1. Introduction
Oculocutaneous albinism (OCA) is an autosomal recessive disease caused by perturbations in the melanogenesis pathway that causes a broad range of pathologies including but not limited to nystagmus, hypopigmentation, and foveal hypoplasia [
1]. This disease is separated into eight subtypes, with each subtype being classified by mutations in a different gene. These subtypes are named OCA type 1–8 and are associated with the following genes in respective order:
TYR,
OCA2 (P gene),
TYRP1,
SLC45A2,
OCA5,
SLC24A5,
C10ORF11, and
DCT (
TYRP2) (OMIM
®, An Online Catalog of Human Genes and Genetic Disorders;
http://www.omim.org; accessed on 3 May 2023). Among these subtypes, the most severe is oculocutaneous albinism type 1 (OCA1), caused by perturbations in the tyrosinase (Tyr) enzyme encoded by the
TYR gene. Patients with this form of albinism can be further described as having OCA1A or OCA1B [
2]. These forms are defined by differences in their severity, as OCA1A causes complete inhibition of melanin production while OCA1B causes a reduction in pigmentation of the hair, skin, and eyes. The phenotype of OCA1A is significant as it is the only OCA subtype to inhibit all levels of pigmentation, highlighting the essential role of Tyr. Over 300 mutations in this enzyme have been identified in OCA1 patients [
3].
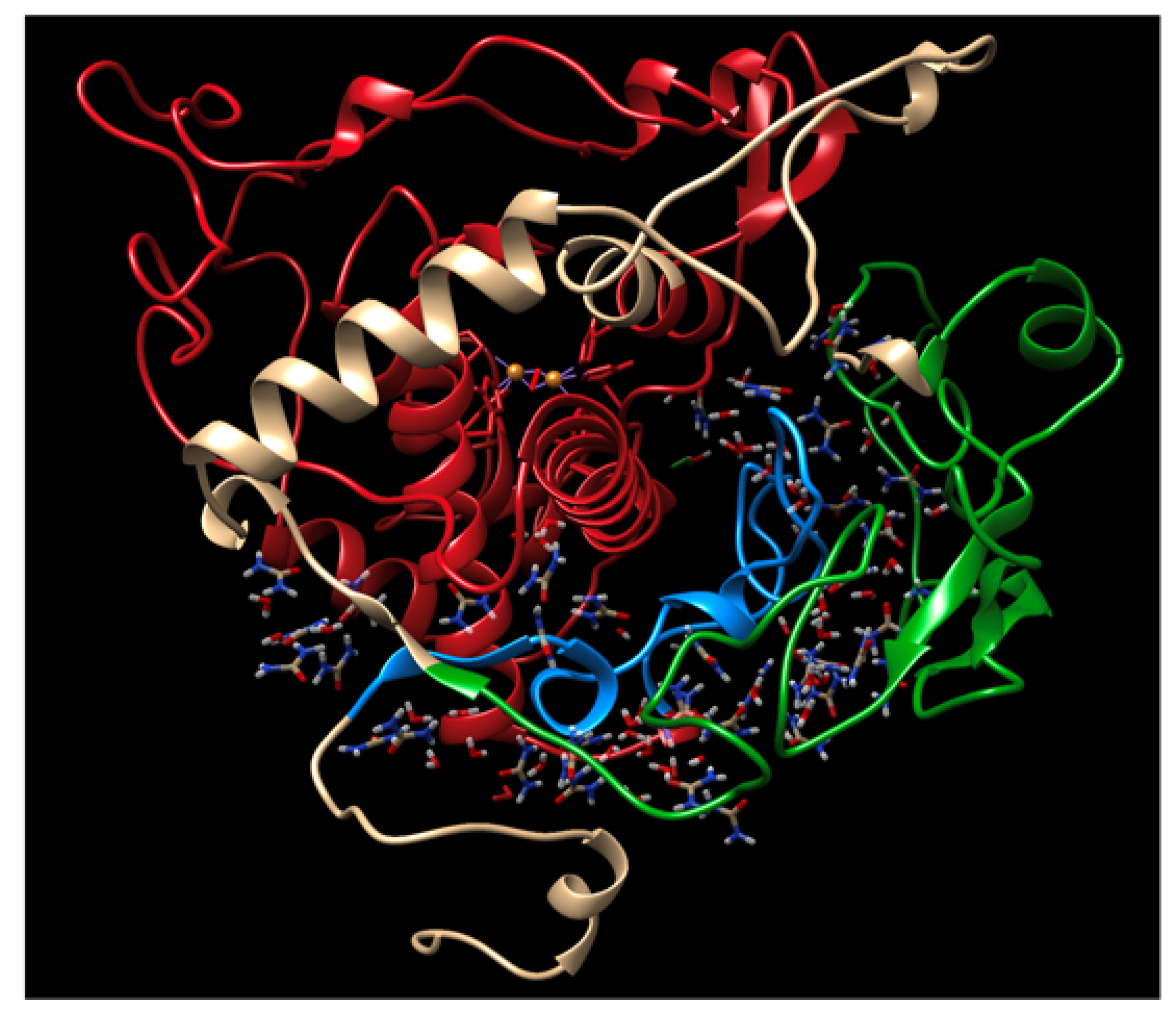
Tyr is a transmembrane metalloenzyme that is essential to produce melanin. The domain architecture of this enzyme consists of a signal peptide (1–23), cysteine-rich subdomain (24–118), catalytic tyrosinase domain (179–409), transmembrane helix (471–501), and cytoplasmic domain (502–519) (
Figure S1). The intramelanosomal body of the enzyme encapsulates the cysteine-rich subdomain and tyrosinase domain, which is essential for enzymatic function. The maturation of this protein is dependent upon the glycosylation of seven asparagine residues, as the enzyme is folded in the endoplasmic reticulum and transported to the melanosome [
4]. Once moved to the melanosome, Tyr remains attached to the organelle membrane where it catalyzes the oxidation of L-tyrosine and L-3,4-dihydroxyphenylalanine (L-DOPA) to dopaquinone [
5]. This reaction is both the first and rate-limiting step in melanin synthesis, making it essential for producing all melanin pigments.
Our group has previously demonstrated that the recombinant intramelanosomal domain of human tyrosinase (Tyr
tr) is required to retain enzymatic activity [
6]. By removing the transmembrane and cytoplasmic regions of Tyr, Tyr
tr is also soluble in an aqueous solution, making it desirable for protein purification. Using this construct, we can move from genetic associations of disease-associated mutation to identifying changes in protein stability and activity that confer these phenotypic changes that occur in OCA1 patients. Two OCA1B-associated mutations (P406L, R402Q) have been shown to cause instability and decreased activity of Tyr
tr relative to the severity of pigmentation defects in vivo [
7]. In vitro urea unfolding experiments have also offered insights into the presence of folded and unfolded states of these variant enzymes but have not brought clarity to the structural determinants of these differences in activity. Through computation, we hoped to investigate the underlying mechanisms behind the heterogenic activity of these hypomorphic mutations.
Homology modeling is a method that takes the amino acid sequence of a protein with an unknown structure and predicts this new structure based on similar solved structures [
8]. This method was previously used to predict the structure of Tyr
tr [
9]. Our homology model of Tyr
tr is composed of two subdomains. The first is an extended EGF-like domain containing a five-membered disulfide bridge network, defining the cysteine-rich subdomain. This network provides structural constraints to the globular domain, increasing its rigidity and stability. The Tyr catalytic subdomain houses a bundle of four embedded helices that make up the Tyr active site. This active site utilizes six histidine residues (H180, H202, H211, H363, H367, H390), the first three coordinating one copper ion (CuA), and the last three coordinating a second (CuB). These ions bind an O
2 molecule to form the complete active site [
10]. To gain information about structural differences in these domains between the wild type and mutants, we also explore the utility of long-duration molecular dynamics (MD) simulations in urea to assist protein destabilization [
11]. At the molecular scale, protein unfolding is a large process that can occur on a timescale of seconds or longer [
12]. Considering the current limitations in computation, and the expensive process of MD, which requires weeks to simulate moderately sized proteins, we aimed to simulate the destabilization of Tyr
tr in urea at the microsecond timescale.
3. Discussion
Tyr is responsible for catalyzing the rate-limiting step in the production of melanin pigments. Genetic perturbations in this enzyme can cause partial or complete inhibition of melanogenesis by destabilizing the native folds of this domain. Clinically, these perturbations manifest as the autosomal recessive disease known as OCA1. This disease has two subtypes, defined as OCA1A and OCA1B. Here, we investigated the mechanisms by which the OCA1B mutations P406L and R402Q destabilize Tyrtr and the specific effects they have on each subdomain of this enzyme. To accomplish this, we built models of each variant from our previously published homology model and simulated these structures for 1 μs of MD in water and 8M urea. Using these simulations in urea, we accelerated the timescale of protein unfolding enough to obtain a glimpse into the molten globule formation of each enzyme variant. With this feat achieved, we were able to observe the unique structural differences between Tyrtr and its mutants to mechanistically describe the destabilizing effects of these amino acid substitutions at the atomic scale. This information is crucial for the downstream development of targeted therapies, which may recover function in these hypomorphic Tyr mutations.
Given the essential role of Tyr in the melanogenesis pathway, it is not surprising that there are over 300 known mutations in the
TYR gene that are associated with OCA1. Studying deleterious mutations, like that of OCA1A-associated mutations, may be more intuitive, as many of these gene products do not reach full maturity and are degraded by the human proteasome, or simply cannot produce a natively folded protein [
7]. In contrast, hypomorphic mutations such as P406L and R402Q allow us to study the roles each of the native amino acids play in the overall stability and activity of these enzymes. With P406 and R402 being located at the periphery of the catalytic domain of Tyr
tr, structural observations alone cannot offer us insights into the roles of these residues. MD simulations function to fill this gap by providing us information on how Tyr
tr behaves differently with each substitution. In addition, simulations in 8M urea allow us to compare and accelerate the timescale at which larger structural changes would occur beyond what is computationally possible to simulate under these conditions.
From this long-duration MD simulation, we can capture events along the unfolding pathway in fine detail. There are four distinct stages in the protein unfolding pathway: the natively folded protein, molten globule, premolten globule, and unfolded protein [
14]. These stages are conserved for both folding and unfolding processes. This framework defines the unique structural identities of each stage in protein folding, providing us a good general sense of the types of protein movements that are significant in the folding process. However, it neglects to pinpoint how the protein transitions from one stage to another. The identification of these transition states is a problem that MD simulation excels at helping us solve [
15]. For Tyr
tr, we can observe a broad range of structural changes that occur before and during equilibration, compaction, and expansion of the globule in different conditions. These subtle structure shifts are difficult, if not impossible to observe in a three-dimensional protein structure in many cases. Studying these changes allows us to target specific motifs within proteins that contribute to instability in natural and mutated states. These motifs may act as targets for future studies aiming to restore function to OCA1-related enzyme variants.
All-atom MD at the μs timescale offers a nuanced view of protein motions that can help us understand structural determinants of protein stability and activity; however, this method is not without its challenges. With our MD configuration, the three-dimensional motion of each atom is calculated based on intrinsic energy characteristics and covalent and noncovalent interactions it formed with other atoms at every femtosecond of simulated time [
16]. Naturally, this method requires massive computational expenditure, which increases with the size of the simulated system. Increasing the timestep, which is the interval at which motions are calculated, reduces the time to solution at the cost of decreasing accuracy [
17]. However, increasing the speed of a simulation without augmenting experimental parameters is possible. Beyond utilizing high-performance compute nodes with powerful graphical processing units (GPUs), it is possible to parallelize a simulation between multiple compute nodes using a message-passing interface [
18,
19]. Unfortunately, our simulation package does not support internode communication for this level of optimization. For these reasons, we were limited to the μs timescale for our simulations (
Table S1).
Despite these computational restraints, we were able to simulate the molten globule transition of Tyr
tr and OCA1-related variants using MD in 8M urea. Molten globule formation is defined by the expansion of the protein and the unpacking of mobile sidechains without destabilization of the native secondary structure [
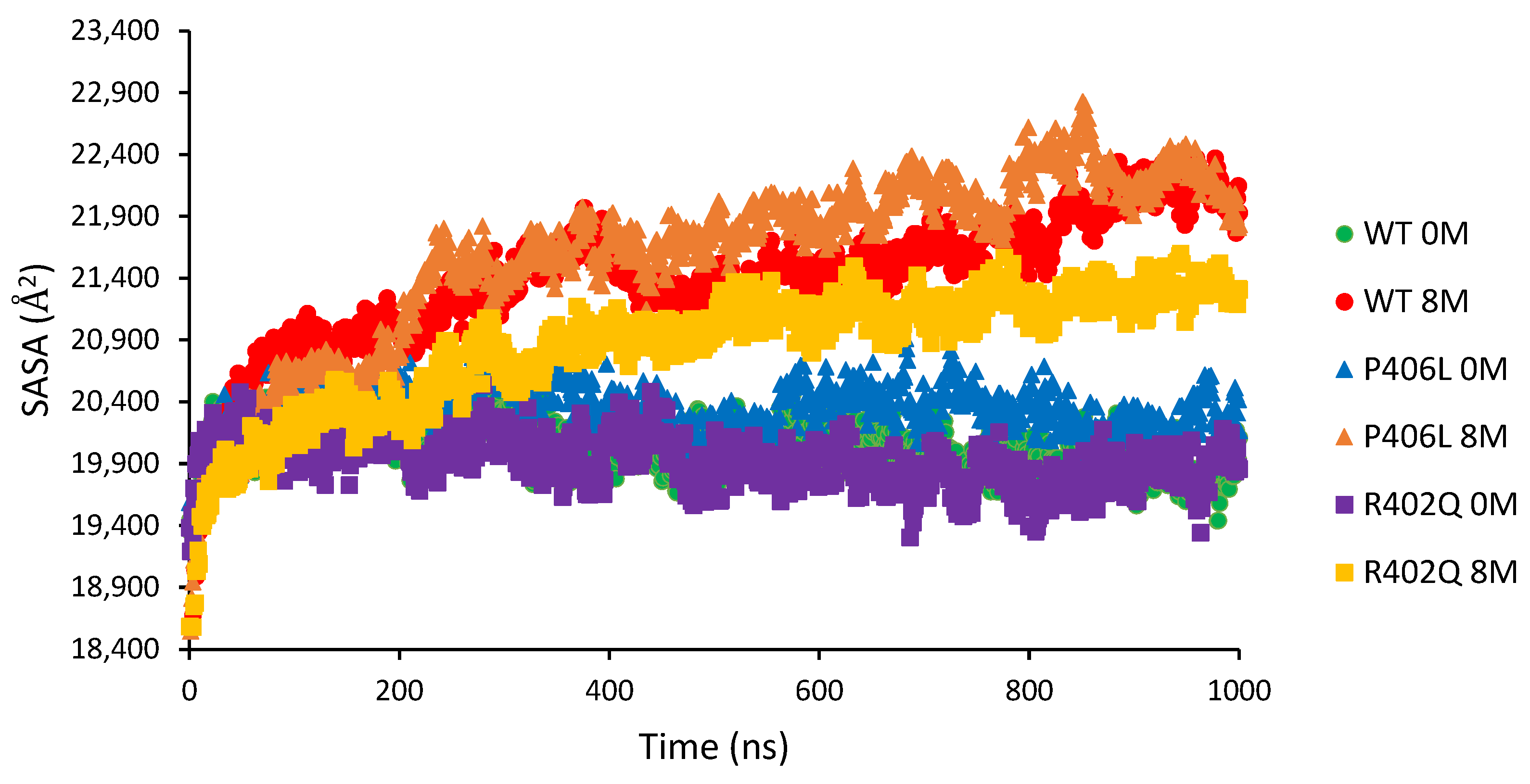
14]. In all 8M urea conditions, we observed increases in SASA and solvent-accessible volumes (
Figure S2). In general, predictions for the changes in P406L were intuitive. The severity of expansion was highest over time for P406L, caused by a decrease in hydrophobic interactions local to the catalytic domain, and a global decrease in hydrogen bonds within the globule. Interestingly, P406L experienced no significant decrease in ionic interactions in the subdomain interface and had the lowest total cavity volumes among all proteins. These results may concern each other, as P406L is shown to have the least stable structure even in water. It is important to note that this structure experiences the largest standard deviations in cavity volume, potentially relating to increased flexibility in sidechains and loops throughout the protein. As P406L expands, the protein’s internal volume may increase as the volume of native cavities on the globule periphery decrease. In essence, the globular structure grows like that of a balloon before more solvent is allowed to enter the protein, triggering the transition from the molten globule to the premolten globule [
14]. The cysteine-rich subdomain does not appear to undergo many, if any, changes for this structure under any condition, whereas the catalytic domain experiences a global increase in structural flexibility reminiscent of the molten globule. Contrary to the P406L mutation, R402Q has rather intricate effects on the entire intramelanosomal domain. Relative to the WT, R402Q is less vulnerable to urea-induced expansion, although it still occurs at a slower rate. We see that R402 is located at the peripheral end of one of the α-helical bundles of the Tyr catalytic domain. When this relatively large amino acid is substituted for glutamine, this appears to increase the flexibility of the local secondary structure enough to extend the helix by one amino acid (H404). This causes local compaction of the helix and adjacent sidechain, increasing the hydrophobic interactions in this area. However, this increased rigidity in the catalytic domain seems to have negative effects on the subdomain interface, as ionic interactions are decreased both in water and urea. It is known that globular proteins often adopt a rigid active site confirmation, which is accompanied by decreases in rigidity in the globule periphery. With additional rigidity in the Tyr
tr periphery from this mutation, it may decrease rigidity in other regions, such as the subdomain interface and cysteine-rich domain, causing a decrease in ionic interactions. Likewise, decreases in rigidity in the catalytic subdomain periphery may increase the rigidity in the subdomain interface, resulting in ionic interactions being sustained in 8M urea for the P406L variant. Indeed, the interplay between local and global changes in protein rigidity for Tyr
tr should be further studied.
Overall, we were able to simulate just a portion of the protein unfolding curve for Tyr
tr, but these data provide us valuable insight into the dynamic transition states in the unfolding pathway. It is known that protein folding/unfolding can occur on the timescale of microseconds to seconds. However, the folding timescale varies based on the size and complexity of the molecule [
12]. Relative to the sigmoidal protein unfolding curve, we expect that our simulation has only reached the very beginning of the upward slope toward full unfolding [
7]. We also predicted that the molten globule forms specifically within the Tyr catalytic subdomain and subdomain interface of this enzyme. In the future, we hope to validate not only the existence of this molten globule in the catalytic subdomain but also the real timescale for these unfolding transitions as well. The use of circular dichroism (CD) and solution nuclear magnetic resonance (NMR) spectroscopy to identify the molten globular states of proteins has been well studied [
20]. This task is partly complicated by the nature of Tyr
tr, as it is not only held together via noncovalent interactions but also with seven total disulfide bonds and a copper–oxygen cluster within the active site. However, these methods may allow us to identify the stages of unfolding for Tyr
tr, complementing the information we gather about the unfolding transition states from our MD studies.
4. Materials and Methods
4.1. Molecular Modeling—WT and Mutants
The Tyrtr homology model was obtained using the structure of homologous protein Tyrp1 (PDB: 5M8L) and accessible from NEI Commons Ocular Proteome Database (
https://neicommons.nei.nih.gov/#/proteomeData, accessed on 1 May 2022). We removed the signal peptide region in UCSF Chimera and extended the C-terminal truncation by 20 residues with the Terminal Extension function within YASARA’s homology modeling package (YASARA Biosciences GmbH, Vienna, Austria, EU). The tyrosinase domain was glycosylated in positions as described previously [
9].
The bonds coordinating copper atoms in the active site were replaced with pseudo bonds maintaining their specified bond lengths and angles throughout the simulations. Charges for the Cu ions were automatically assigned by the YASARA AutoSMILES program. Active site copper cluster geometries were optimized by placing both copper ions 3 Å away from bonded histidine residues, then using semiempirical quantum mechanics via MOPAC 7 [
21]. The implicit solvent was added for the optimization using the COSMO model [
22]. The resulting model was validated using SAVES (
https://www.doe-mbi.ucla.edu/services/; accessed on 3 May 2023). Two OCA1-related mutants (P406L and R402Q) were built from our validated Tyr model using the Edit > Swap > Residue function in YASARA.
4.2. Computer Simulations
For each protein structure, we created a simulation cubic cell of 84 Å × 84 Å × 84 Å extending 10 Å beyond all protein atoms and filled the cell with pure water or aqueous urea using the TIP3P water model. The urea solvent model was built from the 3D conformer SDF file of a single urea molecule from PubChem (
https://pubchem.ncbi.nlm.nih.gov/compound/Urea, accessed on 2 June 2022). The 3D conformer was energy minimized and saved as a PDB file in Chimera then loaded into YASARA. The urea model was parameterized using the AutoSMILES program, creating the final model used for all solvent environments (
Supplementary Table S2). The energy-minimized urea molecule was placed into the empty simulation cell and used to fill the cell. Aqueous urea solvents were generated as described in the documentation of YASARA’s FillCellObj function, in which the concentrations of each solvent component are determined separately using a weighted density value as shown below in Equations (1)–(4).
Here, murea and Vurea are the mass and volume of urea within the simulation cell. ρurea and ρwater are the standard density of urea and water (1.32 g/cm3, 1 g/cm3). ρurea and ρwater are the densities of urea and water within the simulation cell at the target molarity. Vtotal is the total volume within the simulation cell. Murea and curea are the molar mass and molarity of urea in the simulation.
After the solvents were created and energy minimized, the Tyr structure was placed in the center of the simulation cell along with a mass fraction of 0.9% NaCl to neutralize the protein. Solvent environments were energy minimized before each protein structure was added, and the completed simulation system was energy-minimized again before beginning each MD experiment. All solvent molecules within 2 Å of the protein were removed to avoid bumps. Protein equilibration was evaluated using the changes in the accessible surface of each globule. This was shown by sharp changes in surface area values followed by a flattening curve showing that the proteins had adjusted to the simulation environments. This flattening curve was observed in all structures within the first 100 ns of the simulations. The cell neutralization and pKa prediction script within YASARA was used to predict pKa values as a function of electrostatic potentials calculated using Ewald summation, hydrogen bonds, and accessible surface areas of titratable groups.
The resulting systems were energy-minimized before running 1 ms of MD using a modified version of the ‘md_run.mcr’ macro in YASARA. This macro ran MD with the AMBER14 forcefield, with intermolecular forces calculated every 2 fs and intramolecular forces calculated every 1 fs [
23]. Since the default pressure control algorithm is optimized for pure water solvents, and we are approximating the aqueous urea solvent densities, we utilized the built-in Manometer1D pressure control system for all experiments within our modified molecular dynamics script in YASARA. Simulation trajectories were saved every 0.1 ns for subsequent analysis. Simulations for all proteins were run in triplicate using a different random seed for each condition. All simulations were performed on the NIH Biowulf high-performing computation cluster.
4.3. Protein Structure Visualization and Comparisons
All structure alignments and domain visualizations were created in UCSF Chimera. The structures of Tyrtr, P406L, and R406Q were aligned after 1 μs of MD using the MatchMaker tool. The WT was used as the reference and the best-aligning pairs of chains between the reference and match structures were identified using the Needleman–Wunsch algorithm and the blocks substitution matrix (BLOSUM-62). The gap extension penalty was set to 1 and a secondary structure score of 30% was included. Secondary structure assignments were computed for each model, and long atom pairs were matched using iterative pruning until no pair exceeded 2.0 Å. Tyrtr subdomains and the subdomain interface region were identified and compared using the Sequence tool. Embedded solvent visualizations were created in YASARA. This was achieved by loading the MD simulation file of the WT in 8M urea at 1 μs into YASARA, turning off the simulation, and deleting all solvent molecules further than 21 Å from the protein center of mass. To visualize solvent embedded in the subdomain interface, all solvent molecules further than 5 Å from the residues 410–450 (interface region) were removed and the resulting simulation was saved as a PDB file for visualization in UCSF Chimera.
4.4. Unfolding Mutation Screen (UMS)
To further investigate how mutations at these positions of Tyrtr may lead to destabilization, we utilized our global computational mutagenesis pipeline. This pipeline is a collection of programs that allow the user to model protein structures and calculate the destabilizing effects of missense mutations. One of these programs, the unfolding mutation screen (UMS), calculates unfolding propensities from free energy change (ΔΔG) values for all possible missense variants of the structure. Unfolding propensities for each residue position are combined to generate a foldability score ranging from 0 to 19, which indicates the likelihood of a mutation at a certain position resulting in protein unfolding. These foldability scores are mapped to their corresponding residue positions on the structure, providing us with a three-dimensional view of which positions are critical for stability.
4.5. Solvent-Accessible Surface Areas (SASA) and Cavity Volumes
SASA values were calculated for Tyrtr and mutant variants in water and 8M urea for every nanosecond of simulation time using the SurfObj function in YASARA. MOLE 2.5 was used to find the cavity and void volumes of each structure at 0M and 8M urea at 1 μs. This cavity analysis generates an interface that displays each cavity along with its associated volume. These volumes were summed, and the average total cavity volume and standard deviations were compared between structures.
4.6. Intramolecular Noncovalent Interactions
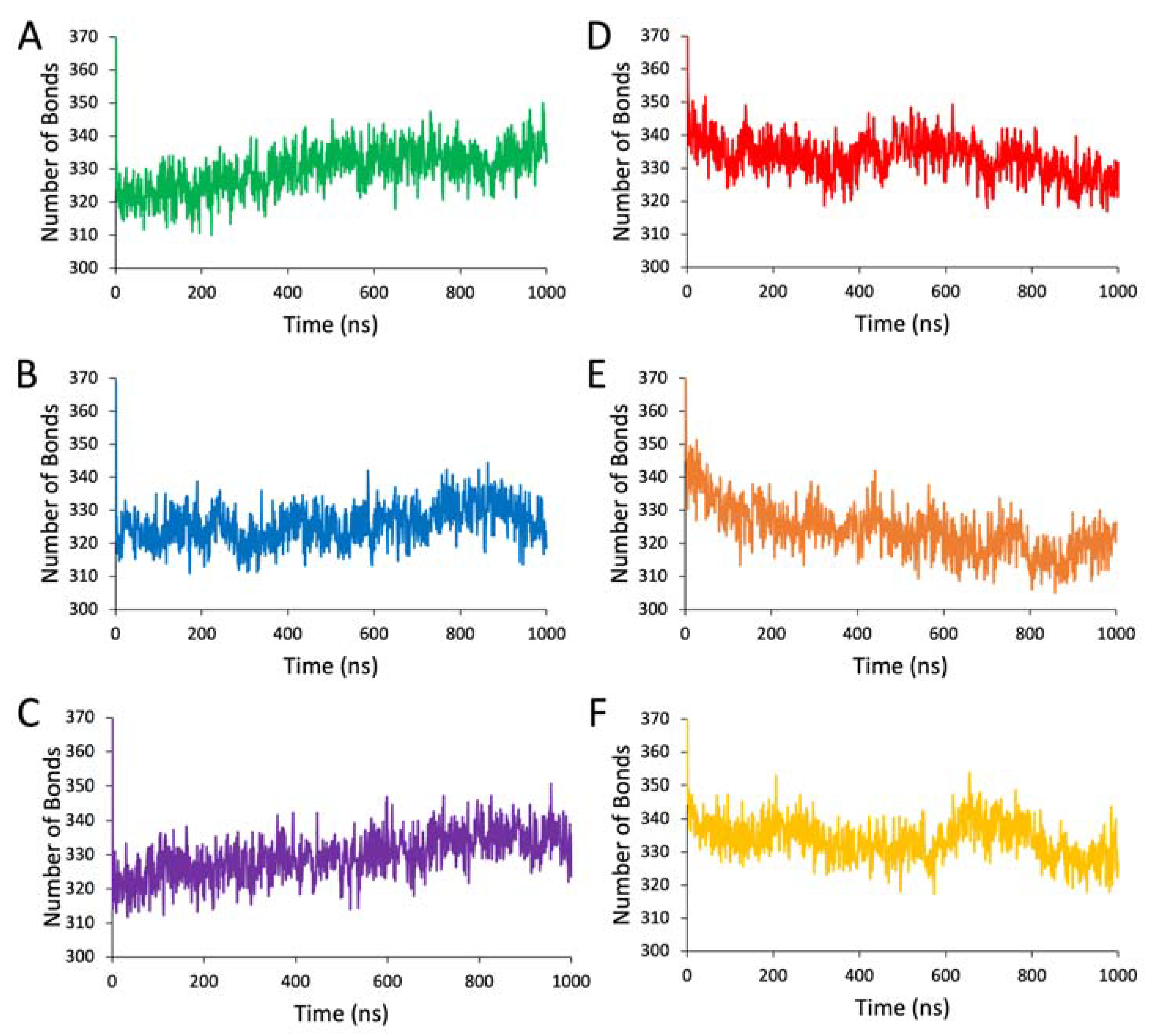
Hydrogen bonding interactions were calculated for each entire protein structure at every nanosecond of the simulations using the ListHboAtom function in YASARA and defining the atom range as the entire protein object. To calculate interatomic hydrophobic contacts with residue 406, all atoms of this residue were selected and designated using the Find Clashes/Contacts tool in UCSF Chimera. Once the atoms are designated and all other atoms are present in the structure, the contact parameters were defined to identify atom pairs with a van der Waals radius overlap less than 0 and greater than −0.4 Å. Intraresidue contacts and contact pairs of 4 or fewer bonds apart were ignored, and intramolecule contacts were included. Ionic interaction energies were derived from the ListInt function in YASARA for specified residue groups within Tyrtr and variants. The distributions of ionic interaction energies were plotted using the seaborn package in Python.
4.7. Measures for MD Quality Assessment
The RMSD of an ensemble of structures from a reference structure was calculated using the RMSD command in YASARA and additional inhouse Python scripts. The ensemble of molecular dynamics snapshots of 1 ns interval was aligned with the reference structure, based on a set of selected Cα atoms, followed by calculations of RMSD values between corresponding atoms of the snapshots and reference structure.
The RMSF was generated using the superimposed molecular dynamics snapshots for every 25 ns interval. These superposed structures were saved as PDB files with their updated coordinates to remove all translational and rotational movements. The RMSF analysis was carried out by loading each superposed structure into YASARA, adding the Cα-atom positions to a calculation table using the AddPosAtom function to calculate the averages and standard deviations using the built-in RMSF command.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}