
Sexual Dimorphism in Cardiometabolic Diseases: The Role of AMPK

Abstract
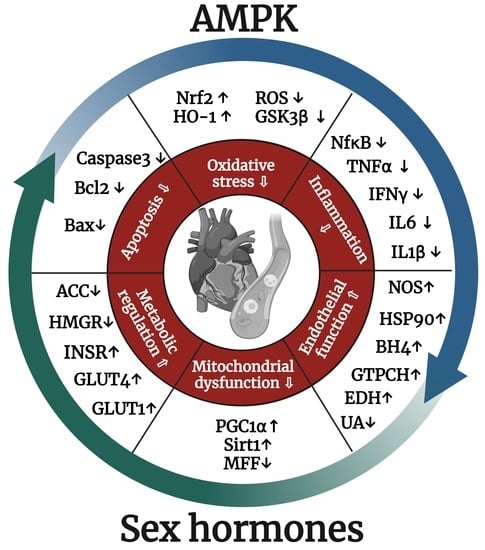
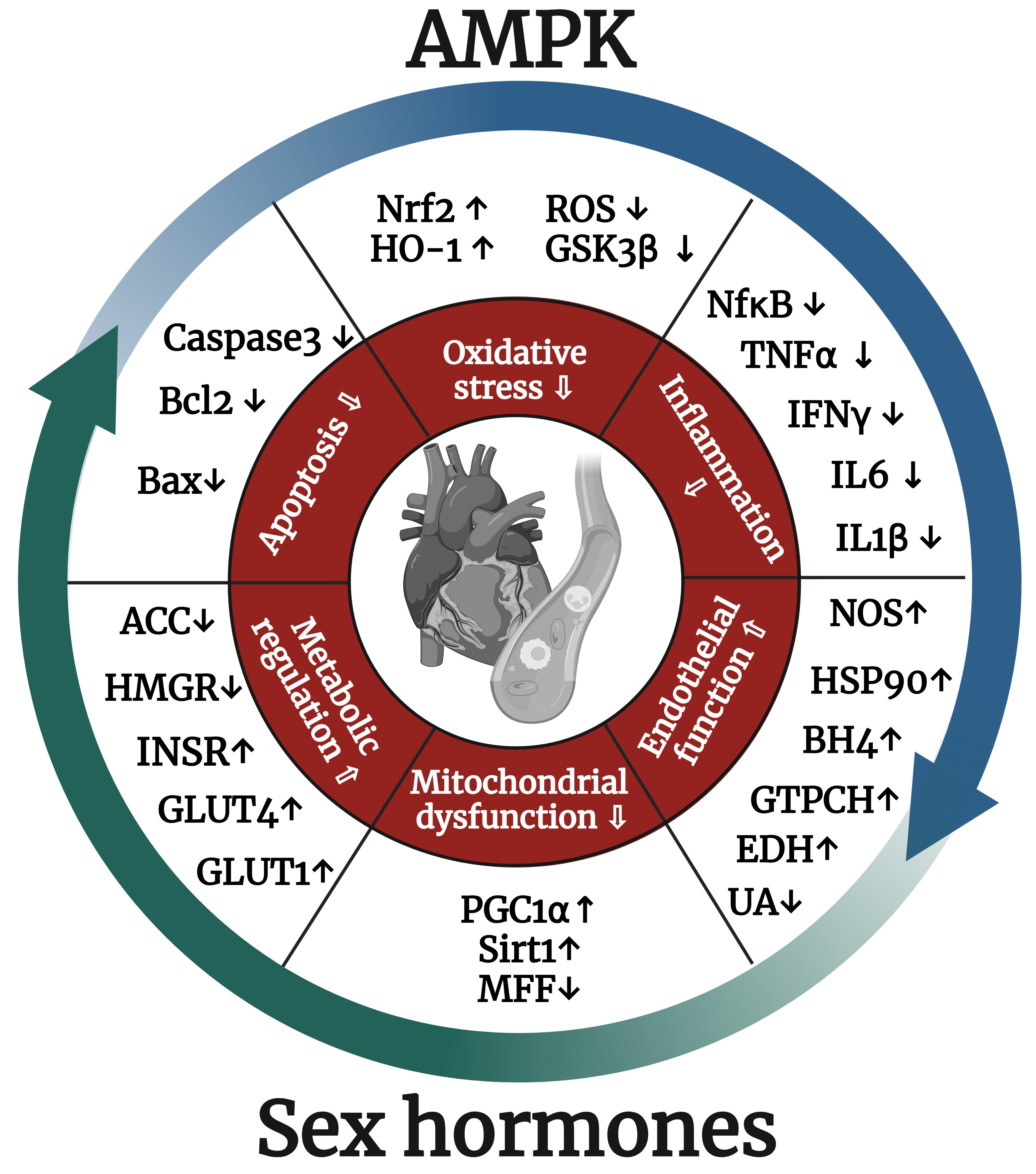
:
{kind=link}
{kind=link}
{kind=link}
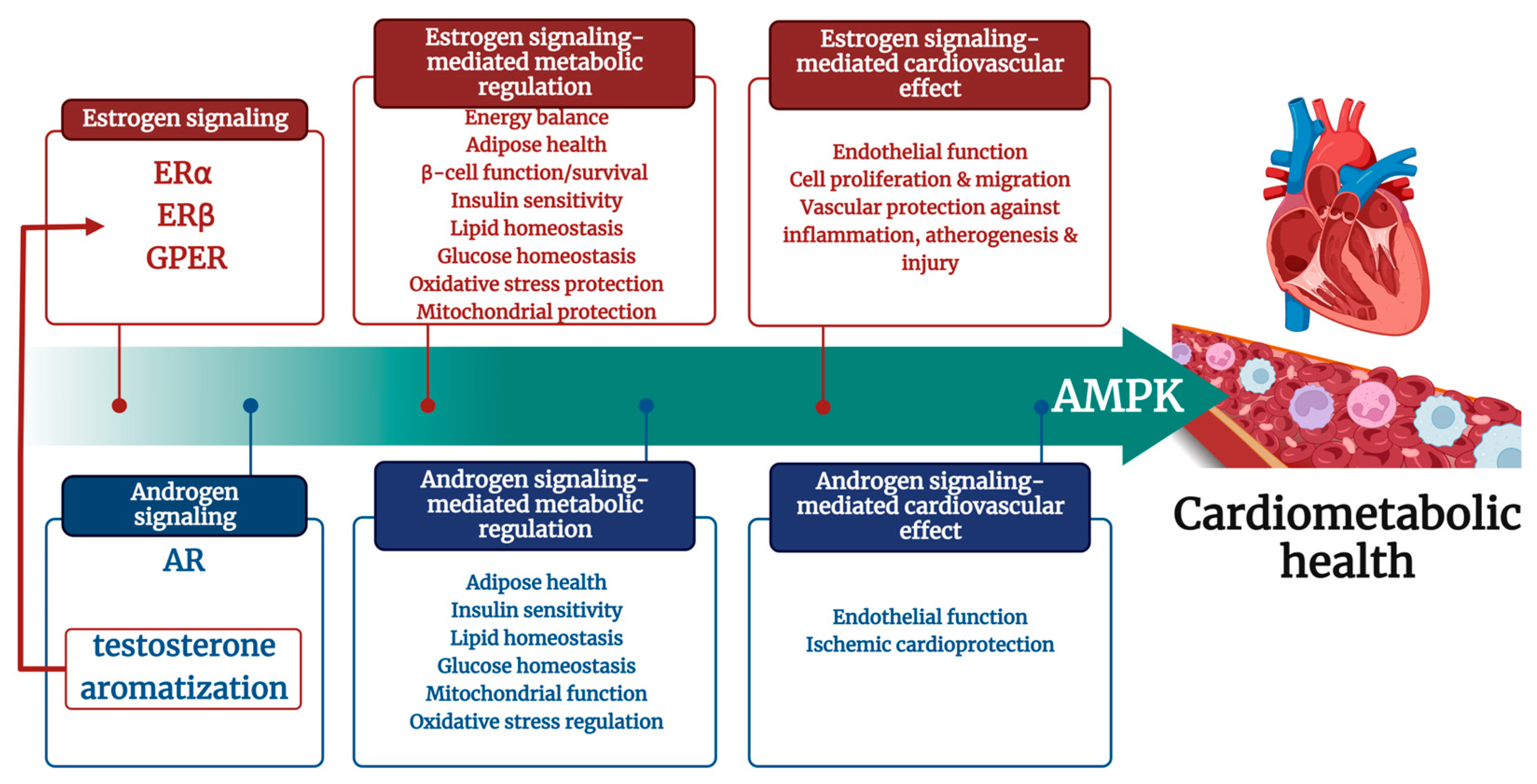{kind=link}
1. Introduction
2. Sexual Dimorphism in Cardiometabolic Diseases
2.1. Insulin Signaling and Sex Hormones
2.2. Insulin Signaling and Metabolic “Master Switch”—AMPK
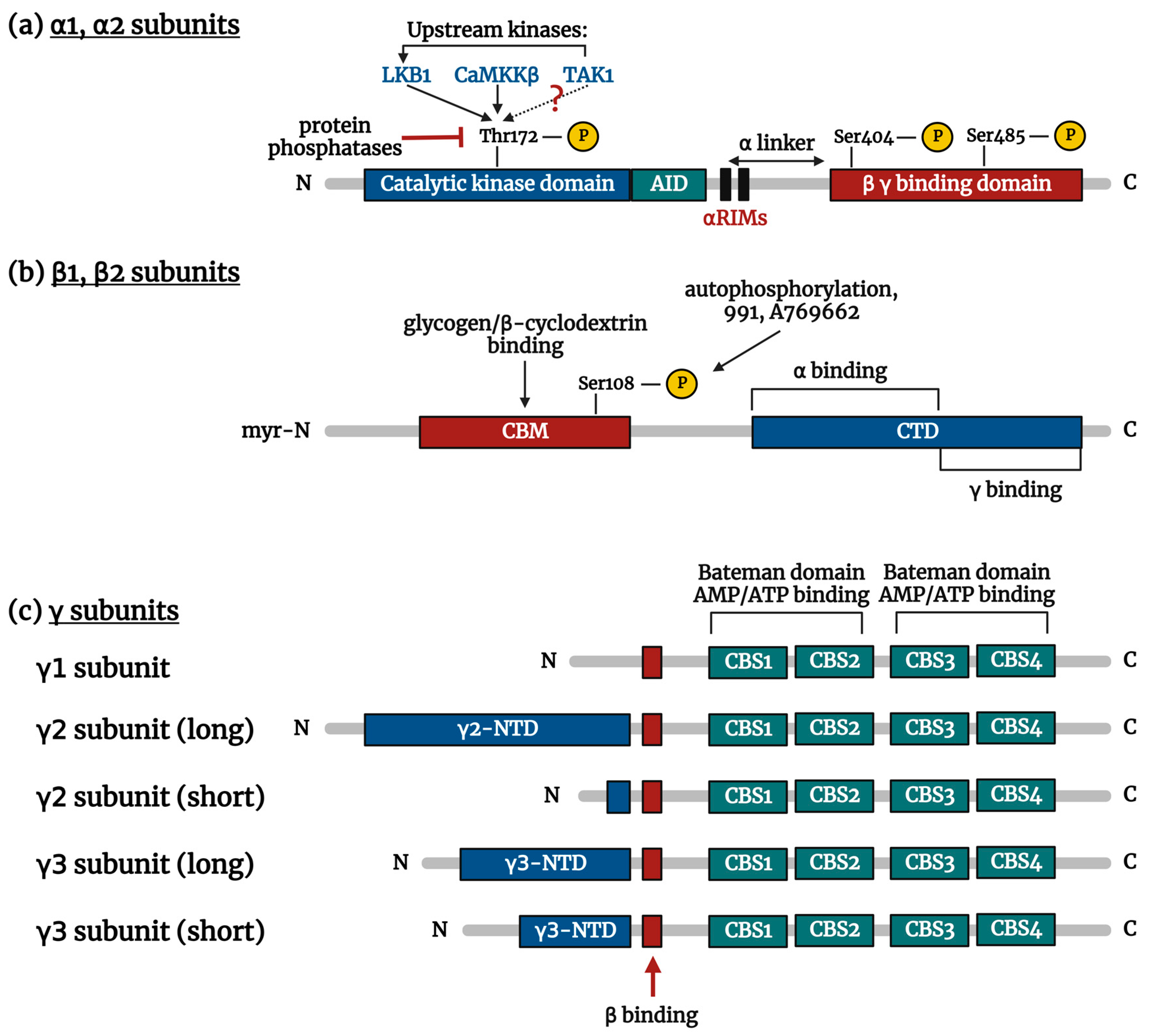
3. AMPK Structure
3.1. α Subunit
3.2. β Subunit
3.3. γ Subunit
4. AMPK Activation
4.1. Physiological Activation
4.2. Pharmacological Activation
4.3. Activation by Upstream Kinases
4.4. Activation by Intracellular Physiological Stimuli
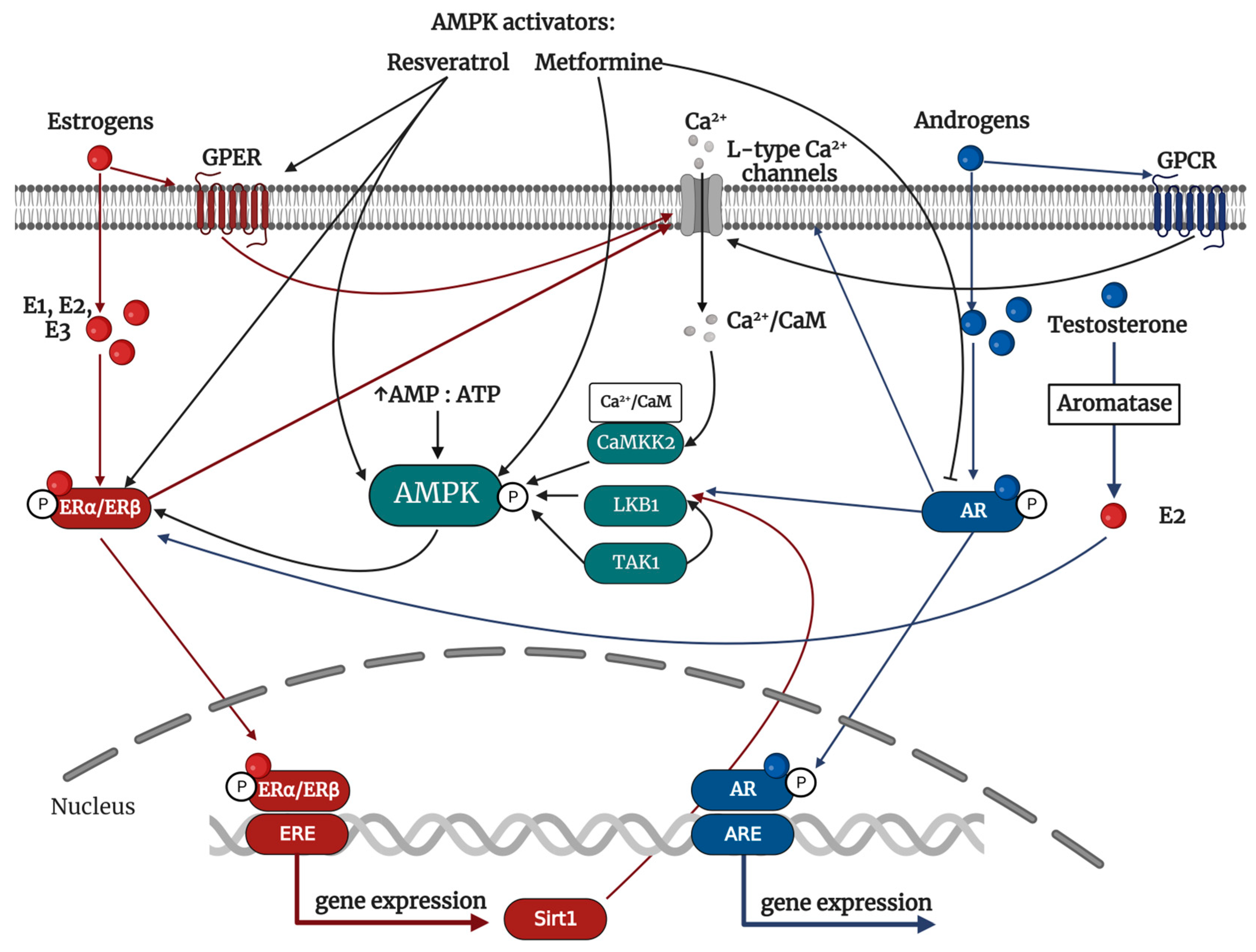
4.5. AMPK as a Mediator of Hormonal Signaling
4.5.1. Female-Related Hormones: Estrogen
4.5.2. Male-Related Hormones: Androgens
4.6. The Regulation of Sex Hormones by AMPK
5. Sexual Dimorphism in AMPK Regulation of Endothelial Function
5.1. Endothelial Function
5.2. AMPK and Endothelial Function
5.2.1. AMPK and Antioxidant Defense as a Protective Tool for Endothelial Homeostasis Maintenance
5.2.2. AMPK and Anti-Inflammatory Actions as a Protective Tool for Endothelial Homeostasis Maintenance
5.2.3. Endothelial Function, Sex Hormones, and Cross-Talk to AMPK
6. Sexual Dimorphism in AMPK Regulation of Cardiac Function
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| αRIMs | α regulatory subunit-interacting motifs |
| ACC | acetyl-CoA carboxylase |
| AID | auto-inhibitory domain |
| AMPK | adenosine monophosphate-dependent protein kinase |
| ARs | androgen receptors |
| ARE | antioxidant response element |
| AT II | angiotensin II |
| Bax | BCL2 associated X |
| Bcl2 | B-cell lymphoma 2 |
| BH4 | tetrahydrobiopterin |
| CaMKKβ | Ca2+-calmodulin-dependent protein kinase kinase beta |
| CBM | carbohydrate-binding module |
| CBS | cystathionine β-synthase |
| CTD | C-terminal domain |
| CVDs | cardiovascular diseases |
| DHT | dihydrotestosterone |
| E1 | estrone |
| E2 | 17β-estradiol |
| E3 | estriol |
| EDH | endothelium-dependent hyperpolarization |
| eNOS | endothelial nitric oxide synthase |
| ER | estrogen receptor |
| ERE | estrogen response elements |
| ETC | electron transport chain |
| GLUT1 | glucose transporter type 1 |
| GLUT4 | glucose transporter type 4 |
| GPCR | G protein-coupled receptors |
| GPER | G-protein-coupled estrogen receptor |
| GSK3β | glycogen synthase kinase-3 beta |
| GTPCH | guanosine triphosphate cyclohydrolase |
| HIF | hypoxia-inducible factor |
| HMGR | 3-hydroxy-3-methylglutaryl-coenzyme A reductase |
| HO-1 | heme oxygenase-1 |
| HSP90 | heat shock protein 90 |
| IL1β | interleukin 1 beta |
| IL6 | interleukin 6 |
| INSR | insulin receptor |
| IR | insulin resistance |
| IRS | insulin receptor substrates |
| LKB1 | liver kinase B1 |
| MAP3K | MAPK kinase |
| MFF | mitochondrial fission factor |
| MO25 | mouse protein-25 |
| MS | metabolic syndrome |
| NFκB | nuclear factor kappa beta |
| NOS | nitric oxide synthase |
| NOX | nicotinamide adenine dinucleotide phosphate oxidases |
| Nrf2 | nuclear factor erythroid 2–related factor 2 |
| NTD | N-terminal domain |
| PGC1α | peroxisome proliferator-activated receptor-gamma coactivator 1 alpha |
| PKB/Cδ | protein kinase B/Cδ |
| ROS | reactive oxygen species |
| Sirt1 | sirtuin 1 |
| SOD | superoxide dismutase |
| STRAD | sterile-20-related adaptor |
| T2DM | type 2 diabetes mellitus |
| TAK1 | transforming growth factor-β-activated kinase 1 |
| TNFα | tumor necrosis factor-alpha |
| UA | uric acid |
| VEGF | vascular endothelial growth factor |
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Mezhal, F.; Ahmad, A.; Abdulle, A.; Leinberger-Jabari, A.; Oulhaj, A.; AlJunaibi, A.; Alnaeemi, A.; Al Dhaheri, A.S.; AlZaabi, E.; Al-Maskari, F.; et al. Metabolic Syndrome in Fasting and Non-Fasting Participants: The UAE Healthy Future Study. Int. J. Environ. Res. Public Health 2022, 19, 13757. [Google Scholar] [CrossRef] [PubMed]
- Ober, C.; Loisel, D.A.; Gilad, Y. Sex-specific genetic architecture of human disease. Nat. Rev. Genet. 2008, 9, 911–922. [Google Scholar]
- Leening, M.J.; Ferket, B.S.; Steyerberg, E.W.; Kavousi, M.; Deckers, J.W.; Nieboer, D.; Heeringa, J.; Portegies, M.L.; Hofman, A.; Ikram, M.A.; et al. Sex differences in lifetime risk and first manifestation of cardiovascular disease: Prospective population based cohort study. BMJ 2014, 349, g5992. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Davidge, S.T. High glucose-induced oxidative stress alters estrogen effects on ERalpha and ERbeta in human endothelial cells: Reversal by AMPK activator. J. Steroid Biochem. Mol. Biol. 2009, 117, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, S.; Foretz, M.; Knorr, M.; Jansen, T.; Hortmann, M.; Wenzel, P.; Oelze, M.; Kleschyov, A.L.; Daiber, A.; Keaney, J.F., Jr.; et al. alpha1AMP-activated protein kinase preserves endothelial function during chronic angiotensin II treatment by limiting Nox2 upregulation. Arter. Thromb. Vasc. Biol. 2011, 31, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Anter, E.; Zou, M.H.; Keaney, J.F., Jr. Estradiol-Mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinase. Circulation 2005, 111, 3473–3480. [Google Scholar] [CrossRef]
- Jansen, T.; Kvandova, M.; Daiber, A.; Stamm, P.; Frenis, K.; Schulz, E.; Munzel, T.; Kroller-Schon, S. The AMP-Activated Protein Kinase Plays a Role in Antioxidant Defense and Regulation of Vascular Inflammation. Antioxidants 2020, 9, 525. [Google Scholar] [CrossRef]
- Rogers, N.H.; Witczak, C.A.; Hirshman, M.F.; Goodyear, L.J.; Greenberg, A.S. Estradiol stimulates Akt, AMP-activated protein kinase (AMPK) and TBC1D1/4, but not glucose uptake in rat soleus. Biochem. Biophys. Res. Commun. 2009, 382, 646–650. [Google Scholar] [CrossRef] [Green Version]
- Troncoso, M.F.; Pavez, M.; Wilson, C.; Lagos, D.; Duran, J.; Ramos, S.; Barrientos, G.; Silva, P.; Llanos, P.; Basualto-Alarcon, C.; et al. Testosterone activates glucose metabolism through AMPK and androgen signaling in cardiomyocyte hypertrophy. Biol. Res. 2021, 54, 3. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, E.; Regitz-Zagrosek, V. Sex differences in cardiometabolic disorders. Nat. Med. 2019, 25, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, J.C. The influence of sex hormones on obesity across the female life span. J. Womens Health 1998, 7, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.H.; Appelman, Y.E. Gender differences in coronary heart disease. Neth. Heart J. 2010, 18, 598–603. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R. Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [Green Version]
- Chella Krishnan, K.; Mehrabian, M.; Lusis, A.J. Sex differences in metabolism and cardiometabolic disorders. Curr. Opin. Lipidol. 2018, 29, 404–410. [Google Scholar] [CrossRef]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef]
- Davis, K.E.; Neinast, M.D.; Sun, K.; Skiles, W.M.; Bills, J.D.; Zehr, J.A.; Zeve, D.; Hahner, L.D.; Cox, D.W.; Gent, L.M. The sexually dimorphic role of adipose and adipocyte estrogen receptors in modulating adipose tissue expansion, inflammation, and fibrosis. Mol. Metab. 2013, 2, 227–242. [Google Scholar] [CrossRef]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models to disease mechanisms. J. Endocrinol. 2014, 220, T1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoleri, D.; Titchenell, P.M. Resolving the paradox of hepatic insulin resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Mikhail, N. The metabolic syndrome: Insulin resistance. Curr. Hypertens. Rep. 2009, 11, 156–158. [Google Scholar] [CrossRef]
- Oya, J.; Nakagami, T.; Yamamoto, Y.; Fukushima, S.; Takeda, M.; Endo, Y.; Uchigata, Y. Effects of age on insulin resistance and secretion in subjects without diabetes. Intern. Med. 2014, 53, 941–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundholm, L.; Bryzgalova, G.; Gao, H.; Portwood, N.; Falt, S.; Berndt, K.; Dicker, A.; Galuska, D.; Zierath, J.; Gustafsson, J. The estrogen receptor a-selective agonist propyl pyrazole triol improves glucose tolerance in ob/ob mice; potential molecular mechanisms. J. Endocrinol. 2008, 199, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Bryzgalova, G.; Gao, H.; Ahrén, B.; Zierath, J.; Galuska, D.; Steiler, T.; Dahlman-Wright, K.; Nilsson, S.; Gustafsson, J.-Å.; Efendic, S. Evidence that oestrogen receptor-α plays an important role in the regulation of glucose homeostasis in mice: Insulin sensitivity in the liver. Diabetologia 2006, 49, 588–597. [Google Scholar] [CrossRef] [Green Version]
- López-Grueso, R.; Gambini, J.; Abdelaziz, K.M.; Monleón, D.; Diaz, A.; El Alami, M.; Bonet-Costa, V.; Borrás, C.; Vina, J. Early, but not late onset estrogen replacement therapy prevents oxidative stress and metabolic alterations caused by ovariectomy. Antioxid. Redox Signal. 2014, 20, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Huidobro, R.I.; Velasco, M.; Larque, C.; Escalona, R.; Hiriart, M. Molecular Insulin Actions Are Sexually Dimorphic in Lipid Metabolism. Front. Endocrinol. 2021, 12, 690484. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Collier, D.; Kassotis, C.; Roepke, T.A.; Kim, M.J.; Blanc, E.; Barouki, R.; Bansal, A.; Cave, M.C.; Chatterjee, S.; et al. Obesity I: Overview and molecular and biochemical mechanisms. Biochem. Pharmacol. 2022, 199, 115012. [Google Scholar] [CrossRef] [PubMed]
- Fagman, J.B.; Wilhelmson, A.S.; Motta, B.M.; Pirazzi, C.; Alexanderson, C.; De Gendt, K.; Verhoeven, G.; Holmäng, A.; Anesten, F.; Jansson, J.-O. The androgen receptor confers protection against diet-induced atherosclerosis, obesity, and dyslipidemia in female mice. FASEB J. 2015, 29, 1540. [Google Scholar] [CrossRef]
- Kelly, D.M.; Jones, T.H. Testosterone and obesity. Obes. Rev. 2015, 16, 581–606. [Google Scholar] [CrossRef]
- Yu, I.C.; Lin, H.Y.; Sparks, J.D.; Yeh, S.; Chang, C. Androgen receptor roles in insulin resistance and obesity in males: The linkage of androgen-deprivation therapy to metabolic syndrome. Diabetes 2014, 63, 3180–3188. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef] [PubMed]
- Prabha, B.; Sini, S.; Sherin, D.; Neethu, S.; Rameshkumar, K.; Manojkumar, T.; Jayamurthy, P.; Radhakrishnan, K. Promalabaricone B from Myristica fatua Houtt. seeds demonstrate antidiabetic potential by modulating glucose uptake via the upregulation of AMPK in L6 myotubes. Nat. Prod. Res. 2021, 35, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Natsume, N.; Yonezawa, T.; Saito, Y.; Woo, J.-T.; Teruya, T. Prenylflavonoids from fruit of Macaranga tanarius promote glucose uptake via AMPK activation in L6 myotubes. J. Nat. Med. 2021, 75, 813–823. [Google Scholar] [PubMed]
- Lee, H.A.; Cho, J.-H.; Afinanisa, Q.; An, G.-H.; Han, J.-G.; Kang, H.J.; Choi, S.H.; Seong, H.-A. Ganoderma lucidum extract reduces insulin resistance by enhancing AMPK activation in high-fat diet-induced obese mice. Nutrients 2020, 12, 3338. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Contreras-Ferrat, A.; Venegas, N.; Osorio-Fuentealba, C.; Pavez, M.; Montoya, K.; Duran, J.; Maass, R.; Lavandero, S.; Estrada, M. Testosterone increases GLUT4-dependent glucose uptake in cardiomyocytes. J. Cell. Physiol. 2013, 228, 2399–2407. [Google Scholar] [CrossRef]
- Mitsuhashi, K.; Senmaru, T.; Fukuda, T.; Yamazaki, M.; Shinomiya, K.; Ueno, M.; Kinoshita, S.; Kitawaki, J.; Katsuyama, M.; Tsujikawa, M.; et al. Testosterone stimulates glucose uptake and GLUT4 translocation through LKB1/AMPK signaling in 3T3-L1 adipocytes. Endocrine 2016, 51, 174–184. [Google Scholar] [CrossRef]
- Yang, S.; Wang, J. Estrogen Activates AMP-Activated Protein Kinase in Human Endothelial Cells via ERbeta/Ca(2+)/Calmodulin-Dependent Protein Kinase Kinase beta Pathway. Cell Biochem. Biophys. 2015, 72, 701–707. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1: Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [Green Version]
- Campagnoli, C.; Berrino, F.; Venturelli, E.; Abba, C.; Biglia, N.; Brucato, T.; Cogliati, P.; Danese, S.; Donadio, M.; Zito, G.; et al. Metformin decreases circulating androgen and estrogen levels in nondiabetic women with breast cancer. Clin. Breast Cancer 2013, 13, 433–438. [Google Scholar] [CrossRef]
- Hirsch, A.; Hahn, D.; Kempna, P.; Hofer, G.; Nuoffer, J.M.; Mullis, P.E.; Fluck, C.E. Metformin inhibits human androgen production by regulating steroidogenic enzymes HSD3B2 and CYP17A1 and complex I activity of the respiratory chain. Endocrinology 2012, 153, 4354–4366. [Google Scholar] [CrossRef] [Green Version]
- Carling, D.; Clarke, P.R.; Zammit, V.A.; Hardie, D.G. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur. J. Biochem. 1989, 186, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.M.; Gillespie, J.G.; Hardie, D.G. Role of the AMP-activated protein kinase in the cellular stress response. Curr. Biol. 1994, 4, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; Ortiz de Montellano, P.R.; Kemp, B.E. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Salt, I.; Scott, J.; Hardie, D.G.; Carling, D. The α1 and α2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett. 1996, 397, 347–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, C.; Snowden, M.A.; Carling, D. Identification of a novel AMP-activated protein kinase β subunit isoform that is highly expressed in skeletal muscle. J. Biol. Chem. 1998, 273, 12443–12450. [Google Scholar] [CrossRef] [Green Version]
- Cheung, P.C.; Salt, I.P.; Davies, S.P.; Hardie, D.G.; Carling, D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem. J. 2000, 346 Pt 3, 659–669. [Google Scholar] [CrossRef]
- Hardie, D.G.; Hawley, S.A.; Scott, J.W. AMP-activated protein kinase—Development of the energy sensor concept. J. Physiol. 2006, 574, 7–15. [Google Scholar] [CrossRef]
- Calabrese, M.F.; Rajamohan, F.; Harris, M.S.; Caspers, N.L.; Magyar, R.; Withka, J.M.; Wang, H.; Borzilleri, K.A.; Sahasrabudhe, P.V.; Hoth, L.R.; et al. Structural basis for AMPK activation: Natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure 2014, 22, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Polekhina, G.; Gupta, A.; Michell, B.J.; van Denderen, B.; Murthy, S.; Feil, S.C.; Jennings, I.G.; Campbell, D.J.; Witters, L.A.; Parker, M.W.; et al. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr. Biol. 2003, 13, 867–871. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [Green Version]
- Oakhill, J.S.; Chen, Z.P.; Scott, J.W.; Steel, R.; Castelli, L.A.; Ling, N.; Macaulay, S.L.; Kemp, B.E. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. USA 2010, 107, 19237–19241. [Google Scholar] [CrossRef]
- Klaus, A.; Zorman, S.; Berthier, A.; Polge, C.; Ramirez, S.; Michelland, S.; Seve, M.; Vertommen, D.; Rider, M.; Lentze, N.; et al. Glutathione S-transferases interact with AMP-activated protein kinase: Evidence for S-glutathionylation and activation in vitro. PLoS ONE 2013, 8, e62497. [Google Scholar] [CrossRef]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Investig. 2004, 113, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Kemp, B.E. Bateman domains and adenosine derivatives form a binding contract. J. Clin. Investig. 2004, 113, 182–184. [Google Scholar] [CrossRef]
- Pinter, K.; Grignani, R.T.; Czibik, G.; Farza, H.; Watkins, H.; Redwood, C. Embryonic expression of AMPK gamma subunits and the identification of a novel gamma2 transcript variant in adult heart. J. Mol. Cell Cardiol. 2012, 53, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2Cα and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar]
- Hardie, D.G.; Salt, I.P.; Davies, S.P. Analysis of the role of the AMP-activated protein kinase in the response to cellular stress. Stress Response Methods Protoc. 2000, 99, 63–74. [Google Scholar]
- Sanders, M.J.; Grondin, P.O.; Hegarty, B.D.; Snowden, M.A.; Carling, D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 2007, 403, 139–148. [Google Scholar] [CrossRef]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ashford, M.L. AMPK: Regulating energy balance at the cellular and whole body levels. Physiology 2014, 29, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [PubMed]
- Naimi, M.; Arous, C.; Van Obberghen, E. Energetic cell sensors: A key to metabolic homeostasis. Trends Endocrinol. Metab. 2010, 21, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, Y.; Sun, T.Q.; Markovtsov, V.; Foretz, M.; Li, W.; Nguyen, H.; Li, Y.; Pan, A.; Uy, G.; Gross, L.; et al. AMPK activation through mitochondrial regulation results in increased substrate oxidation and improved metabolic parameters in models of diabetes. PLoS ONE 2013, 8, e81870. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Hunter, R.W.; Foretz, M.; Bultot, L.; Fullerton, M.D.; Deak, M.; Ross, F.A.; Hawley, S.A.; Shpiro, N.; Viollet, B.; Barron, D. Mechanism of action of compound-13: An α1-selective small molecule activator of AMPK. Chem. Biol. 2014, 21, 866–879. [Google Scholar] [CrossRef] [Green Version]
- Göransson, O.; McBride, A.; Hawley, S.A.; Ross, F.A.; Shpiro, N.; Foretz, M.; Viollet, B.; Hardie, D.G.; Sakamoto, K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 32549–32560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.W.; Ling, N.; Issa, S.M.; Dite, T.A.; O’Brien, M.T.; Chen, Z.-P.; Galic, S.; Langendorf, C.G.; Steinberg, G.R.; Kemp, B.E. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem. Biol. 2014, 21, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Willows, R.; Sanders, M.J.; Xiao, B.; Patel, B.R.; Martin, S.R.; Read, J.; Wilson, J.R.; Hubbard, J.; Gamblin, S.J.; Carling, D. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem. J. 2017, 474, 3059–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, D. Is TAK1 a Direct Upstream Kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412. [Google Scholar] [CrossRef] [Green Version]
- Rostas, J.A.P.; Skelding, K.A. Calcium/Calmodulin-Stimulated Protein Kinase II (CaMKII): Different Functional Outcomes from Activation, Depending on the Cellular Microenvironment. Cells 2023, 12, 401. [Google Scholar] [CrossRef]
- Guerrero-Hernandez, A.; Verkhratsky, A. Calcium signalling in diabetes. Cell Calcium 2014, 56, 297–301. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Alexander, A.; Walker, C.L. The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS Lett. 2011, 585, 952–957. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Lin, H.K. A new mechanism for LKB1 activation. Mol. Cell Oncol. 2018, 5, e1035691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadrich, J.L.; O’Connor, M.B.; Coucouvanis, E. Expression of TAK1, a mediator of TGF-beta and BMP signaling, during mouse embryonic development. Gene Expr. Patterns 2003, 3, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, D.; Dyck, J.R.; Li, Y.; Zhang, H.; Morishima, M.; Mann, D.L.; Taffet, G.E.; Baldini, A.; Khoury, D.S. A pivotal role for endogenous TGF-β-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 17378–17383. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, M.; Liang, B.; Xu, J.; Xie, Z.; Liu, C.; Viollet, B.; Yan, D.; Zou, M.H. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: Role of 26S proteasomes. Circ. Res. 2010, 106, 1117–1128. [Google Scholar] [CrossRef]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [Green Version]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [Green Version]
- Auciello, F.R.; Ross, F.A.; Ikematsu, N.; Hardie, D.G. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 2014, 588, 3361–3366. [Google Scholar] [CrossRef] [Green Version]
- Klimova, T.; Chandel, N. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase: The energy charge hypothesis revisited. Bioessays 2001, 23, 1112–1119. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid. Med. Cell Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.T.; Kola, B.; Korbonits, M. AMPK as a mediator of hormonal signalling. J. Mol. Endocrinol. 2010, 44, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.-B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.J. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016, 49, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Prossnitz, E.R. G-Protein-Coupled Estrogen Receptor (GPER) and Sex-Specific Metabolic Homeostasis. Adv. Exp. Med. Biol. 2017, 1043, 427–453. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Kumar, S.P.S.; Shi, H. Estradiol regulates insulin signaling and inflammation in adipose tissue. Horm. Mol. Biol. Clin. Investig. 2014, 17, 99–107. [Google Scholar] [CrossRef]
- Levin, E.R. Extranuclear steroid receptors are essential for steroid hormone actions. Annu. Rev. Med. 2015, 66, 271–280. [Google Scholar] [CrossRef]
- Simoncini, T.; Mannella, P.; Genazzani, A.R. Rapid estrogen actions in the cardiovascular system. Ann. N. Y. Acad. Sci. 2006, 1089, 424–430. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Hathaway, H.J. What have we learned about GPER function in physiology and disease from knockout mice? J. Steroid Biochem. Mol. Biol. 2015, 153, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Matarrese, P.; Maccari, S.; Vona, R.; Gambardella, L.; Stati, T.; Marano, G. Role of beta-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview. Int. J. Mol. Sci. 2021, 22, 8957. [Google Scholar] [CrossRef]
- Subbamanda, Y.D.; Bhargava, A. Intercommunication between Voltage-Gated Calcium Channels and Estrogen Receptor/Estrogen Signaling: Insights into Physiological and Pathological Conditions. Cells 2022, 11, 3850. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.W.; Wang, J.M.; Chen, S.; Brinton, R.D. 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: A potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience 2005, 135, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, T.; Takei, A.; Tsujikado, K.; Inukai, T. Effects of androgens and estrogens on sirtuin 1 gene expression in human aortic endothelial cells. Saudi Med. J. 2020, 41, 361–368. [Google Scholar] [CrossRef]
- Ueda, K.; Fukuma, N.; Adachi, Y.; Numata, G.; Tokiwa, H.; Toyoda, M.; Otani, A.; Hashimoto, M.; Liu, P.Y.; Takimoto, E. Sex Differences and Regulatory Actions of Estrogen in Cardiovascular System. Front. Physiol. 2021, 12, 738218. [Google Scholar] [CrossRef] [PubMed]
- Salerni, S.; Di Francescomarino, S.; Cadeddu, C.; Acquistapace, F.; Maffei, S.; Gallina, S. The different role of sex hormones on female cardiovascular physiology and function: Not only oestrogens. Eur. J. Clin. Investig. 2015, 45, 634–645. [Google Scholar] [CrossRef]
- Niță, A.-R.; Knock, G.A.; Heads, R.J. Signalling mechanisms in the cardiovascular protective effects of estrogen: With a focus on rapid/membrane signalling. Curr. Res. Physiol. 2021, 4, 103–118. [Google Scholar] [CrossRef]
- Mikkola, T.S.; Clair, R.W.S. Estradiol reduces basal and cytokine induced monocyte adhesion to endothelial cells. Maturitas 2002, 41, 313–319. [Google Scholar] [CrossRef]
- Stallone, J.N.; Oloyo, A.K. Cardiovascular and metabolic actions of the androgens: Is testosterone a Janus-faced molecule? Biochem. Pharmacol. 2023, 208, 115347. [Google Scholar] [CrossRef]
- Schiffer, L.; Kempegowda, P.; Arlt, W.; O’Reilly, M.W. Mechanisms in endocrinology: The sexually dimorphic role of androgens in human metabolic disease. Eur. J. Endocrinol. 2017, 177, R125–R143. [Google Scholar] [CrossRef] [Green Version]
- Goodale, T.; Sadhu, A.; Petak, S.; Robbins, R. Testosterone and the Heart. Methodist Debakey Cardiovasc. J. 2017, 13, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, A.M. Testosterone administration in older men. Endocrinol. Metab. Clin. 2013, 42, 271–286. [Google Scholar]
- Heinlein, C.A.; Chang, C. Androgen receptor (AR) coregulators: An overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, M. Testosterone deficiency, insulin resistance and the metabolic syndrome. Nat. Rev. Endocrinol. 2009, 5, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Holmäng, A.; Björntorp, P. The effects of testosterone on insulin sensitivity in male rats. Acta Physiol. Scand. 1992, 146, 505–510. [Google Scholar] [CrossRef]
- Moghetti, P.; Tosi, F.; Castello, R.; Magnani, C.M.; Negri, C.; Brun, E.; Furlani, L.; Caputo, M.; Muggeo, M. The insulin resistance in women with hyperandrogenism is partially reversed by antiandrogen treatment: Evidence that androgens impair insulin action in women. J. Clin. Endocrinol. Metab. 1996, 81, 952–960. [Google Scholar]
- Kloner, R.A.; Carson, C.; Dobs, A.; Kopecky, S.; Mohler, E.R. Testosterone and cardiovascular disease. J. Am. Coll. Cardiol. 2016, 67, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Farias, J.M.; Tinetti, M.; Khoury, M.; Umpierrez, G.E. Low testosterone concentration and atherosclerotic disease markers in male patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, 4698–4703. [Google Scholar] [CrossRef]
- Sopariwala, D.H.; Rios, A.S.; Park, M.K.; Song, M.S.; Kumar, A.; Narkar, V.A. Estrogen-related receptor alpha is an AMPK-regulated factor that promotes ischemic muscle revascularization and recovery in diet-induced obese mice. FASEB Bioadv. 2022, 4, 602–618. [Google Scholar] [CrossRef]
- Qasem, R.J. The estrogenic activity of resveratrol: A comprehensive review of in vitro and in vivo evidence and the potential for endocrine disruption. Crit. Rev. Toxicol. 2020, 50, 439–462. [Google Scholar] [CrossRef]
- Demir, U.; Koehler, A.; Schneider, R.; Schweiger, S.; Klocker, H. Metformin anti-tumor effect via disruption of the MID1 translational regulator complex and AR downregulation in prostate cancer cells. BMC Cancer 2014, 14, 52. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Kevil, C.G.; Spite, M.R. Regulation and maintenance of vascular tone and patency in cardiovascular health and disease. Int. J. Vasc. Med. 2012, 2012, 396369. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Galis, Z.S. Exploring the Role of Endothelial Cell Resilience in Cardiovascular Health and Disease. Arter. Thromb. Vasc. Biol. 2021, 41, 179–185. [Google Scholar] [CrossRef]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diab. Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, P.; Formica, V.; Roselli, M.; Guadagni, F. Thromboembolic events in patients treated with anti-angiogenic drugs. Curr. Vasc. Pharmacol. 2010, 8, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Stead, R.; Musa, M.G.; Bryant, C.L.; Lanham, S.A.; Johnston, D.A.; Reynolds, R.; Torrens, C.; Fraser, P.A.; Clough, G.F. Developmental conditioning of endothelium-derived hyperpolarizing factor-mediated vasorelaxation. J. Hypertens 2016, 34, 452–463; discussion 463. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Sowers, J.R. Endothelial dysfunction potentially interacts with impaired glucose metabolism to increase cardiovascular risk. Hypertension 2014, 64, 1192–1193. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Liu, X.; Dagda, R.K.; Zhang, Y. How AMPK and PKA Interplay to Regulate Mitochondrial Function and Survival in Models of Ischemia and Diabetes. Oxid. Med. Cell Longev. 2017, 2017, 4353510. [Google Scholar] [CrossRef] [Green Version]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.M.; Fulton, D.; Boo, Y.C.; Sorescu, G.P.; Kemp, B.E.; Jo, H.; Sessa, W.C. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 14841–14849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, T.; Kvandova, M.; Schmal, I.; Kalinovic, S.; Stamm, P.; Kuntic, M.; Foretz, M.; Viollet, B.; Daiber, A.; Oelze, M.; et al. Lack of Endothelial alpha1AMPK Reverses the Vascular Protective Effects of Exercise by Causing eNOS Uncoupling. Antioxidants 2021, 10, 1974. [Google Scholar] [CrossRef] [PubMed]
- Kvandová, M.; Rajlic, S.; Stamm, P.; Schmal, I.; Mihaliková, D.; Kuntic, M.; Bayo Jimenez, M.T.; Hahad, O.; Kollárová, M.; Ubbens, H.; et al. Mitigation of aircraft noise-induced vascular dysfunction and oxidative stress by exercise, fasting, and pharmacological α1AMPK activation: Molecular proof of a protective key role of endothelial α1AMPK against environmental noise exposure. Eur. J. Prev. Cardiol. 2023, zwad075. [Google Scholar] [CrossRef]
- Kroller-Schon, S.; Jansen, T.; Hauptmann, F.; Schuler, A.; Heeren, T.; Hausding, M.; Oelze, M.; Viollet, B.; Keaney, J.F., Jr.; Wenzel, P.; et al. alpha1AMP-activated protein kinase mediates vascular protective effects of exercise. Arter. Thromb. Vasc. Biol. 2012, 32, 1632–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Xu, J.; Song, P.; Viollet, B.; Zou, M.H. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase I. Diabetes 2009, 58, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Enkhjargal, B.; Godo, S.; Sawada, A.; Suvd, N.; Saito, H.; Noda, K.; Satoh, K.; Shimokawa, H. Endothelial AMP-activated protein kinase regulates blood pressure and coronary flow responses through hyperpolarization mechanism in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1505–1513. [Google Scholar] [CrossRef] [Green Version]
- Leung, S.W.; Vanhoutte, P.M. Endothelium-dependent hyperpolarization: Age, gender and blood pressure, do they matter? Acta Physiol. 2017, 219, 108–123. [Google Scholar] [CrossRef]
- Li, P.; Zhang, L.; Zhang, M.; Zhou, C.; Lin, N. Uric acid enhances PKC-dependent eNOS phosphorylation and mediates cellular ER stress: A mechanism for uric acid-induced endothelial dysfunction. Int. J. Mol. Med. 2016, 37, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersch, C.; Palii, S.P.; Kim, K.M.; Angerhofer, A.; Johnson, R.J.; Henderson, G.N. Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucleic Acids 2008, 27, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Lanaspa, M.A.; Cristobal-Garcia, M.; Garcia-Arroyo, F.; Soto, V.; Cruz-Robles, D.; Nakagawa, T.; Yu, M.A.; Kang, D.H.; Johnson, R.J. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp. Nephrol. 2012, 121, e71–e78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andres-Hernando, A.; Ishimoto, T.; Sanchez-Lozada, L.G.; et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS ONE 2012, 7, e48801. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Z.; Chen, Y.; Xie, Y.; Li, Y.; Li, Z. Metformin protects against insulin resistance induced by high uric acid in cardiomyocytes via AMPK signalling pathways in vitro and in vivo. J. Cell Mol. Med. 2021, 25, 6733–6745. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.M.; Lind, L.; Pollare, T.; Berne, C.; Lithell, H. Differences in insulin sensitivity and risk markers due to gender and age in hypertensives. J. Hum. Hypertens 2000, 14, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Zippel, N.; Malik, R.A.; Fromel, T.; Popp, R.; Bess, E.; Strilic, B.; Wettschureck, N.; Fleming, I.; Fisslthaler, B. Transforming growth factor-beta-activated kinase 1 regulates angiogenesis via AMP-activated protein kinase-alpha1 and redox balance in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2792–2799. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Dopheide, J.; Schuhmacher, S.; Thomas, S.R.; Chen, K.; Daiber, A.; Wenzel, P.; Munzel, T.; Keaney, J.F., Jr. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation 2008, 118, 1347–1357. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial thiols in antioxidant protection and redox signaling: Distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal 2012, 16, 476–495. [Google Scholar] [CrossRef]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niture, S.K.; Jain, A.K.; Jaiswal, A.K. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-delta-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J. Cell Sci. 2009, 122, 4452–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, T.; Kroller-Schon, S.; Schonfelder, T.; Foretz, M.; Viollet, B.; Daiber, A.; Oelze, M.; Brandt, M.; Steven, S.; Kvandova, M.; et al. alpha1AMPK deletion in myelomonocytic cells induces a pro-inflammatory phenotype and enhances angiotensin II-induced vascular dysfunction. Cardiovasc. Res. 2018, 114, 1883–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroller-Schon, S.; Jansen, T.; Tran, T.L.P.; Kvandova, M.; Kalinovic, S.; Oelze, M.; Keaney, J.F., Jr.; Foretz, M.; Viollet, B.; Daiber, A.; et al. Endothelial alpha1AMPK modulates angiotensin II-mediated vascular inflammation and dysfunction. Basic Res. Cardiol. 2019, 114, 8. [Google Scholar] [CrossRef]
- Nath, N.; Giri, S.; Prasad, R.; Salem, M.L.; Singh, A.K.; Singh, I. 5-aminoimidazole-4-carboxamide ribonucleoside: A novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J. Immunol. 2005, 175, 566–574. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, J.; Wang, X.; Zhang, Y.; Xia, M. AMP-activated protein kinase suppresses endothelial cell inflammation through phosphorylation of transcriptional coactivator p300. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2897–2908. [Google Scholar] [CrossRef] [Green Version]
- Sader, M.A.; Celermajer, D.S. Endothelial function, vascular reactivity and gender differences in the cardiovascular system. Cardiovasc. Res. 2002, 53, 597–604. [Google Scholar] [CrossRef]
- Mathews, L.; Subramanya, V.; Zhao, D.; Ouyang, P.; Vaidya, D.; Guallar, E.; Yeboah, J.; Herrington, D.; Hays, A.G.; Budoff, M.J.; et al. Endogenous Sex Hormones and Endothelial Function in Postmenopausal Women and Men: The Multi-Ethnic Study of Atherosclerosis. J. Womens Health 2019, 28, 900–909. [Google Scholar] [CrossRef]
- Duckles, S.P.; Miller, V.M. Hormonal modulation of endothelial NO production. Pflug. Arch. 2010, 459, 841–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeill, A.M.; Kim, N.; Duckles, S.P.; Krause, D.N. Chronic estrogen treatment increases levels of endothelial nitric oxide synthase protein in rat cerebral microvessels. Stroke 1999, 30, 2186–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, F.; Guo, Y.; Xu, L.; Hou, N.; Han, F.; Sun, X. Induction of Haemeoxygenase-1 Directly Improves Endothelial Function in Isolated Aortas from Obese Rats through the Ampk-Pi3k/Akt-Enos Pathway. Cell Physiol. Biochem. 2015, 36, 1480–1490. [Google Scholar] [CrossRef]
- Nagata, D.; Mogi, M.; Walsh, K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J. Biol. Chem. 2003, 278, 31000–31006. [Google Scholar] [CrossRef] [Green Version]
- Karwi, Q.G.; Biswas, D.; Pulinilkunnil, T.; Lopaschuk, G.D. Myocardial ketones metabolism in heart failure. J. Card. Fail. 2020, 26, 998–1005. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, X.; Zhou, Q.; Tan, B.; Xu, H.; Yi, Q.; Yan, L.; Xie, M.; Zhang, Y.; Tian, J.; et al. Activation of AMPK Promotes Maturation of Cardiomyocytes Derived from Human Induced Pluripotent Stem Cells. Front. Cell Dev. Biol. 2021, 9, 644667. [Google Scholar] [CrossRef]
- Ha, J.; Guan, K.-L.; Kim, J. AMPK and autophagy in glucose/glycogen metabolism. Mol. Asp. Med. 2015, 46, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol. 2006, 574, 95–112. [Google Scholar] [CrossRef]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R. The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta 2016, 1862, 2199–2210. [Google Scholar] [CrossRef]
- Ikeda, Y.; Sato, K.; Pimentel, D.R.; Sam, F.; Shaw, R.J.; Dyck, J.R.; Walsh, K. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J. Biol. Chem. 2009, 284, 35839–35849. [Google Scholar] [CrossRef] [Green Version]
- Molaei, A.; Molaei, E.; Sadeghnia, H.; Hayes, A.W.; Karimi, G. LKB1: An emerging therapeutic target for cardiovascular diseases. Life Sci. 2022, 306, 120844. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 2019, 176, 4302–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Di Florio, D.N.; Sin, J.; Coronado, M.J.; Atwal, P.S.; Fairweather, D. Sex differences in inflammation, redox biology, mitochondria and autoimmunity. Redox Biol. 2020, 31, 101482. [Google Scholar] [CrossRef]
- Solakidi, S.; Psarra, A.G.; Nikolaropoulos, S.; Sekeris, C.E. Estrogen receptors α and β (ERα and ERβ) and androgen receptor (AR) in human sperm: Localization of ERβ and AR in mitochondria of the midpiece. Hum. Reprod. 2005, 20, 3481–3487. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Clapier, R.; Piquereau, J.; Veksler, V.; Garnier, A. Estrogens, estrogen receptors effects on cardiac and skeletal muscle mitochondria. Front. Endocrinol. 2019, 10, 557. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; McDonald, C.; Petrenko, N.B.; Leblanc, M.; Wang, T.; Giguere, V.; Evans, R.M.; Patel, V.V.; Pei, L. Estrogen-related receptor α (ERRα) and ERRγ are essential coordinators of cardiac metabolism and function. Mol. Cell. Biol. 2015, 35, 1281–1298. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Clapier, R.; Moulin, M.; Piquereau, J.; Lemaire, C.; Mericskay, M.; Veksler, V.; Garnier, A. Mitochondria: A central target for sex differences in pathologies. Clin. Sci. 2017, 131, 803–822. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kvandova, M.; Puzserova, A.; Balis, P. Sexual Dimorphism in Cardiometabolic Diseases: The Role of AMPK. Int. J. Mol. Sci. 2023, 24, 11986. https://doi.org/10.3390/ijms241511986
Kvandova M, Puzserova A, Balis P. Sexual Dimorphism in Cardiometabolic Diseases: The Role of AMPK. International Journal of Molecular Sciences. 2023; 24(15):11986. https://doi.org/10.3390/ijms241511986
Chicago/Turabian StyleKvandova, Miroslava, Angelika Puzserova, and Peter Balis. 2023. "Sexual Dimorphism in Cardiometabolic Diseases: The Role of AMPK" International Journal of Molecular Sciences 24, no. 15: 11986. https://doi.org/10.3390/ijms241511986