
Mitochondrial Dynamics in Neurodegenerative Diseases: Unraveling the Role of Fusion and Fission Processes

 , , , , and
, , , , and
Abstract
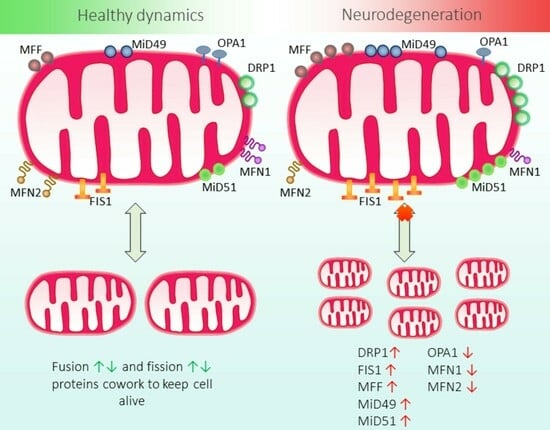
:
{kind=link}
{kind=link}
{kind=link}
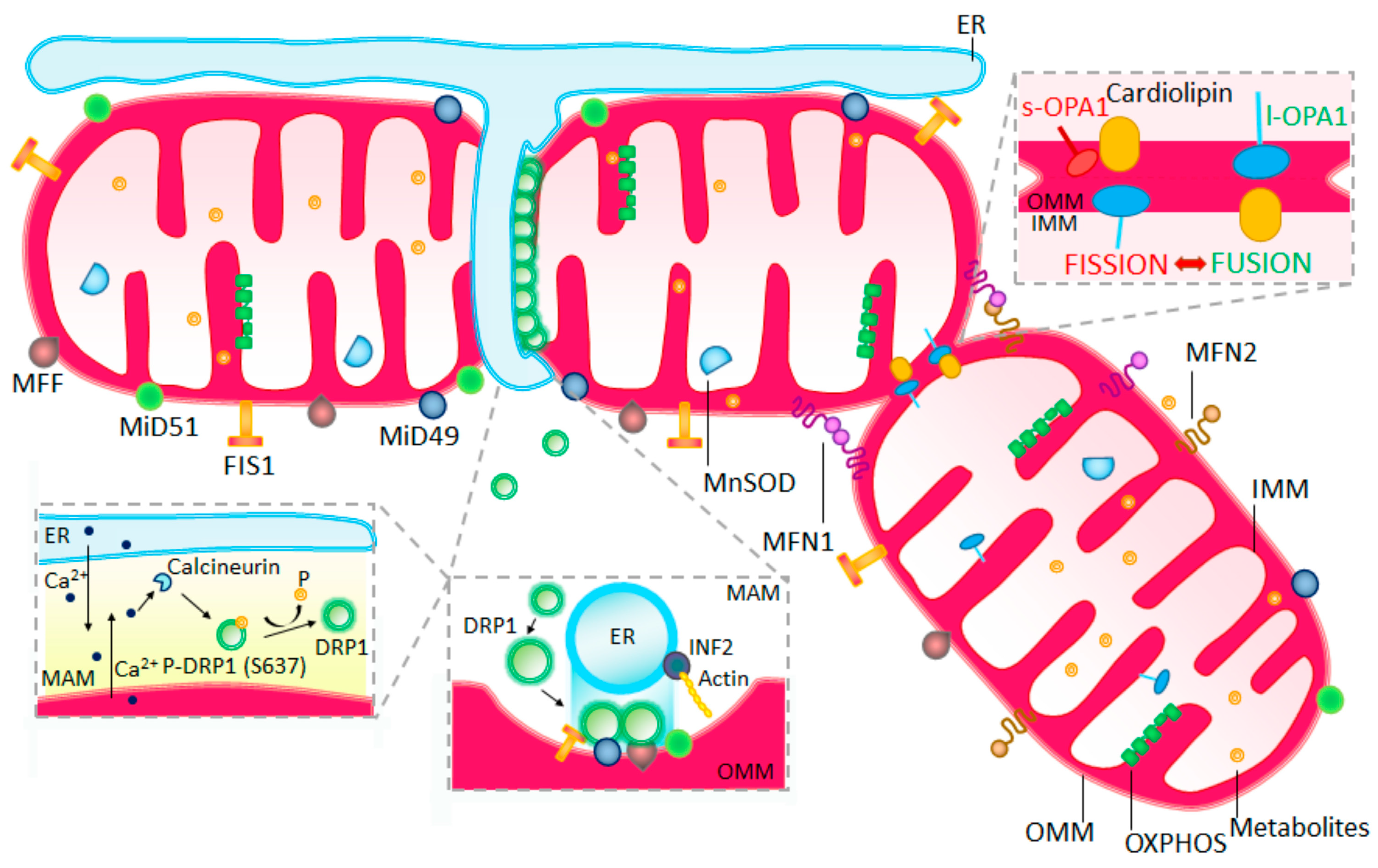{kind=link}
1. Introduction
2. Neurodegenerative Diseases—Gene Mutations
3. Regulatory Proteins in Mitochondrial Fusion and Fission
3.1. Mitochondrial Fission Protein 1 (FIS1)
3.2. Dynamin-Related Protein 1 (DRP1)
3.3. Mitofusins (MFNs)
3.4. Optic Atrophy Protein 1 (OPA1)
4. Mitochondrial Fusion and Fission: Key Tenets of Mitochondrial Quality Control
5. Mitochondrial Fission: Two Pathways
6. Protein Machinery in Mitochondrial Fission
7. Protein Machinery in Mitochondrial Fusion
8. Potential Therapeutic Agents Modulating Mitochondrial Dynamics
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, H.; Zhang, Y.; Yue, J.; Shi, Y.; Xiao, B.; Xiao, W.; Luo, Z. Non-Coding RNAs: The Neuroinflammatory Regulators in Neurodegenerative Diseases. Front. Neurol. 2022, 13, 929290. [Google Scholar] [CrossRef]
- Thomas, E.A. DNA Methylation in Huntington’s Disease: Implications for Transgenerational Effects. Neurosci. Lett. 2016, 625, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Boni, R.; Ali, A.; Shavandi, A.; Clarkson, A.N. Current and Novel Polymeric Biomaterials for Neural Tissue Engineering. J. Biomed. Sci. 2018, 25, 90. [Google Scholar] [CrossRef] [PubMed]
- 2022 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2022, 18, 700–789. [CrossRef]
- Senyilmaz, D.; Virtue, S.; Xu, X.; Tan, C.Y.; Griffin, J.L.; Miller, A.K.; Vidal-Puig, A.; Teleman, A.A. Regulation of Mitochondrial Morphology and Function by Stearoylation of TFR1. Nature 2015, 525, 124–128. [Google Scholar] [CrossRef]
- Strope, T.A.; Birky, C.J.; Wilkins, H.M. The Role of Bioenergetics in Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 9212. [Google Scholar] [CrossRef]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Mégy, C.; Quiles, M.; Cazevieille, C.; et al. ER-Mitochondria Cross-Talk Is Regulated by the Ca2+ Sensor NCS1 and Is Impaired in Wolfram Syndrome. Sci. Signal. 2018, 11, eaaq1380. [Google Scholar] [CrossRef]
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It Is All about Energy. Front. Physiol. 2023, 14, 1114231. [Google Scholar] [CrossRef]
- Patergnani, S.; Morciano, G.; Carinci, M.; Leo, S.; Pinton, P.; Rimessi, A. The “Mitochondrial Stress Responses”: The “Dr. Jekyll and Mr. Hyde” of Neuronal Disorders. Neural Regen. Res. 2022, 17, 2563–2575. [Google Scholar] [CrossRef]
- Lin, M.; Liu, N.; Qin, Z.; Wang, Y. Mitochondrial-Derived Damage-Associated Molecular Patterns Amplify Neuroinflammation in Neurodegenerative Diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.S. Biomarkers in the diagnosis of neurodegenerative diseases. RUDN J. Med. 2022, 26, 431–440. [Google Scholar] [CrossRef]
- Tondo, G.; De Marchi, F. From Biomarkers to Precision Medicine in Neurodegenerative Diseases: Where Are We? J. Clin. Med. 2022, 11, 4515. [Google Scholar] [CrossRef]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Renton, A.E.; Fulton-Howard, B.; Podlesny-Drabiniok, A.; Marcora, E.; Goate, A.M. The Complex Genetic Architecture of Alzheimer’s Disease: Novel Insights and Future Directions. eBioMedicine 2023, 90, 104511. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-Wide Meta-Analysis Identifies New Loci and Functional Pathways Influencing Alzheimer’s Disease Risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Cherian, A.; Divya, K.P.; Vijayaraghavan, A. Parkinson’s Disease—Genetic Cause. Curr. Opin. Neurol. 2023, 36, 292–301. [Google Scholar] [CrossRef]
- Iwaki, H.; Blauwendraat, C.; Leonard, H.L.; Kim, J.J.; Liu, G.; Maple-Grødem, J.; Corvol, J.-C.; Pihlstrøm, L.; van Nimwegen, M.; Hutten, S.J.; et al. Genomewide Association Study of Parkinson’s Disease Clinical Biomarkers in 12 Longitudinal Patients’ Cohorts. Mov. Disord. 2019, 34, 1839–1850. [Google Scholar] [CrossRef]
- Henden, L.; Twine, N.A.; Szul, P.; McCann, E.P.; Nicholson, G.A.; Rowe, D.B.; Kiernan, M.C.; Bauer, D.C.; Blair, I.P.; Williams, K.L. Identity by Descent Analysis Identifies Founder Events and Links SOD1 Familial and Sporadic ALS Cases. NPJ Genom. Med. 2020, 5, 32. [Google Scholar] [CrossRef]
- Fang, T.; Je, G.; Pacut, P.; Keyhanian, K.; Gao, J.; Ghasemi, M. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2066. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of Amyotrophic Lateral Sclerosis: Seeking Therapeutic Targets in the Era of Gene Therapy. J. Hum. Genet. 2023, 68, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Oliveira, S.; Álvarez, I.; Rosas, I.; Menendez-González, M.; Blázquez-Estrada, M.; Aguilar, M.; Corte, D.; Buongiorno, M.; Molina-Porcel, L.; Aldecoa, I.; et al. Intermediate and Expanded HTT Alleles and the Risk for α-Synucleinopathies. Mov. Disord. 2022, 37, 1841–1849. [Google Scholar] [CrossRef]
- Estevez-Fraga, C.; Rodrigues, F.B.; Tabrizi, S.J.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: April 2022. J. Huntingt. Dis. 2022, 11, 105–118. [Google Scholar] [CrossRef]
- Yazdanpanah, N.; Rezaei, N. Chapter 1—Introduction to the Neuroimmunology of Multiple Sclerosis. In Translational Neuroimmunology; Rezaei, N., Yazdanpanah, N., Eds.; Translational Immunology; Academic Press: Cambridge, MA, USA, 2023; Volume 8, pp. 1–9. ISBN 978-0-443-18578-6. [Google Scholar]
- Chiricosta, L.; Blando, S.; D’Angiolini, S.; Gugliandolo, A.; Mazzon, E. A Comprehensive Exploration of the Transcriptomic Landscape in Multiple Sclerosis: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 1448. [Google Scholar] [CrossRef] [PubMed]
- Ferrè, L.; Filippi, M.; Esposito, F. Involvement of Genetic Factors in Multiple Sclerosis. Front. Cell. Neurosci. 2020, 14, 612953. [Google Scholar] [CrossRef]
- Elia, L.; Herting, B.; Alijagic, A.; Buselli, C.; Wong, L.; Morrison, G.; Prado, M.A.; Paulo, J.A.; Gygi, S.P.; Finley, D.; et al. Frontotemporal Dementia Patient Neurons with Progranulin Deficiency Display Protein Dyshomeostasis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Katzeff, J.S.; Bright, F.; Phan, K.; Kril, J.J.; Ittner, L.M.; Kassiou, M.; Hodges, J.R.; Piguet, O.; Kiernan, M.C.; Halliday, G.M.; et al. Biomarker Discovery and Development for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Brain J. Neurol. 2022, 145, 1598–1609. [Google Scholar] [CrossRef]
- Kim, E.-J.; Moon, S.Y.; Kim, H.-J.; Jung, N.-Y.; Lee, S.M.; Kim, Y.E. Semantic Variant Primary Progressive Aphasia with a Pathogenic Variant p.Asp40Gly in the ANXA11 Gene. Eur. J. Neurol. 2022, 29, 3124–3126. [Google Scholar] [CrossRef]
- Kobayashi, A.; Teruya, K.; Matsuura, Y.; Shirai, T.; Nakamura, Y.; Yamada, M.; Mizusawa, H.; Mohri, S.; Kitamoto, T. The Influence of PRNP Polymorphisms on Human Prion Disease Susceptibility: An Update. Acta Neuropathol. 2015, 130, 159–170. [Google Scholar] [CrossRef]
- Bayazid, R.; Orru’, C.; Aslam, R.; Cohen, Y.; Silva-Rohwer, A.; Lee, S.-K.; Occhipinti, R.; Kong, Q.; Shetty, S.; Cohen, M.L.; et al. A Novel Subtype of Sporadic Creutzfeldt–Jakob Disease with PRNP Codon 129MM Genotype and PrP Plaques. Acta Neuropathol. 2023, 146, 121–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.; Tauqir, R. Prion Diseases. Meducator 2023, 1. [Google Scholar] [CrossRef]
- Gareev, I.; Beylerli, O.; Liang, Y.; Lu, E.; Ilyasova, T.; Sufianov, A.; Sufianova, G.; Shi, H.; Ahmad, A.; Yang, G. The Role of Mitochondria-Targeting MiRNAs in Intracerebral Hemorrhage. Curr. Neuropharmacol. 2023, 21, 1065–1080. [Google Scholar] [CrossRef]
- Harrington, J.S.; Ryter, S.W.; Plataki, M.; Price, D.R.; Choi, A.M.K. Mitochondria in Health, Disease, and Aging. Physiol. Rev. 2023, 103, 2349–2422. [Google Scholar] [CrossRef]
- Mishra, Y.; Kaundal, R.K. Role of SIRT3 in Mitochondrial Biology and Its Therapeutic Implications in Neurodegenerative Disorders. Drug Discov. Today 2023, 28, 103583. [Google Scholar] [CrossRef]
- Pinkerton, M.; Barrientos, A. Chapter 24—Diet Restriction-Induced Mitochondrial Signaling and Healthy Aging. In Molecular Nutrition and Mitochondria; Ostojic, S.M., Ed.; Academic Press: Cambridge, MA, USA, 2023; pp. 587–632. ISBN 978-0-323-90256-4. [Google Scholar]
- Zacharioudakis, E.; Gavathiotis, E. Mitochondrial Dynamics Proteins as Emerging Drug Targets. Trends Pharmacol. Sci. 2023, 44, 112–127. [Google Scholar] [CrossRef]
- Yang, B.; Lin, Y.; Shen, Y.-Q. Correcting Abnormal Mitochondrial Dynamics to Facilitate Tumor Treatment. Mitochondrial Commun. 2023, 1, 35–47. [Google Scholar] [CrossRef]
- Mangrulkar, S.V.; Wankhede, N.L.; Kale, M.B.; Upaganlawar, A.B.; Taksande, B.G.; Umekar, M.J.; Anwer, M.K.; Dailah, H.G.; Mohan, S.; Behl, T. Mitochondrial Dysfunction as a Signaling Target for Therapeutic Intervention in Major Neurodegenerative Disease. Neurotox. Res. 2023, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.L.; Tokito, M.K.; Uppala, R.; Sarkar, M.K.; Gudjonsson, J.E.; Holzbaur, E.L.F. NIX Initiates Mitochondrial Fragmentation via DRP1 to Drive Epidermal Differentiation. Cell Rep. 2021, 34, 108689. [Google Scholar] [CrossRef]
- Onoue, K.; Jofuku, A.; Ban-Ishihara, R.; Ishihara, T.; Maeda, M.; Koshiba, T.; Itoh, T.; Fukuda, M.; Otera, H.; Oka, T.; et al. Fis1 Acts as a Mitochondrial Recruitment Factor for TBC1D15 That Is Involved in Regulation of Mitochondrial Morphology. J. Cell Sci. 2013, 126, 176–185. [Google Scholar] [CrossRef]
- Sugioka, R.; Shimizu, S.; Tsujimoto, Y. Fzo1, a Protein Involved in Mitochondrial Fusion, Inhibits Apoptosis*. J. Biol. Chem. 2004, 279, 52726–52734. [Google Scholar] [CrossRef]
- Lee, D.; Kim, J.-E. PDI-Mediated S-Nitrosylation of DRP1 Facilitates DRP1-S616 Phosphorylation and Mitochondrial Fission in CA1 Neurons. Cell Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.L.; Riffle, S.N.; Patel, M.; Marshall, A.; Beasley, H.; Lopez, E.G.; Shao, J.; Vue, Z.; Hinton, A.; Mears, J.A.; et al. DRP1-Mediated Mitochondrial Fission Is Essential to Maintain Cristae Morphology and Bioenergetics. bioRxiv 2022. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible Phosphorylation of Drp1 by Cyclic AMP-Dependent Protein Kinase and Calcineurin Regulates Mitochondrial Fission and Cell Death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial Form and Function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Complementation between Mouse Mfn1 and Mfn2 Protects Mitochondrial Fusion Defects Caused by CMT2A Disease Mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef]
- Hoppins, S.; Edlich, F.; Cleland, M.M.; Banerjee, S.; McCaffery, J.M.; Youle, R.J.; Nunnari, J. The Soluble Form of Bax Regulates Mitochondrial Fusion via MFN2 Homotypic Complexes. Mol. Cell 2011, 41, 150–160. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial Dynamics Regulates Migration and Invasion of Breast Cancer Cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Mann, J.P.; Tabara, L.C.; Alvarez-Guaita, A.; Dong, L.; Haider, A.; Lim, K.; Tandon, P.; Minchin, J.E.N.; O’Rahilly, S.; Patel, S.; et al. Loss of Mfn1 but Not Mfn2 Enhances Adipogenesis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana–Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 Mutations in Charcot–Marie–Tooth Disease Alter Mitochondria-Associated ER Membrane Function but Do Not Impair Bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D. OPA1 Processing Controls Mitochondrial Fusion and Is Regulated by MRNA Splicing, Membrane Potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, Encoding a UGO1-like Protein, Cause an Optic Atrophy Spectrum Disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Piplani, H.; Sin, J.; Sawaged, S.; Hamid, S.M.; Taylor, D.J.; de Freitas Germano, J. At the Heart of Mitochondrial Quality Control: Many Roads to the Top. Cell. Mol. Life Sci. 2021, 78, 3791–3801. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D. Mitochondrial Dynamics and Inheritance during Cell Division, Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Dulac, M.; Leduc-Gaudet, J.-P.; Reynaud, O.; Ayoub, M.-B.; Guérin, A.; Finkelchtein, M.; Hussain, S.N.; Gouspillou, G. Drp1 Knockdown Induces Severe Muscle Atrophy and Remodelling, Mitochondrial Dysfunction, Autophagy Impairment and Denervation. J. Physiol. 2020, 598, 3691–3710. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and Selective Fusion Govern Mitochondrial Segregation and Elimination by Autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Song, Y.; Xu, Y.; Liu, Y.-Y.; Gao, J.; Feng, L.; Zhang, Y.; Shi, L.; Zhang, M.; Guo, D.; Qi, B.; et al. Mitochondrial Quality Control in the Maintenance of Cardiovascular Homeostasis: The Roles and Interregulation of UPS, Mitochondrial Dynamics and Mitophagy. Oxid. Med. Cell. Longev. 2021, 2021, 3960773. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin Is Recruited Selectively to Impaired Mitochondria and Promotes Their Autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jia, L.; Zhao, C.; Wang, H.; Dai, Z.; Jing, Y.; Jiang, B.; Xin, S. Mitochondrial Quality Control in Abdominal Aortic Aneurysm: From Molecular Mechanisms to Therapeutic Strategies. FASEB J. 2023, 37, e22969. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, A.; Oh, K.-J.; Lee, S.C.; Kim, W.K.; Bae, K.-H. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4924. [Google Scholar] [CrossRef]
- Duan, X.; Wei, Y.; Zhang, M.; Zhang, W.; Huang, Y.; Zhang, Y.-H. PI4P-Containing Vesicles from Golgi Contribute to Mitochondrial Division by Coordinating with Polymerized Actin. Int. J. Mol. Sci. 2023, 24, 6593. [Google Scholar] [CrossRef]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Lobet, E.; Willemart, K.; Ninane, N.; Demazy, C.; Sedzicki, J.; Lelubre, C.; De Bolle, X.; Renard, P.; Raes, M.; Dehio, C.; et al. Mitochondrial Fragmentation Affects Neither the Sensitivity to TNFα-Induced Apoptosis of Brucella-Infected Cells nor the Intracellular Replication of the Bacteria. Sci. Rep. 2018, 8, 5173. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Park, G.; Shin, E.; Shin, S.W.; Jana, B.; Jin, S.; Kim, S.; Wang, H.; Kwak, S.K.; Xu, B.; et al. Intramitochondrial Co-Assembly between ATP and Nucleopeptides Induces Cancer Cell Apoptosis. Chem. Sci. 2022, 13, 6197–6204. [Google Scholar] [CrossRef]
- Lee, K.-M.; Yun, J. Mitophagy Stimulation as a Novel Strategy for the Treatment of Mitochondrial Diseases. J. Genet. Med. 2022, 19, 49–56. [Google Scholar] [CrossRef]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct Fission Signatures Predict Mitochondrial Degradation or Biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef]
- Jones, M.D.; Naylor, K. Simple to Complex: The Role of Actin and Microtubules in Mitochondrial Dynamics in Amoeba, Yeast, and Mammalian Cells. Int. J. Mol. Sci. 2022, 23, 9402. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Mercado-Ayón, E.; Clark, E.; Lynch, D.; Lin, H. Drp1-Dependent Peptide Reverse Mitochondrial Fragmentation, a Homeostatic Response in Friedreich Ataxia. Pharmacol. Res. Perspect. 2021, 9, e00755. [Google Scholar] [CrossRef]
- Kalia, R.; Wang, R.Y.-R.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural Basis of Mitochondrial Receptor Binding and Constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 Recruitment in Mitochondrial Fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Mozdy, A.D.; McCaffery, J.M.; Shaw, J.M. Dnm1p Gtpase-Mediated Mitochondrial Fission Is a Multi-Step Process Requiring the Novel Integral Membrane Component Fis1p. J. Cell Biol. 2000, 151, 367–380. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–Lysosome Contacts Regulate Mitochondrial Fission via RAB7 GTP Hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Chang, C.-R.; Blackstone, C. Dynamic Regulation of Mitochondrial Fission through Modification of the Dynamin-Related Protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Peng, X.-D.; Qian, X.-J.; Zhang, K.-M.; Huang, X.; Chen, Y.-H.; Li, Y.-T.; Feng, G.-K.; Zhang, H.-L.; Xu, X.-L.; et al. Fis1 Phosphorylation by Met Promotes Mitochondrial Fission and Hepatocellular Carcinoma Metastasis. Signal Transduct. Target. Ther. 2021, 6, 401. [Google Scholar] [CrossRef] [PubMed]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. Mitochondrial Fission Protein 1: Emerging Roles in Organellar Form and Function in Health and Disease. Front. Endocrinol. 2021, 12, 660095. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-Related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Recasens, A.; Humphrey, S.J.; Ellis, M.; Hoque, M.; Abbassi, R.H.; Chen, B.; Longworth, M.; Needham, E.J.; James, D.E.; Johns, T.G.; et al. Global Phosphoproteomics Reveals DYRK1A Regulates CDK1 Activity in Glioblastoma Cells. Cell Death Discov. 2021, 7, 81. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by Calcineurin Regulates Translocation of Drp1 to Mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Boncompagni, C.; Ramaccini, D.; Pedriali, G.; Bouhamida, E.; Tremoli, E.; Giorgi, C.; Pinton, P. Comprehensive Analysis of Mitochondrial Dynamics Alterations in Heart Diseases. Int. J. Mol. Sci. 2023, 24, 3414. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-β Overproduction Causes Abnormal Mitochondrial Dynamics via Differential Modulation of Mitochondrial Fission/Fusion Proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Mitochondrial Division Inhibitor 1 Reduces Dynamin-Related Protein 1 and Mitochondrial Fission Activity. Hum. Mol. Genet. 2019, 28, 177–199. [Google Scholar] [CrossRef]
- Park, S.; Yang, H.; Go, H.; Kim, H.; Bae, H. Alpha-Synuclein-Specific Regulatory T Cells Ameliorate Parkinson’s Disease Progression in Mice. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W. Mitochondrial Fission and Fusion Factors Reciprocally Orchestrate Mitophagic Culling in Mouse Hearts and Cultured Fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef]
- Shi, W.; Tan, C.; Liu, C.; Chen, D. Mitochondrial Fission Mediated by Drp1-Fis1 Pathway and Neurodegenerative Diseases. Rev. Neurosci. 2023, 34, 275–294. [Google Scholar] [CrossRef]
- Shen, Y.; Jiang, W.-L.; Li, X.; Cao, A.-L.; Li, D.; Li, S.-Z.; Yang, J.; Qian, J. Mitochondrial Dynamics in Neurological Diseases: A Narrative Review. Ann. Transl. Med. 2023, 11, 264. [Google Scholar] [CrossRef]
- Li, Y.; Liang, J.; Tian, X.; Chen, Q.; Zhu, L.; Wang, H.; Liu, Z.; Dai, X.; Bian, C.; Sun, C. Intermittent Fasting Promotes Adipocyte Mitochondrial Fusion through Sirt3-Mediated Deacetylation of Mdh2. Br. J. Nutr. 2023, 1–14. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of Fusion Results in Mitochondrial Heterogeneity and Dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dang, X.; Franco, A.; Dorn, G.W. Reciprocal Regulation of Mitofusin 2-Mediated Mitophagy and Mitochondrial Fusion by Different PINK1 Phosphorylation Events. Front. Cell Dev. Biol. 2022, 10, 868465. [Google Scholar] [CrossRef] [PubMed]
- Pyakurel, A.; Savoia, C.; Hess, D.; Scorrano, L. Extracellular Regulated Kinase Phosphorylates Mitofusin 1 to Control Mitochondrial Morphology and Apoptosis. Mol. Cell 2015, 58, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-L.; Lee, H.-L.; Yang, S.-F.; Wang, S.-W.; Lin, C.-P.; Hsieh, Y.-H.; Chiou, H.-L. Protodioscin Induces Mitochondrial Apoptosis of Human Hepatocellular Carcinoma Cells Through Eliciting ER Stress-Mediated IP3R Targeting Mfn1/Bak Expression. J. Hepatocell. Carcinoma 2022, 9, 327–341. [Google Scholar] [CrossRef]
- Sloat, S.R.; Whitley, B.N.; Engelhart, E.A.; Hoppins, S. Identification of a Mitofusin Specificity Region That Confers Unique Activities to Mfn1 and Mfn2. Mol. Biol. Cell 2019, 30, 2309–2319. [Google Scholar] [CrossRef]
- Lee, J.E.; Seo, B.J.; Han, M.J.; Hong, Y.J.; Hong, K.; Song, H.; Lee, J.W.; Do, J.T. Changes in the Expression of Mitochondrial Morphology-Related Genes during the Differentiation of Murine Embryonic Stem Cells. Stem Cells Int. 2020, 2020, e9369268. [Google Scholar] [CrossRef]
- Cao, Y.-L.; Meng, S.; Chen, Y.; Feng, J.-X.; Gu, D.-D.; Yu, B.; Li, Y.-J.; Yang, J.-Y.; Liao, S.; Chan, D.C.; et al. MFN1 Structures Reveal Nucleotide-Triggered Dimerization Critical for Mitochondrial Fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-Dependent Cristae Modulation Is Essential for Cellular Adaptation to Metabolic Demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two Forms of Opa1 Cooperate to Complete Fusion of the Mitochondrial Inner-Membrane. eLife 2020, 9, e50973. [Google Scholar] [CrossRef]
- Wang, R.; Mishra, P.; Garbis, S.D.; Moradian, A.; Sweredoski, M.J.; Chan, D.C. Identification of New OPA1 Cleavage Site Reveals That Short Isoforms Regulate Mitochondrial Fusion. Mol. Biol. Cell 2021, 32, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Liu, X.; Liesa, M.; Wikstrom, J.D.; Molina, A.J.A.; Las, G.; Yaniv, G.; Hajnóczky, G.; Shirihai, O.S. Biophysical Properties of Mitochondrial Fusion Events in Pancreatic β-Cells and Cardiac Cells Unravel Potential Control Mechanisms of Its Selectivity. Am. J. Physiol.-Cell Physiol. 2010, 299, C477–C487. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vázquez, J.; Enríquez, J.A.; Gerle, C.; et al. The Cristae Modulator Optic Atrophy 1 Requires Mitochondrial ATP Synthase Oligomers to Safeguard Mitochondrial Function. Nat. Commun. 2018, 9, 3399. [Google Scholar] [CrossRef] [PubMed]
- Saita, S.; Ishihara, T.; Maeda, M.; Iemura, S.; Natsume, T.; Mihara, K.; Ishihara, N. Distinct Types of Protease Systems Are Involved in Homeostasis Regulation of Mitochondrial Morphology via Balanced Fusion and Fission. Genes Cells 2016, 21, 408–424. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and Roles of Mitophagy in Neurodegenerative Diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xue, H.; Yue, Y.; Hao, S.; Huang, S.-H.; Zhang, Z. Role of Mitophagy in the Neurodegenerative Diseases and Its Pharmacological Advances: A Review. Front. Mol. Neurosci. 2022, 15, 1014251. [Google Scholar] [CrossRef]
- Lin, L.; Lee, J.H.; Bongmba, O.Y.N.; Ma, X.; Zhu, X.; Sheikh-Hamad, D.; Sun, Y. The Suppression of Ghrelin Signaling Mitigates Age-Associated Thermogenic Impairment. Aging 2014, 6, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sibilla, C.; Liu, R.; Yun, J.; Hay, B.A.; Blackstone, C.; Chan, D.C.; Harvey, R.J.; Guo, M. Clueless/CLUH Regulates Mitochondrial Fission by Promoting Recruitment of Drp1 to Mitochondria. Nat. Commun. 2022, 13, 1582. [Google Scholar] [CrossRef]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Guan, P.; Ye, X.; Lu, Y.; Hang, Y.; Su, Y.; Hu, W. SOCS6 Promotes Mitochondrial Fission and Cardiomyocyte Apoptosis and Is Negatively Regulated by Quaking-Mediated MiR-19b. Oxid. Med. Cell. Longev. 2022, 2022, 1121323. [Google Scholar] [CrossRef] [PubMed]
- Colitti, M.; Montanari, T. Brain-Derived Neurotrophic Factor Modulates Mitochondrial Dynamics and Thermogenic Phenotype on 3T3-L1 Adipocytes. Tissue Cell 2020, 66, 101388. [Google Scholar] [CrossRef]
- Kim, T.W.; Jeong, J.-H.; Hong, S.-C. The Impact of Sleep and Circadian Disturbance on Hormones and Metabolism. Int. J. Endocrinol. 2015, 2015, e591729. [Google Scholar] [CrossRef]
- Sun, X.; Ran, D.; Zhao, X.; Huang, Y.; Long, S.; Liang, F.; Guo, W.; Nucifora, F.C.; Gu, H.; Lu, X.; et al. Melatonin Attenuates HLRRK2-Induced Sleep Disturbances and Synaptic Dysfunction in a Drosophila Model of Parkinson’s Disease. Mol. Med. Rep. 2016, 13, 3936–3944. [Google Scholar] [CrossRef]
- Mitochondrial Dynamics in the Rat Dorsal Raphe Nucleus Underlie Sleep Pressure and Sleep Structure Regulation. Available online: https://www.researchsquare.com (accessed on 1 August 2023).
- Ding, M.; Feng, N.; Tang, D.; Feng, J.; Li, Z.; Jia, M.; Liu, Z.; Gu, X.; Wang, Y.; Fu, F.; et al. Melatonin Prevents Drp1-Mediated Mitochondrial Fission in Diabetic Hearts through SIRT1-PGC1α Pathway. J. Pineal Res. 2018, 65, e12491. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin Attenuates Myocardial Ischemia-Reperfusion Injury via Improving Mitochondrial Fusion/Mitophagy and Activating the AMPK-OPA1 Signaling Pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef]
- Arribas, R.L.; Romero, A.; Egea, J.; de los Ríos, C. Modulation of Serine/Threonine Phosphatases by Melatonin: Therapeutic Approaches in Neurodegenerative Diseases. Br. J. Pharmacol. 2018, 175, 3220–3229. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, C.; Wang, J.; Huang, X.; Yu, H.; Li, S.; Li, S.; Zhang, Z.; Liu, J.; Yang, X.; et al. Melatonin Ameliorates Cognitive Deficits through Improving Mitophagy in a Mouse Model of Alzheimer’s Disease. J. Pineal Res. 2021, 71, e12774. [Google Scholar] [CrossRef]
- Mihajlovic, M.; Vinken, M. Mitochondria as the Target of Hepatotoxicity and Drug-Induced Liver Injury: Molecular Mechanisms and Detection Methods. Int. J. Mol. Sci. 2022, 23, 3315. [Google Scholar] [CrossRef]
- Li, R.-L.; Wang, L.-Y.; Duan, H.-X.; Zhang, Q.; Guo, X.; Wu, C.; Peng, W. Regulation of Mitochondrial Dysfunction Induced Cell Apoptosis Is a Potential Therapeutic Strategy for Herbal Medicine to Treat Neurodegenerative Diseases. Front. Pharmacol. 2022, 13, 937289. [Google Scholar] [CrossRef] [PubMed]
- Garza, S.; Chen, L.; Galano, M.; Cheung, G.; Sottas, C.; Li, L.; Li, Y.; Zirkin, B.R.; Papadopoulos, V. Mitochondrial Dynamics, Leydig Cell Function, and Age-Related Testosterone Deficiency. FASEB J. 2022, 36, e22637. [Google Scholar] [CrossRef]
- Au, N.P.B.; Chand, R.; Kumar, G.; Asthana, P.; Tam, W.Y.; Tang, K.M.; Ko, C.-C.; Ma, C.H.E. A Small Molecule M1 Promotes Optic Nerve Regeneration to Restore Target-Specific Neural Activity and Visual Function. Proc. Natl. Acad. Sci. USA 2022, 119, e2121273119. [Google Scholar] [CrossRef]
- Budi, Y.P.; Hsu, M.-C.; Lin, Y.-C.; Lee, Y.-J.; Chiu, H.-Y.; Chiu, C.-H.; Jiang, Y.-F. The Injections of Mitochondrial Fusion Promoter M1 during Proestrus Disrupt the Progesterone Secretion and the Estrous Cycle in the Mouse. Sci. Rep. 2023, 13, 2392. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Sumegi, K.; Fekete, K.; Hocsak, E.; Debreceni, B.; Setalo, G.; Kovacs, K.; Deres, L.; Kengyel, A.; Kovacs, D.; et al. Activation of Mitochondrial Fusion Provides a New Treatment for Mitochondria-Related Diseases. Biochem. Pharmacol. 2018, 150, 86–96. [Google Scholar] [CrossRef]
- Zeng, K.-W.; Wang, J.-K.; Wang, L.-C.; Guo, Q.; Liu, T.-T.; Wang, F.-J.; Feng, N.; Zhang, X.-W.; Liao, L.-X.; Zhao, M.-M.; et al. Small Molecule Induces Mitochondrial Fusion for Neuroprotection via Targeting CK2 without Affecting Its Conventional Kinase Activity. Signal Transduct. Target. Ther. 2021, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an Activator of Sirtuin-3 (SIRT3) Preserves Mitochondria and Protects the Heart from Doxorubicin-Induced Cardiomyopathy in Mice. Oncotarget 2017, 8, 34082–34098. [Google Scholar] [CrossRef]
- Liu, C.; Han, Y.; Gu, X.; Li, M.; Du, Y.; Feng, N.; Li, J.; Zhang, S.; Maslov, L.N.; Wang, G.; et al. Paeonol Promotes Opa1-Mediated Mitochondrial Fusion via Activating the CK2α-Stat3 Pathway in Diabetic Cardiomyopathy. Redox Biol. 2021, 46, 102098. [Google Scholar] [CrossRef]
- Qian, Y.; Zhao, M.; Han, Q.; Wang, J.; Liao, L.; Yang, H.; Liu, D.; Tu, P.; Liang, H.; Zeng, K. Pharmacologically Targeting Molecular Motor Promotes Mitochondrial Fission for Anti-Cancer. Acta Pharm. Sin. B 2021, 11, 1853–1866. [Google Scholar] [CrossRef]
- Chidipi, B.; Shah, S.I.; Reiser, M.; Kanithi, M.; Garces, A.; Cha, B.J.; Ullah, G.; Noujaim, S.F. All-Trans Retinoic Acid Increases DRP1 Levels and Promotes Mitochondrial Fission. Cells 2021, 10, 1202. [Google Scholar] [CrossRef]
- Xu, H.; Liu, Y.-Y.; Li, L.-S.; Liu, Y.-S. Sirtuins at the Crossroads between Mitochondrial Quality Control and Neurodegenerative Diseases: Structure, Regulation, Modifications, and Modulators. Aging Dis. 2023, 14, 794–824. [Google Scholar] [CrossRef] [PubMed]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a Cell-Permeable Inhibitor of Dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef]
- Shen, F.; Gai, J.; Xing, J.; Guan, J.; Fu, L.; Li, Q. Dynasore Suppresses Proliferation and Induces Apoptosis of the Non-Small-Cell Lung Cancer Cell Line A549. Biochem. Biophys. Res. Commun. 2018, 495, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chai, E.; Mou, Y.; Roda, R.H.; Blackstone, C.; Li, X.-J. Inhibiting Mitochondrial Fission Rescues Degeneration in Hereditary Spastic Paraplegia Neurons. Brain J. Neurol. 2022, 145, 4016–4031. [Google Scholar] [CrossRef] [PubMed]
- Rios, L.; Pokhrel, S.; Li, S.-J.; Heo, G.; Haileselassie, B.; Mochly-Rosen, D. Targeting an Allosteric Site in Dynamin-Related Protein 1 to Inhibit Fis1-Mediated Mitochondrial Dysfunction. Nat. Commun. 2023, 14, 4356. [Google Scholar] [CrossRef]
- Yang, H.; Wang, L.; Zang, C.; Yang, X.; Bao, X.; Shang, J.; Zhang, Z.; Liu, H.; Ju, C.; Li, F.; et al. Squamosamide Derivative FLZ Diminishes Aberrant Mitochondrial Fission by Inhibiting Dynamin-Related Protein 1. Front. Pharmacol. 2021, 12, 588003. [Google Scholar] [CrossRef]
- Yin, X.; Li, Z.; Lyu, C.; Wang, Y.; Ding, S.; Ma, C.; Wang, J.; Cui, S.; Wang, J.; Guo, D.; et al. Induced Effect of Zinc Oxide Nanoparticles on Human Acute Myeloid Leukemia Cell Apoptosis by Regulating Mitochondrial Division. IUBMB Life 2022, 74, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 (Mdivi-1) Protects Neurons against Excitotoxicity through the Modulation of Mitochondrial Function and Intracellular Ca2+ Signaling. Front. Mol. Neurosci. 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Song, L.; Yu, J.; Huang, F.; Li, Y.; Ma, C. Mdivi-1: A Promising Drug and Its Underlying Mechanisms in the Treatment of Neurodegenerative Diseases. Histol. Histopathol. 2022, 37, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor Mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor That Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef]
- Bordt, E.A.; Zhang, N.; Waddell, J.; Polster, B.M. The Non-Specific Drp1 Inhibitor Mdivi-1 Has Modest Biochemical Antioxidant Activity. Antioxidants 2022, 11, 450. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; You, M.; Fan, C.; Rong, R.; Li, H.; Xia, X. Pathologically High Intraocular Pressure Induces Mitochondrial Dysfunction through Drp1 and Leads to Retinal Ganglion Cell PANoptosis in Glaucoma. Redox Biol. 2023, 62, 102687. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics–Fusion, Fission, Movement, and Mitophagy–in Neurodegenerative Diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 Inhibitor Diminishes Aberrant Mitochondrial Fission and Neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef]
- Hung, C.H.-L.; Cheng, S.S.-Y.; Cheung, Y.-T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.-S.; Lee, S.M.-Y.; Chang, R.C.-C. A Reciprocal Relationship between Reactive Oxygen Species and Mitochondrial Dynamics in Neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef]
- Downs, E.; Bottrell, A.D.; Naylor, K. Identifying the Effects of Reactive Oxygen Species on Mitochondrial Dynamics and Cytoskeleton Stability in Dictyostelium Discoideum. Cells 2021, 10, 2147. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grel, H.; Woznica, D.; Ratajczak, K.; Kalwarczyk, E.; Anchimowicz, J.; Switlik, W.; Olejnik, P.; Zielonka, P.; Stobiecka, M.; Jakiela, S. Mitochondrial Dynamics in Neurodegenerative Diseases: Unraveling the Role of Fusion and Fission Processes. Int. J. Mol. Sci. 2023, 24, 13033. https://doi.org/10.3390/ijms241713033
Grel H, Woznica D, Ratajczak K, Kalwarczyk E, Anchimowicz J, Switlik W, Olejnik P, Zielonka P, Stobiecka M, Jakiela S. Mitochondrial Dynamics in Neurodegenerative Diseases: Unraveling the Role of Fusion and Fission Processes. International Journal of Molecular Sciences. 2023; 24(17):13033. https://doi.org/10.3390/ijms241713033
Chicago/Turabian StyleGrel, Hubert, Damian Woznica, Katarzyna Ratajczak, Ewelina Kalwarczyk, Julia Anchimowicz, Weronika Switlik, Piotr Olejnik, Piotr Zielonka, Magdalena Stobiecka, and Slawomir Jakiela. 2023. "Mitochondrial Dynamics in Neurodegenerative Diseases: Unraveling the Role of Fusion and Fission Processes" International Journal of Molecular Sciences 24, no. 17: 13033. https://doi.org/10.3390/ijms241713033