; Influences AR-regulated transcriptome partly by facilitating the recruitment of AR to the chromatin | [196] |
| LINC00675 | Up | Upregulates AR protein | Stabilizes the AR protein by prohibiting AR from ubiquitination | [58] |
| LncRNA HORAS | Up | Upregulates AR | Likely due to a posttranscriptional regulatory manner | [59] |
| LncRNA LBCS | Down | Reduces AR protein | Guilds hnRNPK to the 5′-UTR of AR mRNA and thus inhibits AR translation efficiency | [186] |
| LncRNA NXTAR | Down | Reduces AR protein | Recruits EZH2 to promoter region of AR mRNA | [117] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.; Tan, H.; Yu, G.; Zhan, M.; Xu, B. Molecular Mechanisms of Noncoding RNA in the Occurrence of Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 1305. https://doi.org/10.3390/ijms24021305
Lin Y, Tan H, Yu G, Zhan M, Xu B. Molecular Mechanisms of Noncoding RNA in the Occurrence of Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences. 2023; 24(2):1305. https://doi.org/10.3390/ijms24021305
Chicago/Turabian StyleLin, Yu, Haisong Tan, Guopeng Yu, Ming Zhan, and Bin Xu. 2023. "Molecular Mechanisms of Noncoding RNA in the Occurrence of Castration-Resistant Prostate Cancer" International Journal of Molecular Sciences 24, no. 2: 1305. https://doi.org/10.3390/ijms24021305