
Enamel Matrix Derivative Suppresses Chemokine Expression in Oral Epithelial Cells

 ,
, 
Abstract
:1. Introduction
2. Result
2.1. TNFα but Not LPS Increases Chemokine Expression in HSC2 Cells
2.2. EMD Causes Phosphorylation of smad3 and Translocation of smad2/3 in HSC2 Cells
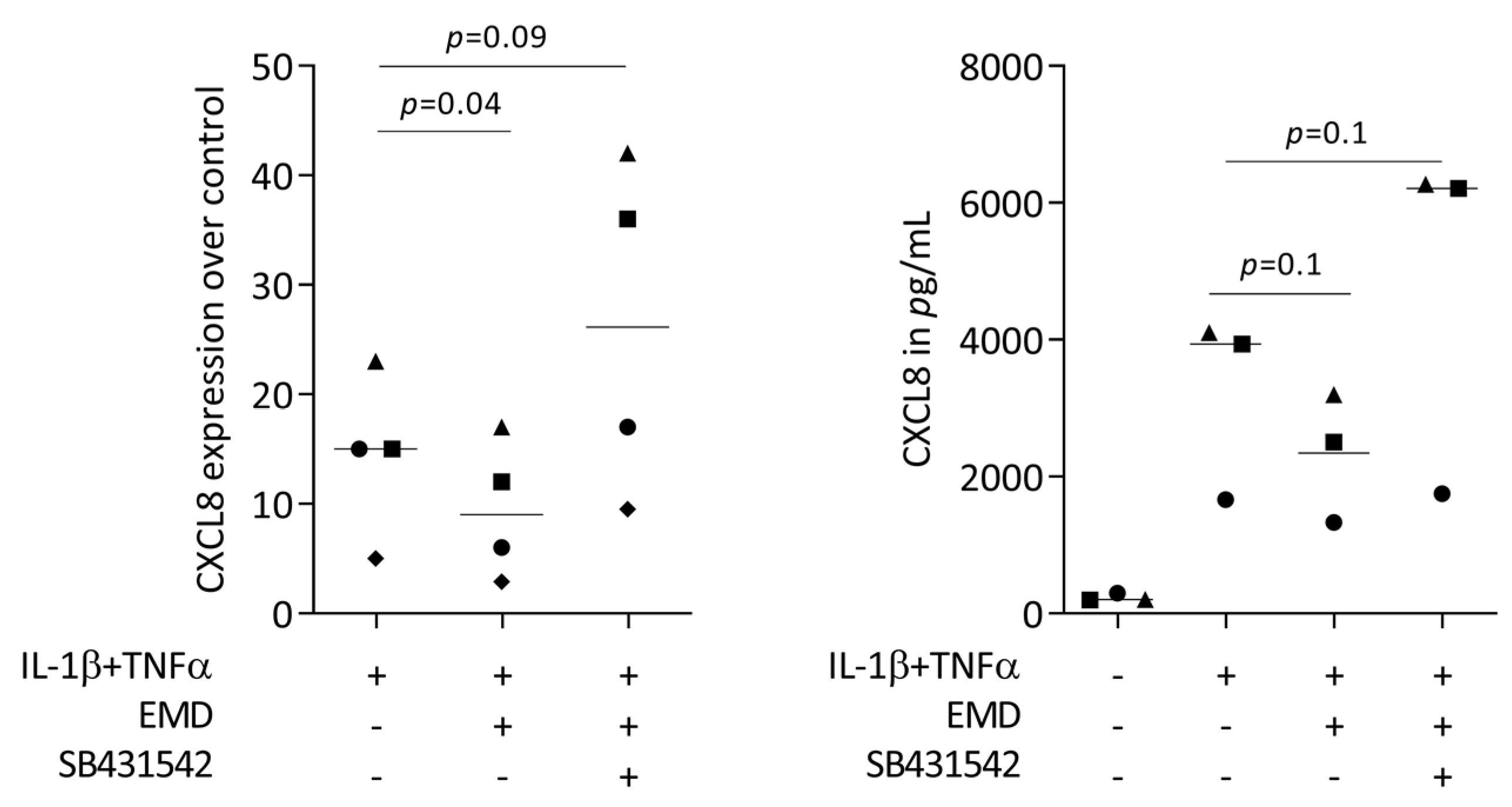
2.3. Blocking the TGF-β Signaling Reverses the Ability of EMD to Reduce CXCL8 Expression in HSC2 Cells
2.4. Primary Oral Epithelial Cells Are Comparable with HSC2 Cells with Respect to CXCL8 Expression
3. Discussion
4. Materials and Methods
4.1. HSC2 Cells and Primary Oral Epithelial Cells
4.2. RNAseq
4.3. Real-Time Polymerase Chain Reaction (RT-PCR) and Immunoassay
4.4. Immunofluorescence Analysis
4.5. Western Blot Analysis
4.6. Statistical Analysis and Acronyms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moutsopoulos, N.M.; Konkel, J.E. Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol. 2018, 39, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.M.; Doyle, A.D.; Greenwell-Wild, T.; Dutzan, N.; Tran, C.L.; Abusleme, L.; Juang, L.J.; Leung, J.; Chun, E.M.; Lum, A.G.; et al. Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science 2021, 374, eabl5450. [Google Scholar] [CrossRef] [PubMed]
- Gorr, S.U. Antimicrobial peptides in periodontal innate defense. Front. Oral Biol. 2012, 15, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Dutzan, N.; Abusleme, L.; Bridgeman, H.; Greenwell-Wild, T.; Zangerle-Murray, T.; Fife, M.E.; Bouladoux, N.; Linley, H.; Brenchley, L.; Wemyss, K.; et al. On-going Mechanical Damage from Mastication Drives Homeostatic Th17 Cell Responses at the Oral Barrier. Immunity 2017, 46, 133–147. [Google Scholar] [CrossRef]
- Yuan, X.; Liu, B.; Cuevas, P.; Brunski, J.; Aellos, F.; Petersen, J.; Koehne, T.; Broer, S.; Gruber, R.; LeBlanc, A.; et al. Linking the Mechanics of Chewing to Biology of the Junctional Epithelium. J. Dent. Res. 2023, 220345231185288. [Google Scholar] [CrossRef]
- Yamamoto, M.; Aizawa, R. Maintaining a protective state for human periodontal tissue. Periodontol. 2000 2021, 86, 142–156. [Google Scholar] [CrossRef]
- Bosshardt, D.D. The periodontal pocket: Pathogenesis, histopathology and consequences. Periodontol. 2000 2018, 76, 43–50. [Google Scholar] [CrossRef]
- Bosshardt, D.D.; Sculean, A. Does periodontal tissue regeneration really work? Periodontol. 2000 2009, 51, 208–219. [Google Scholar] [CrossRef]
- Bosshardt, D.D.; Lang, N.P. The junctional epithelium: From health to disease. J. Dent. Res. 2005, 84, 9–20. [Google Scholar] [CrossRef]
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Primers 2017, 3, 17038. [Google Scholar] [CrossRef]
- Demkovych, A.; Kalashnikov, D.; Hasiuk, P.; Zubchenko, S.; Vorobets, A. The influence of microbiota on the development and course of inflammatory diseases of periodontal tissues. Front. Oral Health 2023, 4, 1237448. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.J.; Hajishengallis, G.; Koo, H. Social networking at the microbiome-host interface. Infect. Immun. 2023, e0012423. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Illuminating the oral microbiome and its host interactions: Animal models of disease. FEMS Microbiol. Rev. 2023, 47, fuad018. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lamont, R.J.; Koo, H. Oral polymicrobial communities: Assembly, function, and impact on diseases. Cell Host Microbe 2023, 31, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Belibasakis, G.N.; Belstrom, D.; Eick, S.; Gursoy, U.K.; Johansson, A.; Kononen, E. Periodontal microbiology and microbial etiology of periodontal diseases: Historical concepts and contemporary perspectives. Periodontol. 2000 2023. [Google Scholar] [CrossRef]
- Jepsen, K.; Sculean, A.; Jepsen, S. Complications and treatment errors related to regenerative periodontal surgery. Periodontol. 2000 2023, 92, 120–134. [Google Scholar] [CrossRef]
- Tomasi, C.; Abrahamsson, K.H.; Apatzidou, D. Subgingival instrumentation. Periodontol. 2000 2023. [Google Scholar] [CrossRef]
- Golub, L.M.; Lee, H.M. Periodontal therapeutics: Current host-modulation agents and future directions. Periodontol. 2000 2020, 82, 186–204. [Google Scholar] [CrossRef]
- Mombelli, A. Maintenance therapy for teeth and implants. Periodontol. 2000 2019, 79, 190–199. [Google Scholar] [CrossRef]
- Williams, D.W.; Greenwell-Wild, T.; Brenchley, L.; Dutzan, N.; Overmiller, A.; Sawaya, A.P.; Webb, S.; Martin, D.; Genomics, N.N.; Computational Biology, C.; et al. Human oral mucosa cell atlas reveals a stromal-neutrophil axis regulating tissue immunity. Cell 2021, 184, 4090–4104. [Google Scholar] [CrossRef]
- Kobayashi, Y. The role of chemokines in neutrophil biology. Front. Biosci. 2008, 13, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Silva, L.M.; Theofilou, V.I.; Greenwell-Wild, T.; Li, L.; Williams, D.W.; Ikeuchi, T.; Brenchley, L.; Genomics, N.N.; Computational Biology, C.; et al. Neutrophil extracellular traps and extracellular histones potentiate IL-17 inflammation in periodontitis. J. Exp. Med. 2023, 220. [Google Scholar] [CrossRef] [PubMed]
- Gestrelius, S.; Lyngstadaas, S.P.; Hammarstrom, L. Emdogain--periodontal regeneration based on biomimicry. Clin. Oral Investig. 2000, 4, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Gestrelius, S.; Andersson, C.; Johansson, A.C.; Persson, E.; Brodin, A.; Rydhag, L.; Hammarstrom, L. Formulation of enamel matrix derivative for surface coating. Kinetics and cell colonization. J. Clin. Periodontol. 1997, 24, 678–684. [Google Scholar] [CrossRef]
- Hammarstrom, L.; Heijl, L.; Gestrelius, S. Periodontal regeneration in a buccal dehiscence model in monkeys after application of enamel matrix proteins. J. Clin. Periodontol. 1997, 24, 669–677. [Google Scholar] [CrossRef]
- Hammarstrom, L. Enamel matrix, cementum development and regeneration. J. Clin. Periodontol. 1997, 24, 658–668. [Google Scholar] [CrossRef]
- Suarez-Lopez Del Amo, F.; Monje, A. Efficacy of biologics for alveolar ridge preservation/reconstruction and implant site development: An American Academy of Periodontology best evidence systematic review. J. Periodontol. 2022, 93, 1827–1847. [Google Scholar] [CrossRef]
- Tavelli, L.; Chen, C.J.; Barootchi, S.; Kim, D.M. Efficacy of biologics for the treatment of periodontal infrabony defects: An American Academy of Periodontology best evidence systematic review and network meta-analysis. J. Periodontol. 2022, 93, 1803–1826. [Google Scholar] [CrossRef]
- Moldovan, R.; Mester, A.; Piciu, A.; Bran, S.; Onisor, F. Clinical Outcomes of Enamel Matrix Derivate Used in Surgical and Non-Surgical Treatment of Peri-Implantitis: A Systematic Review of Clinical Studies. Medicina 2022, 58, 1819. [Google Scholar] [CrossRef]
- Roccuzzo, A.; Imber, J.C.; Stahli, A.; Kloukos, D.; Salvi, G.E.; Sculean, A. Enamel matrix derivative as adjunctive to non-surgical periodontal therapy: A systematic review and meta-analysis of randomized controlled trials. Clin. Oral Investig. 2022, 26, 4263–4280. [Google Scholar] [CrossRef]
- Miron, R.J.; Sculean, A.; Cochran, D.L.; Froum, S.; Zucchelli, G.; Nemcovsky, C.; Donos, N.; Lyngstadaas, S.P.; Deschner, J.; Dard, M.; et al. Twenty years of enamel matrix derivative: The past, the present and the future. J. Clin. Periodontol. 2016, 43, 668–683. [Google Scholar] [CrossRef] [PubMed]
- Panahipour, L.; Sordi, M.B.; Kargarpour, Z.; Gruber, R. TGF-beta Signalling Mediates the Anti-Inflammatory Activity of Enamel Matrix Derivative In Vitro. Int. J. Mol. Sci. 2022, 23, 9778. [Google Scholar] [CrossRef]
- Sordi, M.B.; Cabral da Cruz, A.C.; Panahipour, L.; Gruber, R. Enamel Matrix Derivative Decreases Pyroptosis-Related Genes in Macrophages. Int. J. Mol. Sci. 2022, 23, 5078. [Google Scholar] [CrossRef] [PubMed]
- Gruber, R.; Roos, G.; Caballe-Serrano, J.; Miron, R.; Bosshardt, D.D.; Sculean, A. TGF-betaRI kinase activity mediates Emdogain-stimulated in vitro osteoclastogenesis. Clin. Oral Investig. 2014, 18, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Gruber, R.; Bosshardt, D.D.; Miron, R.J.; Gemperli, A.C.; Buser, D.; Sculean, A. Enamel matrix derivative inhibits adipocyte differentiation of 3T3-L1 cells via activation of TGF-betaRI kinase activity. PLoS ONE 2013, 8, e71046. [Google Scholar] [CrossRef]
- Stahli, A.; Bosshardt, D.; Sculean, A.; Gruber, R. Emdogain-regulated gene expression in palatal fibroblasts requires TGF-betaRI kinase signaling. PLoS ONE 2014, 9, e105672. [Google Scholar] [CrossRef]
- Kapferer, I.; Schmidt, S.; Gstir, R.; Durstberger, G.; Huber, L.A.; Vietor, I. Gene-expression profiles of epithelial cells treated with EMD in vitro: Analysis using complementary DNA arrays. J. Periodontal Res. 2011, 46, 118–125. [Google Scholar] [CrossRef]
- Kawase, T.; Okuda, K.; Momose, M.; Kato, Y.; Yoshie, H.; Burns, D.M. Enamel matrix derivative (EMDOGAIN) rapidly stimulates phosphorylation of the MAP kinase family and nuclear accumulation of smad2 in both oral epithelial and fibroblastic human cells. J. Periodontal Res. 2001, 36, 367–376. [Google Scholar] [CrossRef]
- Fang, W.B.; Mafuvadze, B.; Yao, M.; Zou, A.; Portsche, M.; Cheng, N. TGF-beta Negatively Regulates CXCL1 Chemokine Expression in Mammary Fibroblasts through Enhancement of Smad2/3 and Suppression of HGF/c-Met Signaling Mechanisms. PLoS ONE 2015, 10, e0135063. [Google Scholar] [CrossRef]
- Zou, A.; Lambert, D.; Yeh, H.; Yasukawa, K.; Behbod, F.; Fan, F.; Cheng, N. Elevated CXCL1 expression in breast cancer stroma predicts poor prognosis and is inversely associated with expression of TGF-beta signaling proteins. BMC Cancer 2014, 14, 781. [Google Scholar] [CrossRef]
- Wang, Q.; Lin, W.; Zhou, X.; Lei, K.; Xu, R.; Zhang, X.; Xiong, Q.; Sheng, R.; Song, W.; Liu, W.; et al. Single-Cell Transcriptomic Atlas of Gingival Mucosa in Type 2 Diabetes. J. Dent. Res. 2022, 101, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Finoti, L.S.; Nepomuceno, R.; Pigossi, S.C.; Corbi, S.C.; Secolin, R.; Scarel-Caminaga, R.M. Association between interleukin-8 levels and chronic periodontal disease: A PRISMA-compliant systematic review and meta-analysis. Medicine 2017, 96, e6932. [Google Scholar] [CrossRef] [PubMed]
- Aldahlawi, S.; Youssef, A.R.; Shahabuddin, S. Evaluation of chemokine CXCL10 in human gingival crevicular fluid, saliva, and serum as periodontitis biomarker. J. Inflamm. Res. 2018, 11, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Havens, A.M.; Chiu, E.; Taba, M.; Wang, J.; Shiozawa, Y.; Jung, Y.; Taichman, L.S.; D’Silva, N.J.; Gopalakrishnan, R.; Wang, C.; et al. Stromal-derived factor-1alpha (CXCL12) levels increase in periodontal disease. J. Periodontol. 2008, 79, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Gamonal, J.; Bascones, A.; Jorge, O.; Silva, A. Chemokine RANTES in gingival crevicular fluid of adult patients with periodontitis. J. Clin. Periodontol. 2000, 27, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Zhou, Z.; Huang, Y.; Huang, S.; Lin, Z.; He, F.; Song, Z. Exploration of the shared gene signatures and molecular mechanisms between periodontitis and inflammatory bowel disease: Evidence from transcriptome data. Gastroenterol. Rep. 2023, 11, goad041. [Google Scholar] [CrossRef]
- Shinjo, T.; Onizuka, S.; Zaitsu, Y.; Ishikado, A.; Park, K.; Li, Q.; Yokomizo, H.; Zeze, T.; Sato, K.; St-Louis, R.; et al. Dysregulation of CXCL1 Expression and Neutrophil Recruitment in Insulin Resistance and Diabetes-Related Periodontitis in Male Mice. Diabetes 2023, 72, 986–998. [Google Scholar] [CrossRef]
- Santos, A.F.P.; Cervantes, L.C.C.; Panahipour, L.; Souza, F.A.; Gruber, R. Proof-of-Principle Study Suggesting Potential Anti-Inflammatory Activity of Butyrate and Propionate in Periodontal Cells. Int. J. Mol. Sci. 2022, 23, 11006. [Google Scholar] [CrossRef]
- Rath-Deschner, B.; Memmert, S.; Damanaki, A.; Nokhbehsaim, M.; Eick, S.; Cirelli, J.A.; Gotz, W.; Deschner, J.; Jager, A.; Nogueira, A.V.B. CXCL1, CCL2, and CCL5 modulation by microbial and biomechanical signals in periodontal cells and tissues-in vitro and in vivo studies. Clin. Oral Investig. 2020, 24, 3661–3670. [Google Scholar] [CrossRef]
- Ramage, G.; Lappin, D.F.; Millhouse, E.; Malcolm, J.; Jose, A.; Yang, J.; Bradshaw, D.J.; Pratten, J.R.; Culshaw, S. The epithelial cell response to health and disease associated oral biofilm models. J. Periodontal Res. 2017, 52, 325–333. [Google Scholar] [CrossRef]
- Greer, A.; Irie, K.; Hashim, A.; Leroux, B.G.; Chang, A.M.; Curtis, M.A.; Darveau, R.P. Site-Specific Neutrophil Migration and CXCL2 Expression in Periodontal Tissue. J. Dent. Res. 2016, 95, 946–952. [Google Scholar] [CrossRef]
- Dommisch, H.; Staufenbiel, I.; Schulze, K.; Stiesch, M.; Winkel, A.; Fimmers, R.; Dommisch, J.; Jepsen, S.; Miosge, N.; Adam, K.; et al. Expression of antimicrobial peptides and interleukin-8 during early stages of inflammation: An experimental gingivitis study. J. Periodontal Res. 2015, 50, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wang, J.; Wang, K.; Li, C.; Zhang, S.; Jing, D.; Xu, C.; Wang, X.; Zhao, H.; Feng, J.Q. Axin2(+)-Mesenchymal PDL Cells, Instead of K14(+) Epithelial Cells, Play a Key Role in Rapid Cementum Growth. J. Dent. Res. 2019, 98, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Dong, X.; Ma, Y.; Yao, J. Chemokine (C-X-C motif) ligand 1 maintains the immune surveillance function of natural killer cells via the PDK2/mTOR signaling pathway. Cell Biol. Toxicol. 2022. [Google Scholar] [CrossRef]
- Nii, T.; Yumoto, H.; Hirota, K.; Miyake, Y. Anti-inflammatory effects of olanexidine gluconate on oral epithelial cells. BMC Oral Health 2019, 19, 239. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, W.; Wang, Q. 1,25-dihydroxyvitamin D(3) suppresses lipopolysaccharide-induced interleukin-6 production through aryl hydrocarbon receptor/nuclear factor-kappaB signaling in oral epithelial cells. BMC Oral Health 2019, 19, 236. [Google Scholar] [CrossRef]
- Panahipour, L.; Oladzad Abbasabadi, A.; Gruber, R. Oral Cell Lysates Reduce the Inflammatory Response of Activated Macrophages. J. Clin. Med. 2023, 12, 1701. [Google Scholar] [CrossRef]
- An, N.; Holl, J.; Wang, X.; Rausch, M.A.; Andrukhov, O.; Rausch-Fan, X. Potential Suppressive Effect of Nicotine on the Inflammatory Response in Oral Epithelial Cells: An In Vitro Study. Int. J. Environ. Res. Public Health 2021, 18, 483. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Uehara, A.; Sugawara, S.; Takada, H. Priming of human oral epithelial cells by interferon-gamma to secrete cytokines in response to lipopolysaccharides, lipoteichoic acids and peptidoglycans. J. Med. Microbiol. 2002, 51, 626–634. [Google Scholar] [CrossRef]
- Tamai, R.; Sugawara, S.; Takeuchi, O.; Akira, S.; Takada, H. Synergistic effects of lipopolysaccharide and interferon-gamma in inducing interleukin-8 production in human monocytic THP-1 cells is accompanied by up-regulation of CD14, Toll-like receptor 4, MD-2 and MyD88 expression. J. Endotoxin Res. 2003, 9, 145–153. [Google Scholar] [CrossRef]
- Pourgonabadi, S.; Muller, H.D.; Mendes, J.R.; Gruber, R. Saliva initiates the formation of pro-inflammatory macrophages in vitro. Arch. Oral Biol. 2017, 73, 295–301. [Google Scholar] [CrossRef]
- Muller, H.D.; Cvikl, B.B.; Lussi, A.A.; Gruber, R.R. Salivary pellets induce a pro-inflammatory response involving the TLR4-NF-kB pathway in gingival fibroblasts. BMC Oral Health 2016, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, L.; Cervino, G.; Herford, A.S.; Lauritano, F.; D’Amico, C.; Lo Giudice, R.; Laino, L.; Troiano, G.; Crimi, S.; Cicciu, M. Interferon Crevicular Fluid Profile and Correlation with Periodontal Disease and Wound Healing: A Systemic Review of Recent Data. Int. J. Mol. Sci. 2018, 19, 1908. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Pandit, B. IL32: The multifaceted and unconventional cytokine. Hum. Immunol. 2021, 82, 659–667. [Google Scholar] [CrossRef]
- Johnson, D.; Carbonetti, N. Roles and Effects of Interferon Lambda Signaling in the Context of Bacterial Infections. J. Interferon Cytokine Res. 2023. [Google Scholar] [CrossRef]
- Manivasagam, S.; Klein, R.S. Type III Interferons: Emerging Roles in Autoimmunity. Front. Immunol. 2021, 12, 764062. [Google Scholar] [CrossRef]
- Goel, R.R.; Kotenko, S.V.; Kaplan, M.J. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2021, 17, 349–362. [Google Scholar] [CrossRef]
- Deng, Z.; Hu, W.; Ai, H.; Chen, Y.; Dong, S. The Dramatic Role of IFN Family in Aberrant Inflammatory Osteolysis. Curr. Gene Ther. 2021, 21, 112–129. [Google Scholar] [CrossRef]
- Wang, J.M.; Huang, A.F.; Xu, W.D.; Su, L.C. Insights into IL-29: Emerging role in inflammatory autoimmune diseases. J. Cell. Mol. Med. 2019, 23, 7926–7932. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Sack, G.H., Jr. Serum Amyloid A (SAA) Proteins. Subcell. Biochem. 2020, 94, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Heinz, L.X.; Rebsamen, M.; Rossi, D.C.; Staehli, F.; Schroder, K.; Quadroni, M.; Gross, O.; Schneider, P.; Tschopp, J. The death domain-containing protein Unc5CL is a novel MyD88-independent activator of the pro-inflammatory IRAK signaling cascade. Cell Death Differ. 2012, 19, 722–731. [Google Scholar] [CrossRef]
- Hoshikawa, H.; Goto, R.; Mori, T.; Mitani, T.; Mori, N. Expression of prostaglandin E2 receptors in oral squamous cell carcinomas and growth inhibitory effects of an EP3 selective antagonist, ONO-AE3-240. Int. J. Oncol. 2009, 34, 847–852. [Google Scholar] [CrossRef]
- Osawa, K.; Umemura, M.; Nakakaji, R.; Tanaka, R.; Islam, R.M.; Nagasako, A.; Fujita, T.; Yokoyama, U.; Koizumi, T.; Mitsudo, K.; et al. Prostaglandin E(2) receptor EP4 regulates cell migration through Orai1. Cancer Sci. 2020, 111, 160–174. [Google Scholar] [CrossRef]
- Cho, Y.J.; Liang, P. Killin is a p53-regulated nuclear inhibitor of DNA synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 5396–5401. [Google Scholar] [CrossRef]
- Kargarpour, Z.; Nasirzade, J.; Panahipour, L.; Miron, R.J.; Gruber, R. Platelet-Rich Fibrin Decreases the Inflammatory Response of Mesenchymal Cells. Int. J. Mol. Sci. 2021, 22, 11333. [Google Scholar] [CrossRef]
- Di Summa, F.; Kargarpour, Z.; Nasirzade, J.; Stahli, A.; Mitulovic, G.; Panic-Jankovic, T.; Koller, V.; Kaltenbach, C.; Muller, H.; Panahipour, L.; et al. TGFbeta activity released from platelet-rich fibrin adsorbs to titanium surface and collagen membranes. Sci. Rep. 2020, 10, 10203. [Google Scholar] [CrossRef]
- Luo, Q.; Liu, Y.; Shi, K.; Shen, X.; Yang, Y.; Liang, X.; Lu, L.; Qiao, W.; Chen, A.; Hong, D.; et al. An autonomous activation of interleukin-17 receptor signaling sustains inflammation and promotes disease progression. Immunity 2023. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Wu, S.; Li, J.; Fu, W.; He, W.; Liu, X.; Slutsky, A.S.; Zhang, H.; Li, Y. Human alveolar epithelial type II cells in primary culture. Physiol. Rep. 2015, 3, 12288. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Peng, J.; Sun, T.; Li, N.; Zhang, L.; Ren, J.; Yuan, H.; Kan, S.; Pan, Q.; Li, X.; et al. Epithelial EZH2 serves as an epigenetic determinant in experimental colitis by inhibiting TNFalpha-mediated inflammation and apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3796–E3805. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Sequence | Reverse Sequence |
|---|---|---|
| CXCL1 [80] | TCCTGCATCCCCCATAGTTA | CTTCAGGAACAGCCACCAGT |
| CXCL2 [81] | CCCATGGTTAAGAAAATCATCG | CTTCAGGAACAGCCACCAAT |
| CXCL8 [82] | AACTTCTCCACAACCCTCTG | TTGGCAGCCTTCCTGATTTC |
| CCL20 [83] | GCTGCTTTGATGTCAGTGCT | GCAGTCAAAGTTGCTTGCTG |
| GAPDH | AAGCCACATCGCTCAGACAC | GCCCAATACGACCAAATCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panahipour, L.; Botta, S.; Abbasabadi, A.O.; Afradi, Z.; Gruber, R. Enamel Matrix Derivative Suppresses Chemokine Expression in Oral Epithelial Cells. Int. J. Mol. Sci. 2023, 24, 13991. https://doi.org/10.3390/ijms241813991
Panahipour L, Botta S, Abbasabadi AO, Afradi Z, Gruber R. Enamel Matrix Derivative Suppresses Chemokine Expression in Oral Epithelial Cells. International Journal of Molecular Sciences. 2023; 24(18):13991. https://doi.org/10.3390/ijms241813991
Chicago/Turabian StylePanahipour, Layla, Sara Botta, Azarakhsh Oladzad Abbasabadi, Zohreh Afradi, and Reinhard Gruber. 2023. "Enamel Matrix Derivative Suppresses Chemokine Expression in Oral Epithelial Cells" International Journal of Molecular Sciences 24, no. 18: 13991. https://doi.org/10.3390/ijms241813991