
The First Genome-Wide Mildew Locus O Genes Characterization in the Lamiaceae Plant Family

Abstract
:1. Introduction
2. Results
2.1. MLO Gene Annotation in Eleven Lamiaceae Species
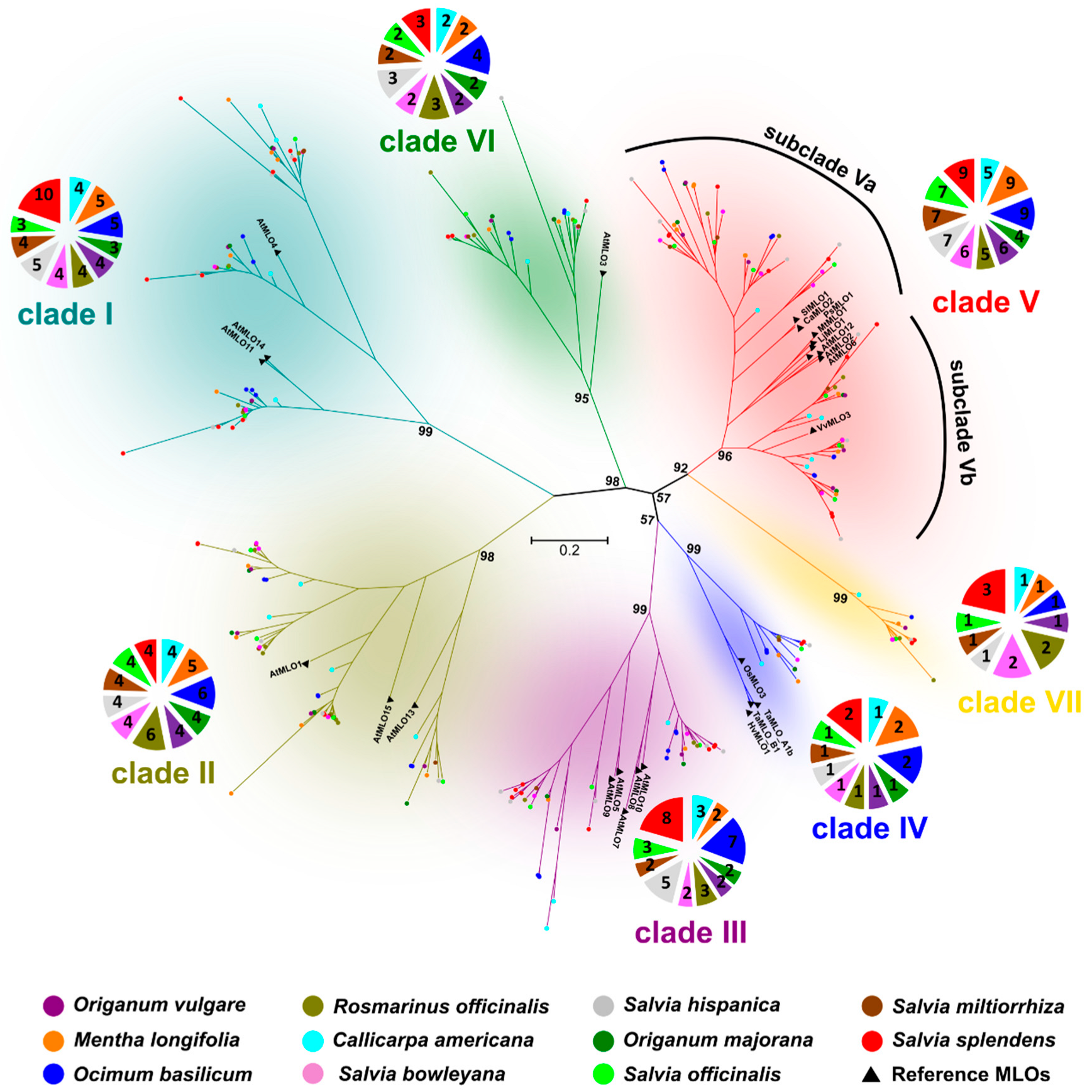
2.2. Identification of MLO Genes Associated to PM Susceptibility
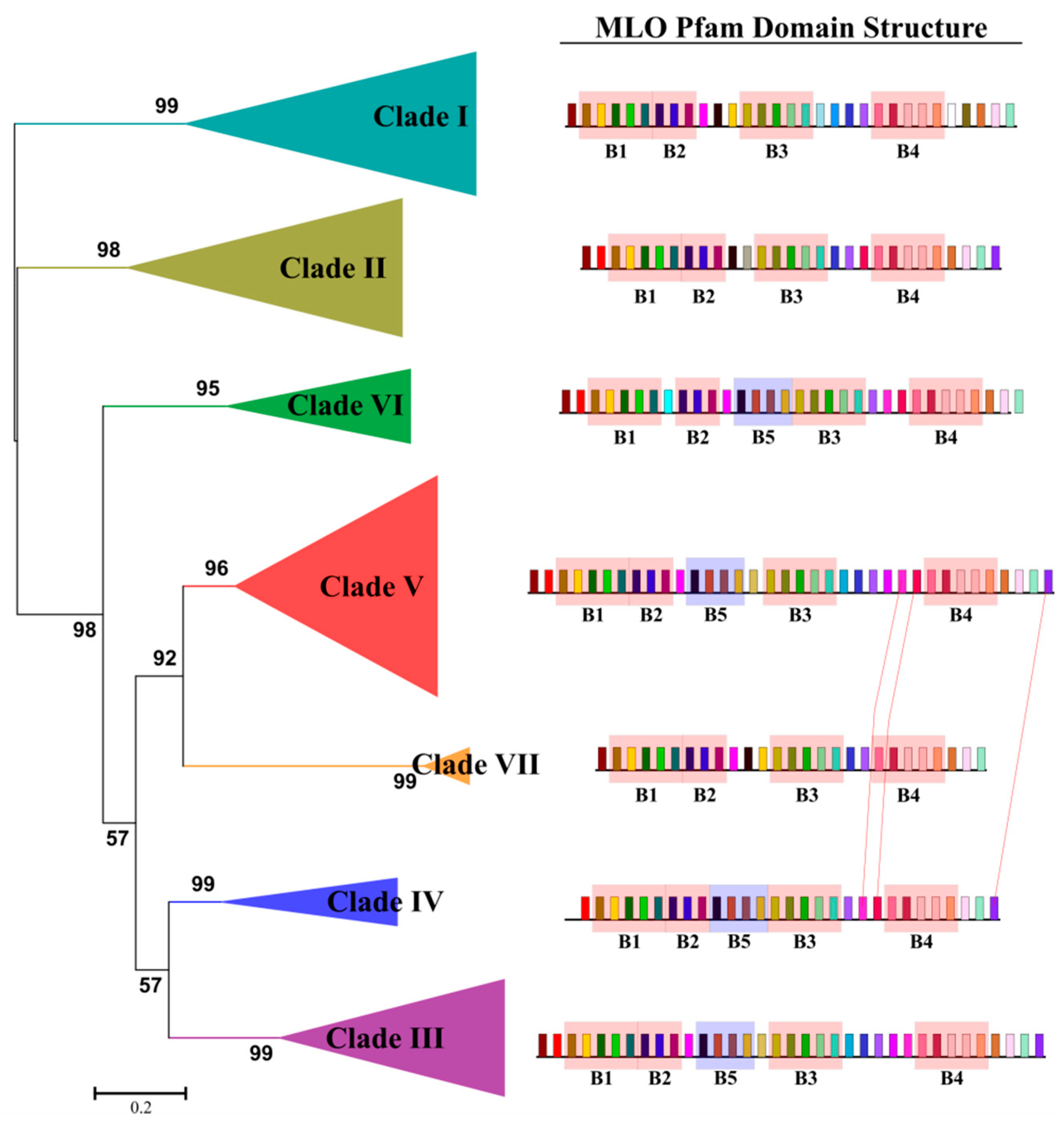
2.3. Characterization of Clade-Specific Amino Acid Motifs
2.4. Selection Pressure Acting on Lamiaceae MLO Phylogenetic Clades
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Taxa Data Set
5.2. Identification and Manual Curation of Eleven Lamiacaeae MLO Gene Families
5.3. Multiple Sequence Alignments and Phylogenetic Analysis
5.4. De Novo Prediction of MLO-Encoding Genes Motifs
5.5. Evolution Rates at Codon Sites
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MLO | mildew locus O |
| EO | essential oil |
| PM | powdery mildew |
| MEME | multiple EM for motif elicitation |
| SLAC | single-likelihood ancestor counting |
| PSC | positively selected codon |
| NSC | negatively selected codon |
| MAST | motif alignment and search tool |
| BIC | Bayesian information criterion |
| Pfam | protein family database |
References
- Tamokou, J.D.D.; Mbaveng, A.T.; Kuete, V. Antimicrobial Activities of African Medicinal Spices and Vegetables. In Medicinal Spices and Vegetables from Africa; Elsevier: Amsterdam, The Netherlands, 2017; pp. 207–237. ISBN 9780128094419. [Google Scholar]
- Medeiros-Neves, B.; Teixeira, H.F.; von Poser, G.L. The Genus Pterocaulon (Asteraceae)—A Review on Traditional Medicinal Uses, Chemical Constituents and Biological Properties. J. Ethnopharmacol. 2018, 224, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Tongnuanchan, P.; Benjakul, S. Essential Oils: Extraction, Bioactivities, and Their Uses for Food Preservation. J. Food Sci. 2014, 79, R1231–R1249. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, F. Lamiaceae in the Treatment of Cardiovascular Diseases. Front. Biosci. 2021, 26, 4909. [Google Scholar] [CrossRef]
- Nieto, G. Biological Activities of Three Essential Oils of the Lamiaceae Family. Medicines 2017, 4, 63. [Google Scholar] [CrossRef]
- Lange, B.M.; Mahmoud, S.S.; Wildung, M.R.; Turner, G.W.; Davis, E.M.; Lange, I.; Baker, R.C.; Boydston, R.A.; Croteau, R.B. Improving Peppermint Essential Oil Yield and Composition by Metabolic Engineering. Proc. Natl. Acad. Sci. USA 2011, 108, 16944–16949. [Google Scholar] [CrossRef] [PubMed]
- Wichura, A. Golovinomyces orontii and Other Powdery Mildews on Rosmarinus officinalis. Plant Pathol. Quar. 2012, 2, 162–166. [Google Scholar] [CrossRef]
- Szpyrka, E.; Słowik-Borowiec, M. Consumer Health Risk to Pesticide Residues in Salvia officinalis L. and Its Infusions. J. Environ. Sci. Health Part B Pestic. Food Contam. Agric. Wastes 2019, 54, 14–19. [Google Scholar] [CrossRef]
- Zheng, Z.; Nonomura, T.; Appiano, M.; Pavan, S.; Matsuda, Y.; Toyoda, H.; Wolters, A.M.A.; Visser, R.G.F.; Bai, Y. Loss of Function in Mlo Orthologs Reduces Susceptibility of Pepper and Tomato to Powdery Mildew Disease Caused by Leveillula taurica. PLoS ONE 2013, 8, e70723. [Google Scholar] [CrossRef]
- Nekrasov, V.; Wang, C.; Win, J.; Lanz, C.; Weigel, D.; Kamoun, S. Rapid Generation of a Transgene-Free Powdery Mildew Resistant Tomato by Genome Deletion. Sci. Rep. 2017, 7, 482. [Google Scholar] [CrossRef]
- Ingvardsen, C.R.; Massange-Sánchez, J.A.; Borum, F.; Uauy, C.; Gregersen, P.L. Development of Mlo-Based Resistance in Tetraploid Wheat against Wheat Powdery Mildew. Theor. Appl. Genet. 2019, 132, 3009–3022. [Google Scholar] [CrossRef]
- Kim, M.C.; Lee, S.H.; Kim, J.K.; Chun, H.J.; Choi, M.S.; Chung, W.S.; Moon, B.C.; Kang, C.H.; Park, C.Y.; Yoo, J.H.; et al. Mlo, a Modulator of Plant Defense and Cell Death, Is a Novel Calmodulin-Binding Protein. J. Biol. Chem. 2002, 277, 19304–19314. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, Y.; Lu, P.; Feng, C.; Niu, Q.; Lin, G.; Kong, D.; Liu, L.; Luan, S.; Li, L.; et al. Arabidopsis MLO4 Functions as a Ca2+ Channel Essential for Mechanosensing in Root Tips. bioRxiv 2022. bioRxiv:2022.06.05.494847. [Google Scholar] [CrossRef]
- Iovieno, P.; Bracuto, V.; Pavan, S.; Lotti, C.; Ricciardi, L.; Andolfo, G. Identification and Functional Inference on the MLO-Family in Viridiplantae. J. Plant Pathol. 2016, 98, 587–594. [Google Scholar] [CrossRef]
- Iovieno, P.; Andolfo, G.; Schiavulli, A.; Catalano, D.; Ricciardi, L.; Frusciante, L.; Ercolano, M.R.; Pavan, S. Structure, Evolution and Functional Inference on the Mildew Locus O (MLO) Gene Family in Three Cultivated Cucurbitaceae spp. BMC Genom. 2015, 16, 1112. [Google Scholar] [CrossRef]
- Pépin, N.; Hebert, F.O.; Joly, D.L. Genome-Wide Characterization of the MLO Gene Family in Cannabis sativa Reveals Two Genes as Strong Candidates for Powdery Mildew Susceptibility. Front. Plant Sci. 2021, 12, 729261. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Pavan, S.; Zheng, Z.; Zappel, N.F.; Reinstädler, A.; Lotti, C.; De Giovanni, C.; Ricciardi, L.; Lindhout, P.; Visser, R.; et al. Naturally Occurring Broad-Spectrum Powdery Mildew Resistance in a Central American Tomato Accession Is Caused by Loss of Mlo Function. Mol. Plant-Microbe Interact. 2008, 21, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Humphry, M.; Reinstädler, A.; Ivanov, S.; Bisseling, T.; Panstruga, R. Durable Broad-Spectrum Powdery Mildew Resistance in Pea Er1 Plants Is Conferred by Natural Loss-of-Function Mutations in PsMLO1. Mol. Plant Pathol. 2011, 12, 866–878. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Garcia, J.; Kusch, S.; Panstruga, R. Magical Mystery Tour: MLO Proteins in Plant Immunity and Beyond. New Phytol. 2014, 204, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.-Y.; Guo, Y.; Cheng, Y.; Hu, Y.; Xiao, S.; Wang, Y.; Wen, Y.-Q. CRISPR/Cas9-Mediated Mutagenesis of VvMLO3 Results in Enhanced Resistance to Powdery Mildew in Grapevine (Vitis vinifera). Hortic. Res. 2020, 7, 116. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, X.; Shan, Q.; Zhang, Y.; Liu, J.; Gao, C.; Qiu, J.-L. Simultaneous Editing of Three Homoeoalleles in Hexaploid Bread Wheat Confers Heritable Resistance to Powdery Mildew. Nat. Biotechnol. 2014, 32, 947–951. [Google Scholar] [CrossRef]
- Wang, L.; Lee, M.; Sun, F.; Song, Z.; Yang, Z.; Yue, G.H. A Chromosome-Level Genome Assembly of Chia Provides Insights into High Omega-3 Content and Coat Color Variation of Its Seeds. Plant Commun. 2022, 3, 100326. [Google Scholar] [CrossRef] [PubMed]
- Vining, K.J.; Pandelova, I.; Lange, I.; Parrish, A.N.; Lefors, A.; Kronmiller, B.; Liachko, I.; Kronenberg, Z.; Srividya, N.; Lange, B.M. Chromosome-Level Genome Assembly of Mentha longifolia L. Reveals Gene Organization Underlying Disease Resistance and Essential Oil Traits. G3 Genes Genomes Genet. 2022, 12, jkac112. [Google Scholar] [CrossRef]
- Song, Z.; Lin, C.; Xing, P.; Fen, Y.; Jin, H.; Zhou, C.; Gu, Y.Q.; Wang, J.; Li, X. A High-quality Reference Genome Sequence of Salvia miltiorrhiza Provides Insights into Tanshinone Synthesis in Its Red Rhizomes. Plant Genome 2020, 13, e20041. [Google Scholar] [CrossRef]
- Bornowski, N.; Hamilton, J.P.; Liao, P.; Wood, J.C.; Dudareva, N.; Buell, C.R. Genome Sequencing of Four Culinary Herbs Reveals Terpenoid Genes Underlying Chemodiversity in the Nepetoideae. DNA Res. 2020, 27, dsaa016. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P.; Godden, G.T.; Lanier, E.; Bhat, W.W.; Kinser, T.J.; Vaillancourt, B.; Wang, H.; Wood, J.C.; Jiang, J.; Soltis, P.S.; et al. Generation of a Chromosome-Scale Genome Assembly of the Insect-Repellent Terpenoid-Producing Lamiaceae Species, Callicarpa americana. Gigascience 2020, 9, giaa093. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.-H.; Liu, H.; Zhang, R.-G.; Xu, J.; Zhou, S.-S.; Jiao, S.-Q.; Yan, X.-M.; Tian, X.-C.; Shi, T.-L.; Luo, H.; et al. Chromosome-Scale Assembly and Evolution of the Tetraploid Salvia splendens (Lamiaceae) Genome. Hortic. Res. 2021, 8, 177. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, D.; Chen, B.; Liang, L.; Huang, Z.; Fan, W.; Chen, J.; He, W.; Chen, H.; Huang, L.; et al. Insights into Salvianolic Acid B Biosynthesis from Chromosome-scale Assembly of the Salvia bowleyana Genome. J. Integr. Plant Biol. 2021, 63, 1309–1323. [Google Scholar] [CrossRef]
- Li, C.-Y.; Yang, L.; Liu, Y.; Xu, Z.-G.; Gao, J.; Huang, Y.-B.; Xu, J.-J.; Fan, H.; Kong, Y.; Wei, Y.-K.; et al. The Sage Genome Provides Insight into the Evolutionary Dynamics of Diterpene Biosynthesis Gene Cluster in Plants. Cell Rep. 2022, 40, 111236. [Google Scholar] [CrossRef]
- Keller, O.; Kollmar, M.; Stanke, M.; Waack, S. A Novel Hybrid Gene Prediction Method Employing Protein Multiple Sequence Alignments. Bioinformatics 2011, 27, 757–763. [Google Scholar] [CrossRef]
- Devoto, A.; Piffanelli, P.; Nilsson, I.; Wallin, E.; Panstruga, R.; von Heijne, G.; Schulze-Lefert, P. Topology, Subcellular Localization, and Sequence Diversity of the Mlo Family in Plants. J. Biol. Chem. 1999, 274, 34993–35004. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Devoto, A.; Hartmann, H.A.; Piffanelli, P.; Elliott, C.; Simmons, C.; Taramino, G.; Goh, C.S.; Cohen, F.E.; Emerson, B.C.; Schulze-Lefert, P.; et al. Molecular Phylogeny and Evolution of the Plant-Specific Seven-Transmembrane MLO Family. J. Mol. Evol. 2003, 56, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Datamonkey: Rapid Detection of Selective Pressure on Individual Sites of Codon Alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Carović-Stanko, K.; Liber, Z.; Besendorfer, V.; Javornik, B.; Bohanec, B.; Kolak, I.; Satovic, Z. Genetic Relations among Basil Taxa (Ocimum L.) Based on Molecular Markers, Nuclear DNA Content, and Chromosome Number. Plant Syst. Evol. 2010, 285, 13–22. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.H. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef]
- Appiano, M.; Catalano, D.; Santillán Martínez, M.; Lotti, C.; Zheng, Z.; Visser, R.G.F.; Ricciardi, L.; Bai, Y.; Pavan, S. Monocot and Dicot MLO Powdery Mildew Susceptibility Factors Are Functionally Conserved in Spite of the Evolution of Class-Specific Molecular Features. BMC Plant Biol. 2015, 15, 257. [Google Scholar] [CrossRef]
- Kusch, S.; Pesch, L.; Panstruga, R. Comprehensive Phylogenetic Analysis Sheds Light on the Diversity and Origin of the MLO Family of Integral Membrane Proteins. Genome Biol. Evol. 2016, 8, 878–895. [Google Scholar] [CrossRef]
- Lin, Y.; Cheng, Y.; Jin, J.; Jin, X.; Jiang, H.; Yan, H.; Cheng, B. Genome Duplication and Gene Loss Affect the Evolution of Heat Shock Transcription Factor Genes in Legumes. PLoS ONE 2014, 9, e102825. [Google Scholar] [CrossRef]
- Andolfo, G.; Dohm, J.C.; Himmelbauer, H. Prediction of NB-LRR Resistance Genes Based on Full-length Sequence Homology. Plant J. 2022, 110, 1592–1602. [Google Scholar] [CrossRef]
- Andolfo, G.; Sánchez, C.S.; Cañizares, J.; Pico, M.B.; Ercolano, M.R. Large-Scale Gene Gains and Losses Molded the NLR Defense Arsenal during the Cucurbita Evolution. Planta 2021, 254, 82. [Google Scholar] [CrossRef]
- Elliott, C.; Müller, J.; Miklis, M.; Bhat, R.A.; Schulze-Lefert, P.; Panstruga, R. Conserved Extracellular Cysteine Residues and Cytoplasmic Loop–Loop Interplay Are Required for Functionality of the Heptahelical MLO Protein. Biochem. J. 2005, 385, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Panstruga, R. Discovery of Novel Conserved Peptide Domains by Ortholog Comparison within Plant Multi-Protein Families. Plant Mol. Biol. 2005, 59, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, Y.; Wang, S. Point Mutations with Positive Selection Were a Major Force during the Evolution of a Receptor-Kinase Resistance Gene Family of Rice. Plant Physiol. 2006, 140, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, C.G.; D’Esposito, D.; Cigliano, R.A.; Ercolano, M.R. Comparison of Tomato Transcriptomic Profiles Reveals Overlapping Patterns in Abiotic and Biotic Stress Responses. Int. J. Mol. Sci. 2023, 24, 4061. [Google Scholar] [CrossRef]
- Andolfo, G.; Di Donato, A.; Darrudi, R.; Errico, A.; Aiese Cigliano, R.; Ercolano, M.R. Draft of Zucchini (Cucurbita pepo L.) Proteome: A Resource for Genetic and Genomic Studies. Front. Genet. 2017, 8, 181. [Google Scholar] [CrossRef]
- Terauchi, R.; Yoshida, K. Towards Population Genomics of Effector–Effector Target Interactions. New Phytol. 2010, 187, 929–939. [Google Scholar] [CrossRef]
- Andolfo, G.; Ercolano, M.R. Plant Innate Immunity Multicomponent Model. Front. Plant Sci. 2015, 6, 987. [Google Scholar] [CrossRef]
- Ercolano, M.R.; D’Esposito, D.; Andolfo, G.; Frusciante, L. Multilevel Evolution Shapes the Function of NB-LRR Encoding Genes in Plant Innate Immunity. Front. Plant Sci. 2022, 13, 1007288. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional Classification of Proteins via Subfamily Domain Architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A General Empirical Model of Protein Evolution Derived from Multiple Protein Families Using a Maximum-Likelihood Approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Species | Common Name | Genome Size (Mb) | Predicted Gene Set | Number of MLO Loci | Reference Genome |
|---|---|---|---|---|---|
| Callicarpa americana | American beautyberry | 506 | 32,164 | 23 | Hamilton et al., 2020 [26] |
| Mentha longifolia | Silver mint | 470 | 52,520 * | 33 | Vining et al., 2016 [23] |
| Ocimum basilicum | Basil | 2068 | 78,990 | 45 | Bornowski et al., 2020 [25] |
| Origanum majorana | Marjoram | 761 | 33,929 | 21 | |
| Origanum vulgare | Oregano | 630 | 32,623 | 20 | |
| Rosmarinus officinalis | Rosemary | 1014 | 51,389 | 29 | |
| Salvia bowleyana | Nan-dan-shen | 462 | 44,044 | 22 | Zheng et al., 2021 [28] |
| Salvia hispanica | Chia | 348 | 31,069 | 28 | Wang et al., 2022 [22] |
| Salvia miltiorrhiza | Dan-shen | 595 | 32,483 | 21 | Song et al., 2020 [24] |
| Salvia officinalis | Sage | 480 | 50,957 * | 28 | Li et al., 2022 [29] |
| Salvia splendens | Scarlet sage | 808 | 56,267 | 54 | Jia et al., 2021 [27] |
| Total | 324 | ||||
| Species | Genomic Length (bp) | Number of Exons for Locus | Protein Length (aa) | Domain Length (aa) * |
|---|---|---|---|---|
| Callicarpa americana | 5084 | 12 | 458 | 387 |
| Mentha longifolia | 4763 | 12 | 507 | 334 |
| Ocimum basilicum | 4101 | 11 | 407 | 335 |
| Origanum majorana | 3960 | 11 | 433 | 348 |
| Origanum vulgare | 4215 | 13 | 482 | 404 |
| Rosmarinus officinalis | 3591 | 11 | 412 | 341 |
| Salvia bowleyana | 5105 | 13 | 436 | 344 |
| Salvia hispanica | 3958 | 13 | 494 | 406 |
| Salvia miltiorrhiza | 5008 | 14 | 520 | 449 |
| Salvia officinalis | 4167 | 12 | 474 | 344 |
| Salvia splendens | 4082 | 11 | 454 | 343 |
| Aligned Coding Sequences | MLO Loci (n.) | Average Identity (%) | Π * | ω | PSCs | NSCs |
|---|---|---|---|---|---|---|
| C. americana MLO family | 23 | 39.2 | 0.364 | 0.223 | 0 | 247 |
| M. longifolia MLO family | 33 | 25.7 | 0.428 | 0.349 | 0 | 192 |
| O. basilicum MLO family | 45 | 29.9 | 0.398 | 0.250 | 0 | 243 |
| O. majorana MLO family | 21 | 33.2 | 0.374 | 0.251 | 2 | 292 |
| O. vulgare MLO family | 20 | 37.9 | 0.41 | 0.212 | 0 | 250 |
| R. officinalis MLO family | 29 | 34.3 | 0.369 | 0.249 | 2 | 180 |
| S. bowleyana MLO family | 22 | 36.7 | 0.369 | 0.259 | 0 | 182 |
| S. hispanica MLO family | 28 | 35.9 | 0.407 | 0.257 | 1 | 253 |
| S. miltiorrhiza MLO family | 21 | 45.1 | 0.431 | 0.221 | 2 | 264 |
| S. officinalis MLO family | 28 | 33.7 | 0.409 | 0.256 | 0 | 264 |
| S. splendens MLO family | 54 | 29.9 | 0.342 | 0.311 | 4 | 267 |
| Lamiaceae MLO clade I | 51 | 48.9 | 0.314 | 0.307 | 0 | 279 |
| Lamiaceae MLO clade II | 49 | 55.6 | 0.307 | 0.267 | 0 | 311 |
| Lamiaceae MLO clade III | 39 | 50.8 | 0.268 | 0.291 | 0 | 255 |
| Lamiaceae MLO clade IV | 14 | 68.3 | 0.146 | 0.236 | 0 | 96 |
| Lamiaceae MLO clade V | 73 | 44.2 | 0.254 | 0.375 | 4 | 371 |
| Lamiaceae MLO clade VI | 27 | 53.6 | 0.252 | 0.369 | 0 | 178 |
| Lamiaceae MLO clade VII | 13 | 73 | 0.115 | 0.216 | 0 | 65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuseppe, A.; Raffaella, E.M. The First Genome-Wide Mildew Locus O Genes Characterization in the Lamiaceae Plant Family. Int. J. Mol. Sci. 2023, 24, 13627. https://doi.org/10.3390/ijms241713627
Giuseppe A, Raffaella EM. The First Genome-Wide Mildew Locus O Genes Characterization in the Lamiaceae Plant Family. International Journal of Molecular Sciences. 2023; 24(17):13627. https://doi.org/10.3390/ijms241713627
Chicago/Turabian StyleGiuseppe, Andolfo, and Ercolano Maria Raffaella. 2023. "The First Genome-Wide Mildew Locus O Genes Characterization in the Lamiaceae Plant Family" International Journal of Molecular Sciences 24, no. 17: 13627. https://doi.org/10.3390/ijms241713627