
Elucidating the Racemization Mechanism of Aliphatic and Aromatic Amino Acids by In Silico Tools
 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
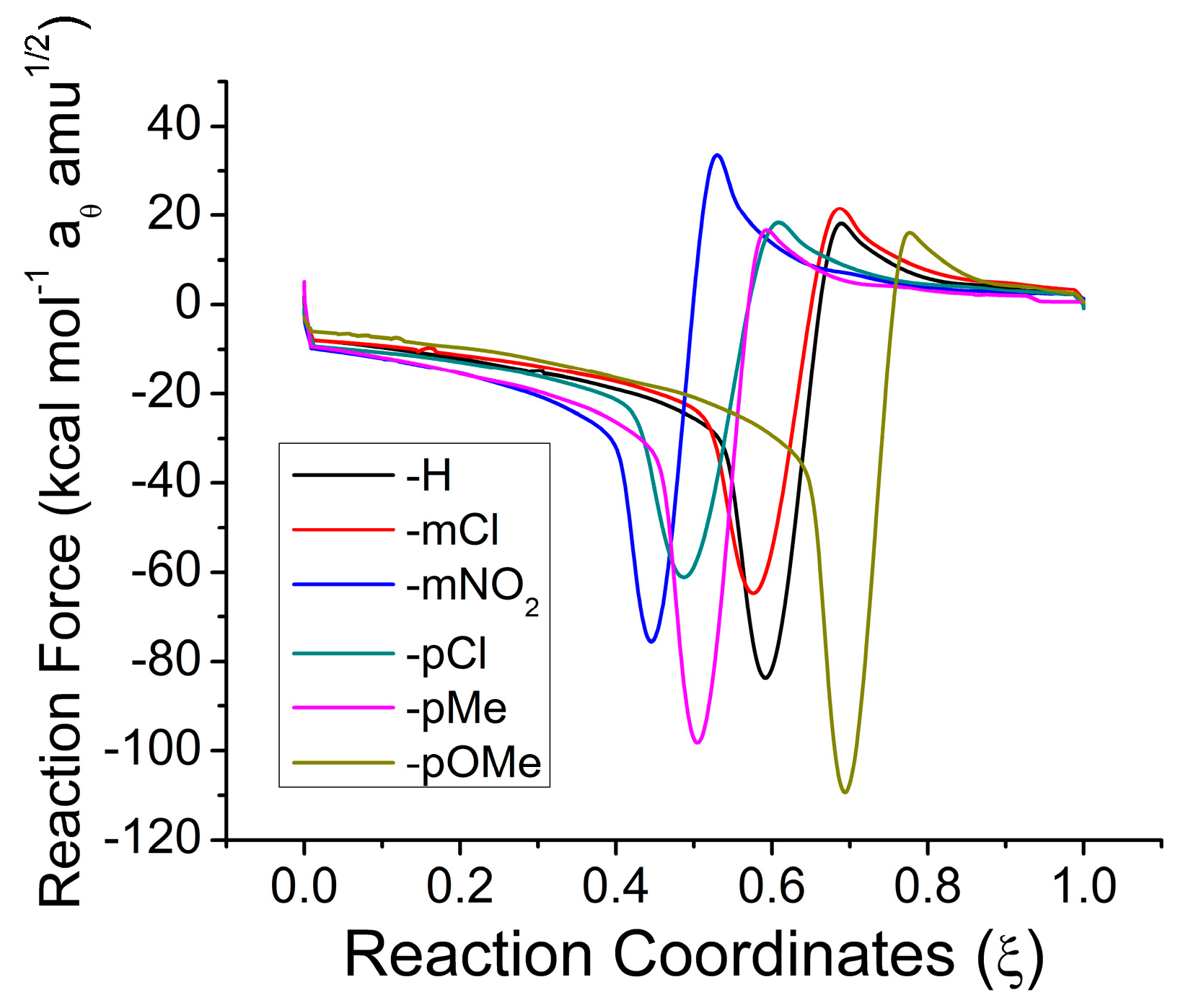
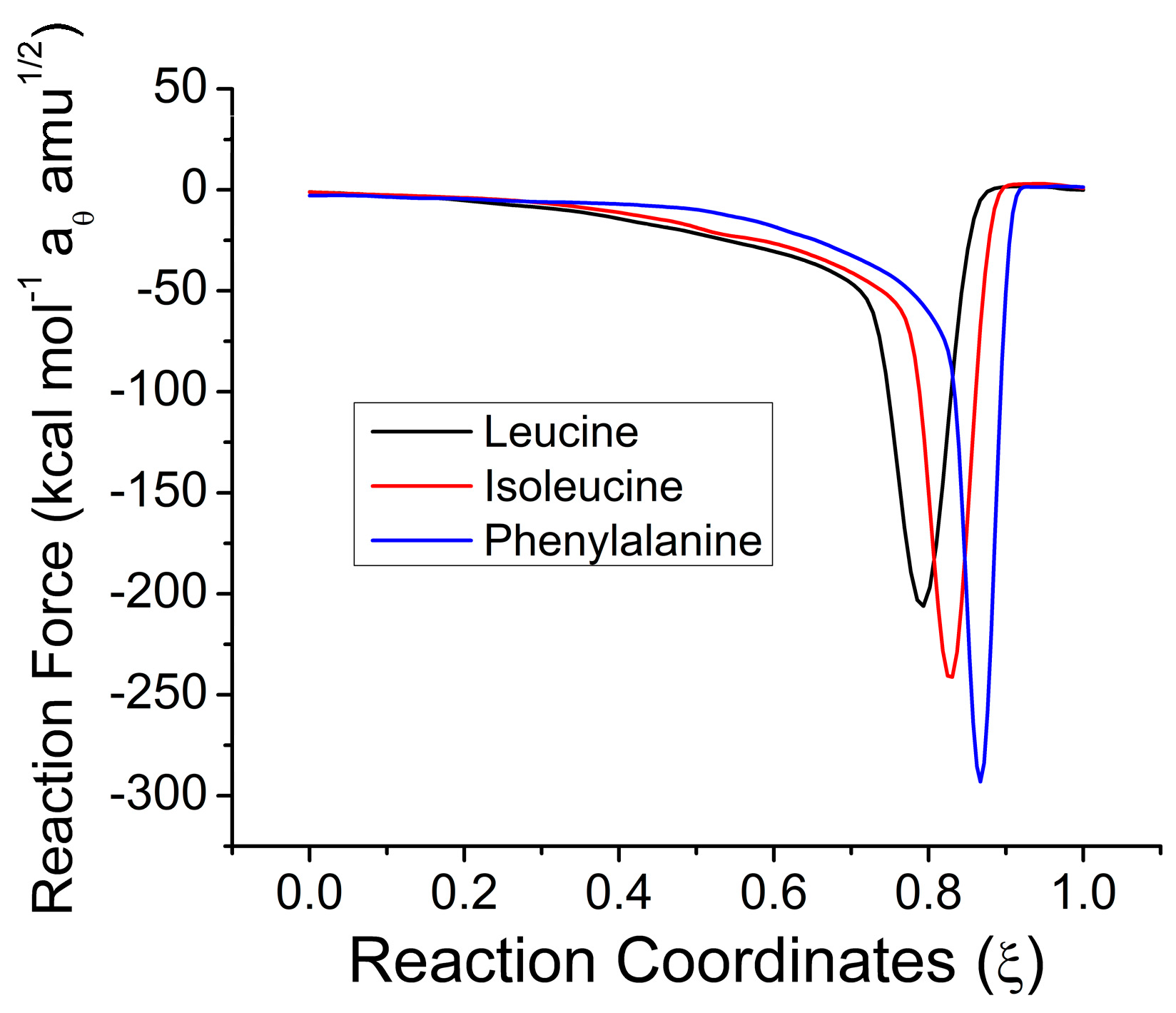
2.1. Reaction Energy Results
2.2. Geometric Results
2.3. Electronic Results
3. Discussion
4. Materials and Methods
4.1. Data Set: Aromatic and Aliphatic Amino Acids
4.2. Computational Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ballard, A.; Narduolo, S.; Ahmad, H.O.; Cosgrove, D.A.; Leach, A.G.; Buurma, N.J. The problem of racemization in drug discovery and tools to predict it. Expert Opin. Drug Discov. 2019, 14, 527–539. [Google Scholar] [CrossRef]
- Marcone, G.L.; Rosini, E.; Crespi, E.; Pollegioni, L. D-amino acids in foods. Appl. Microbiol. Biotechnol. 2020, 104, 555–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Leinisch, F.; Greco, I.; Zhang, W.; Shu, N.; Chuang, C.Y.; Lund, M.N.; Davies, M.J. Characterisation and quantification of protein oxidative modifications and amino acid racemisation in powdered infant milk formula. Free Radic. Res. 2019, 53, 68–81. [Google Scholar] [CrossRef]
- Smith, S.W. Chiral toxicology: It’s the same thing only different. Toxicol. Sci. 2009, 110, 4–30. [Google Scholar] [CrossRef]
- Bastings, J.J.A.J.; van Eijk, H.M.; Damink, S.W.O.; Rensen, S.S. D-amino acids in health and disease: A focus on cancer. Nutrients 2019, 11, 2205. [Google Scholar] [CrossRef] [Green Version]
- Mann, L.; Lang, M.; Schulze, P.; Halz, J.H.; Csuk, R.; Hoenke, S.; Seidel, R.W.; Richter, A. Racemization-free synthesis of Nα-2-thiophenoyl-phenylalanine-2-morpholinoanilide enantiomers and their antimycobacterial activity. Amino Acids 2021, 53, 1187–1196. [Google Scholar] [CrossRef]
- Yan, T.; Feringa, B.L.; Barta, K. Direct catalytic N-alkylation of α-amino acid esters and amides using alcohols with high retention of stereochemistry. ChemSusChem 2021, 14, 2303–2307. [Google Scholar] [CrossRef]
- Hättasch, T.; Schmuck, C.; Niemeyer, J. Triazole groups as biomimetic amide groups in peptides can trigger racemization. Arkivoc 2021, 2021, 185–196. [Google Scholar] [CrossRef]
- Harriehausen, I.; Bollmann, J.; Carneiro, T.; Bettenbrock, K.; Seidel-Morgenstern, A. Characterization of an immobilized amino acid racemase for potential application in enantioselective chromatographic resolution processes. Catalysts 2021, 11, 726. [Google Scholar] [CrossRef]
- Huang, H.; Jin, Y.; Shirbhate, M.E.; Kang, D.; Choi, M.; Chen, Q.; Kim, Y.; Kim, S.J.; Byun, I.S.; Wang, M.; et al. Enantioselective extraction of unprotected amino acids coupled with racemization. Nat. Commun. 2021, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Dyakin, V.V.; Wisniewski, T.M.; Lajtha, A. Racemization in post-translational modifications relevance to protein aging, aggregation and neurodegeneration: Tip of the iceberg. Symmetry 2021, 13, 455. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, S.A.; Rincón, L.; Torres, F.J.; Rodríguez, V.; Mora, J.R. A computational study of the reaction mechanism involved in the fast cleavage of an unconstrained amide bond assisted by an amine intramolecular nucleophilic attack. J. Comput. Chem. 2021, 42, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Meinard, C.; Bruneau, P.; Perronnet, J. High-performance liquid chromatograph coupled with two detectors: A UV spectrometer and a polarimeter. Example in the field of pyrethroids: Identification of enantiomers. J. Chromatogr. 1985, 349, 109–116. [Google Scholar] [CrossRef]
- Broberg, A.; Nord, C.; Levenfors, J.J.; Bjerketorp, J.; Guss, B.; Öberg, B. In-peptide amino acid racemization via inter-residue oxazoline intermediates during acidic hydrolysis. Amino Acids 2021, 53, 323–331. [Google Scholar] [CrossRef]
- Brewer, G.; Brewer, C.; Butcher, R.J.; Zemba, M. Structural evidence of a ketimine as the product of an amino acid with an aldehyde. An intermediate in the racemization and transamination of amino acids. Inorg. Chem. Commun. 2016, 64, 35–38. [Google Scholar] [CrossRef]
- Smith, G.G.; Sivakua, T. Mechanism of the racemization of amino acids. Kinetics of racemization of arylglycines. J. Org. Chem. 1983, 48, 627–634. [Google Scholar] [CrossRef]
- Dudek, W.M.; Ostrowski, S.; Dobrowolski, J.C. On aromaticity of the aromatic α-amino acids and tuning of the NICS indices to find the aromaticity order. J. Phys. Chem. A 2022, 126, 3433–3444. [Google Scholar] [CrossRef]
- Toussaint, F.C.; Defrance, T.; Decouvreur, S.; Carly, N.; Merschaert, A. Intensification of free-radical racemization for a non-activated amine in a continuous flow reactor. Synthesis 2020, 52, 3389–3396. [Google Scholar] [CrossRef]
- Ruszczycky, M.W.; Liu, H.W. Distinguishing concerted versus stepwise mechanisms using isotope effects on isotope effects. Biochemistry 2021, 60, 3416–3418. [Google Scholar] [CrossRef]
- Dougherty, D.A. Cation-p interactions involving aromatic amino acids. J. Nutr. 2007, 137, 1504S–1508S. [Google Scholar] [CrossRef] [Green Version]
- Cotzias, G.C.; Van Woert, M.H.; Schiffer, L.M. Aromatic amino acids and modification of parkinsonism. J. Med. 1967, 276, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Himmelreich, N.; Montioli, R.; Bertoldi, M.; Carducci, C.; Leuzzi, V.; Gemperle, C.; Berner, T.; Hyland, K.; Thöny, B.; Hoffmann, G.F.; et al. Aromatic amino acid decarboxylase deficiency: Molecular and metabolic basis and therapeutic outlook. Mol. Genet. Metab. 2019, 127, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Xue, B.; Rehak, P.; Jacoby, G.; Ji, W.; Shimon, L.J.W.; Beck, R.; Král, P.; Cao, Y.; Gazit, E. Self-assembly of aromatic amino acid enantiomers into supramolecular materials of high rigidity. ACS Nano 2020, 14, 1694–1706. [Google Scholar] [CrossRef]
- Planas, F.; McLeish, M.J.; Himo, F. Enzymatic stetter reaction: Computational study of the reaction mechanism of MenD. ACS Catal. 2021, 11, 12355–12366. [Google Scholar] [CrossRef]
- Javierre, G.; Fortrie, R.; Jean, M.; Moraleda, D.; Naubron, J.V.; Fotiadu, F. Racemization and transesterification of alkyl hydrogeno-phenylphosphinates. J. Mol. Model. 2017, 23, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Zhang, B.; Wang, M. Estimation of the racemization rate constants for α-amino acids using density functional theory. Comput. Theor. Chem. 2017, 1104, 43–46. [Google Scholar] [CrossRef]
- Pokluda, J.; Černý, M.; Šob, M.; Umeno, Y. Ab initio calculations of mechanical properties: Methods and applications. Prog. Mater. Sci. 2015, 73, 127–158. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; He, Z.; Xu, Y.; Yu, W. Theoretical investigations on charge transport properties of tetrabenzo[a,d,j,m]coronene derivatives using different density functional theory functionals (B3LYP, M06-2X, and WB97XD). J. Chem. Res. 2019, 48, 293–303. [Google Scholar] [CrossRef]
- Cuesta, S.A.; Torres, F.J.; Rincón, L.; Paz, J.L.; Márquez, E.A.; Mora, J.R. Effect of the nucleophile’s nature on chloroacetanilide herbicides cleavage reaction mechanism. A dft study. Int. J. Mol. Sci. 2021, 22, 6876. [Google Scholar] [CrossRef]
- Inostroza-Rivera, R.; Herrera, B.; Toro-Labbé, A. Using the reaction force and the reaction electronic flux on the proton transfer of formamide derived systems. Phys. Chem. Chem. Phys. 2014, 16, 14489–14495. [Google Scholar] [CrossRef]
- Cuesta, S.A.; Mora, J.R.; Meneses, L.M.; Márquez, E.A.; Flores-Morales, V.; Rincón, L.; Torres, F.J.; Zambrano, C.H. Unveiling the structure-reactivity relationship involved in the reaction mechanism of the HCl-catalyzed alkyl t-butyl ethers thermal decomposition. A computational study. Int. J. Quantum Chem. 2022, 122, e26915. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goerigk, L.; Sharma, R. The INV24 test set: How well do quantum-chemical methods describe inversion and racemization barriers? Can. J. Chem. 2016, 94, 1133–1143. [Google Scholar] [CrossRef]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Racemization of the succinimide intermediate formed in proteins and peptides: A computational study of the mechanism catalyzed by dihydrogen phosphate ion. Int. J. Mol. Sci. 2016, 17, 1698. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, G.; Basavaprabhu; Narendra, N.; Vishwanatha, T.M.; Sureshbabu, V.V. Amino acid chlorides: A journey from instability and racemization toward broader utility in organic synthesis including peptides and their mimetics. Tetrahedron 2015, 71, 2785–2832. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Performance of SM6, SM8, and SMD on the SAMPL1 test set for the prediction of small-molecule solvation free energies. J. Phys. Chem. B 2009, 113, 4538–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Cuesta, S.A.; Mora, J.; Márquez, E. Gas phase decomposition of t-butyl methyl ether catalyzed by different hydrogen halides: A dft study. infoANALÍTICA 2022, 10, 65–79. [Google Scholar] [CrossRef]
- Türker, L.; Atalar, T.; Gümüş, S.; Çamur, Y. A dft study on nitrotriazines. J. Hazard. Mater. 2009, 167, 440–448. [Google Scholar] [CrossRef]
- Da, L.G.; He, J.; Hu, L.F. DFT Study on the structure and racemization mechanism of 2-amino-2′-hydroxy-1,1′-binaphthyl. J. Phys. Org. Chem. 2019, 32, e3900. [Google Scholar] [CrossRef]
- De Souza, M.A.F.; Ventura, E.; Do Monte, S.A.; Riveros, J.M.; Longo, R.L. Revisiting the concept of the (a)synchronicity of diels-alder reactions based on the dynamics of quasiclassical trajectories. J. Comput. Chem. 2016, 37, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Novikov, A.S.; Kuznetsov, M.L.; Pombeiro, A.J.L. Theory of the formation and decomposition of N-heterocyclic aminooxycarbenes through metal-assisted [2+3]-dipolar cycloaddition/retro- cycloaddition. Chem.—A Eur. J. 2013, 19, 2874–2888. [Google Scholar] [CrossRef] [PubMed]
- Novikov, A.S.; Kuznetsov, M.L. Theoretical study of Re(IV) and Ru(II) bis-isocyanide complexes and their reactivity in cycloaddition reactions with nitrones. Inorg. Chim. Acta 2012, 380, 78–89. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L.; Pombeiro, A.J.L.; Bokach, N.A.; Shul’Pin, G.B. Generation of HO• radical from hydrogen peroxide catalyzed by aqua complexes of the group III metals [M(H2O)n]3+ (M = Ga, In, Sc, Y, or La): A theoretical study. ACS Catal. 2013, 3, 1195–1208. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| This Work | Experimental * | |||||
|---|---|---|---|---|---|---|
| X | Ea (kcal/mol) | ΔH‡ (kcal/mol) | ΔG‡ (kcal/mol) | ΔS‡ (cal/mol K) | Ea (kcal/mol) | |
| Aromatic | H | 20.20 | 18.67 | 24.35 | −14.80 | 20.7 |
| mCl | 18.51 | 16.98 | 21.96 | −12.97 | 20.7 | |
| mNO2 | 17.25 | 15.72 | 21.58 | −15.28 | 20.8 | |
| pCl | 19.19 | 17.66 | 23.82 | −16.05 | 20.3 | |
| pMe | 20.98 | 19.46 | 25.55 | −15.90 | 21.1 | |
| pOMe | 21.29 | 19.76 | 24.88 | −13.36 | 21.1 | |
| Aliphatic | Leucine | 27.27 | 26.57 | 33.10 | −17.04 | 27.9 |
| Isoleucine | 28.09 | 25.74 | 32.35 | −16.43 | 28.1 | |
| Phenylalanine | 25.87 | 24.35 | 30.39 | −15.75 | 24.0 | |
| X | W1 | W2 | W3 | W4 | W14 | W23 | |
|---|---|---|---|---|---|---|---|
| Aromatic | H | 6.9307 | 2.9724 | 0.5943 | 4.3796 | 11.3104 | 3.5667 |
| mCl | 5.4205 | 2.3746 | 0.8609 | 4.9446 | 10.3651 | 3.2354 | |
| mNO2 | 4.9695 | 2.0323 | 1.0508 | 8.1109 | 13.0804 | 3.0831 | |
| pCl | 4.4025 | 2.4017 | 0.8704 | 6.0197 | 10.4222 | 3.2721 | |
| pMe | 6.7781 | 3.2663 | 0.5146 | 5.6673 | 12.4454 | 3.7809 | |
| pOMe | 9.2743 | 3.7475 | 0.3897 | 2.6983 | 11.9726 | 4.1373 | |
| Aliphatic | Leu | 21.0125 | 7.4794 | 0.0588 | 0.0694 | 21.0819 | 7.5381 |
| IsoLeu | 21.6972 | 6.6868 | 0.1123 | 0.1332 | 21.8304 | 6.7990 | |
| PheAla | 20.2607 | 6.4456 | 0.0742 | 0.0489 | 20.3096 | 6.5198 |
| Compound | Phenylglycine | mCl-Phenylglycine | mNO2-Phenylglycine | |||
|---|---|---|---|---|---|---|
| Bond | C1-H5 | H5-O6 | C1-H5 | H5-O6 | C1-H5 | H5-O6 |
| Reactant | 0.8114 | 0.0446 | 0.7943 | 0.0544 | 0.7899 | 0.0566 |
| T. State | 0.3396 | 0.4344 | 0.3546 | 0.4171 | 0.3675 | 0.4059 |
| Product | 0.0809 | 0.6602 | 0.0782 | 0.6639 | 0.0549 | 0.6878 |
| %Ev | 64.59 | 63.32 | 61.40 | 59.51 | 57.47 | 55.34 |
| Sy | 0.9901 | 0.9843 | 0.9811 | |||
| Compound | pCl-Phenylglycine | pMe-Phenylglycine | pOMe-Phenylglycine | |||
| Bond | C1-H5 | H5-O6 | C1-H5 | H5-O6 | C1-H5 | H5-O6 |
| Reactant | 0.7751 | 0.0666 | 0.8092 | 0.0463 | 0.8408 | 0.0278 |
| T. State | 0.3505 | 0.4203 | 0.3305 | 0.4424 | 0.3254 | 0.4472 |
| Product | 0.0781 | 0.6625 | 0.0787 | 0.6619 | 0.0891 | 0.6521 |
| %Ev | 60.92 | 59.36 | 65.53 | 64.34 | 68.56 | 67.18 |
| Sy | 0.9870 | 0.9909 | 0.9898 | |||
| Compound | Leucine | Isoleucine | Phenylalanine | |||
|---|---|---|---|---|---|---|
| Bond | C1-H5 | H5-O6 | C1-H5 | H5-O6 | C1-H5 | H5-O6 |
| Reactant | 0.8856 | 0.0058 | 0.8874 | 0.0055 | 0.8967 | 0.0040 |
| T. State | 0.2220 | 0.5332 | 0.2394 | 0.5112 | 0.2453 | 0.5213 |
| Product | 0.1113 | 0.6239 | 0.1060 | 0.6285 | 0.1206 | 0.6271 |
| %Ev | 85.70 | 85.33 | 82.93 | 81.17 | 83.93 | 83.02 |
| Sy | 0.9978 | 0.9893 | 0.9945 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andino, M.S.; Mora, J.R.; Paz, J.L.; Márquez, E.A.; Perez-Castillo, Y.; Agüero-Chapin, G. Elucidating the Racemization Mechanism of Aliphatic and Aromatic Amino Acids by In Silico Tools. Int. J. Mol. Sci. 2023, 24, 11877. https://doi.org/10.3390/ijms241511877
Andino MS, Mora JR, Paz JL, Márquez EA, Perez-Castillo Y, Agüero-Chapin G. Elucidating the Racemization Mechanism of Aliphatic and Aromatic Amino Acids by In Silico Tools. International Journal of Molecular Sciences. 2023; 24(15):11877. https://doi.org/10.3390/ijms241511877
Chicago/Turabian StyleAndino, Mateo S., José R. Mora, José L. Paz, Edgar A. Márquez, Yunierkis Perez-Castillo, and Guillermin Agüero-Chapin. 2023. "Elucidating the Racemization Mechanism of Aliphatic and Aromatic Amino Acids by In Silico Tools" International Journal of Molecular Sciences 24, no. 15: 11877. https://doi.org/10.3390/ijms241511877