
High Daytime Temperature Responsive MicroRNA Profiles in Developing Grains of Rice Varieties with Contrasting Chalkiness


Abstract
:1. Introduction
2. Results
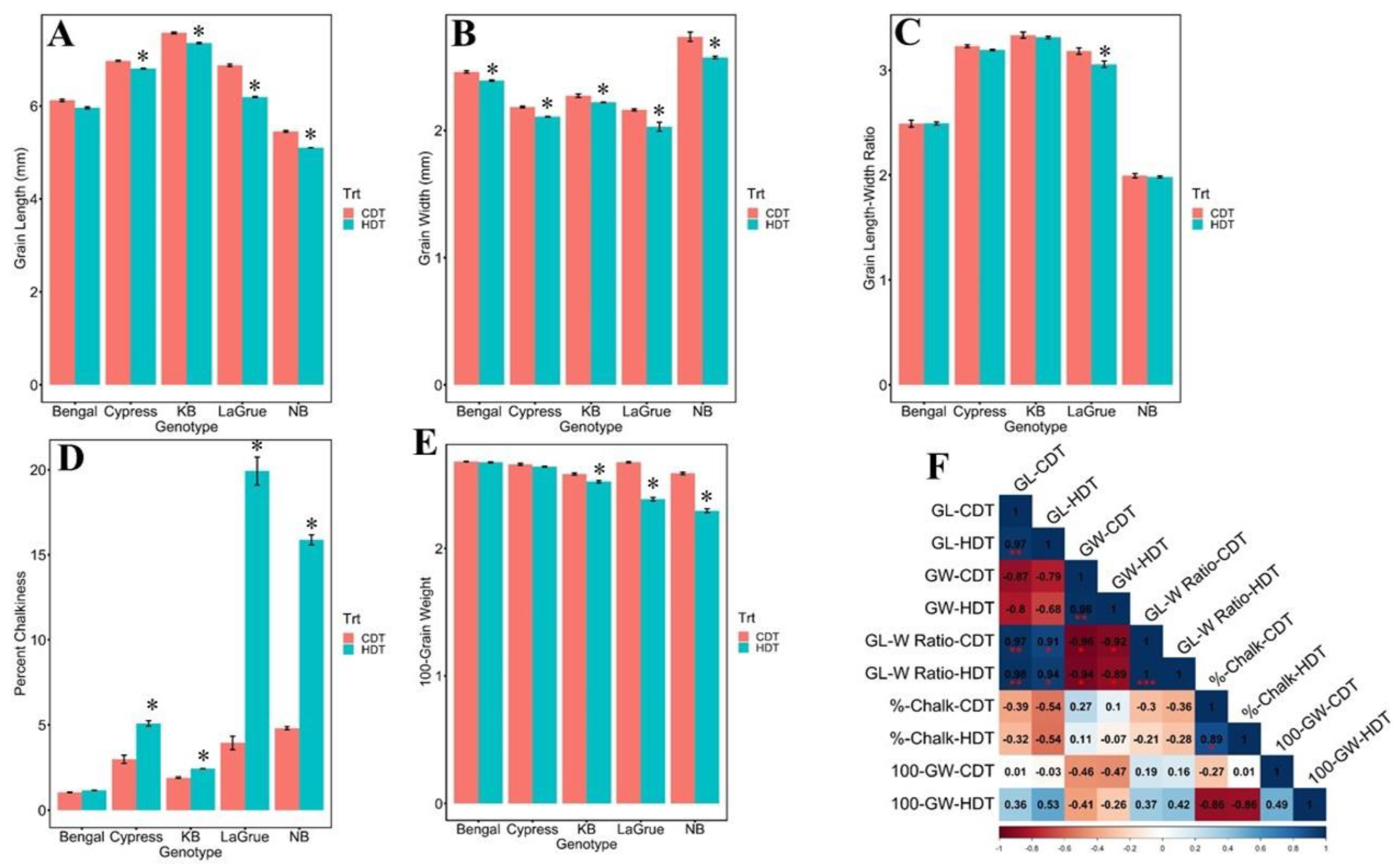
2.1. Phenotypic Variation and Trait Correlation Analyses
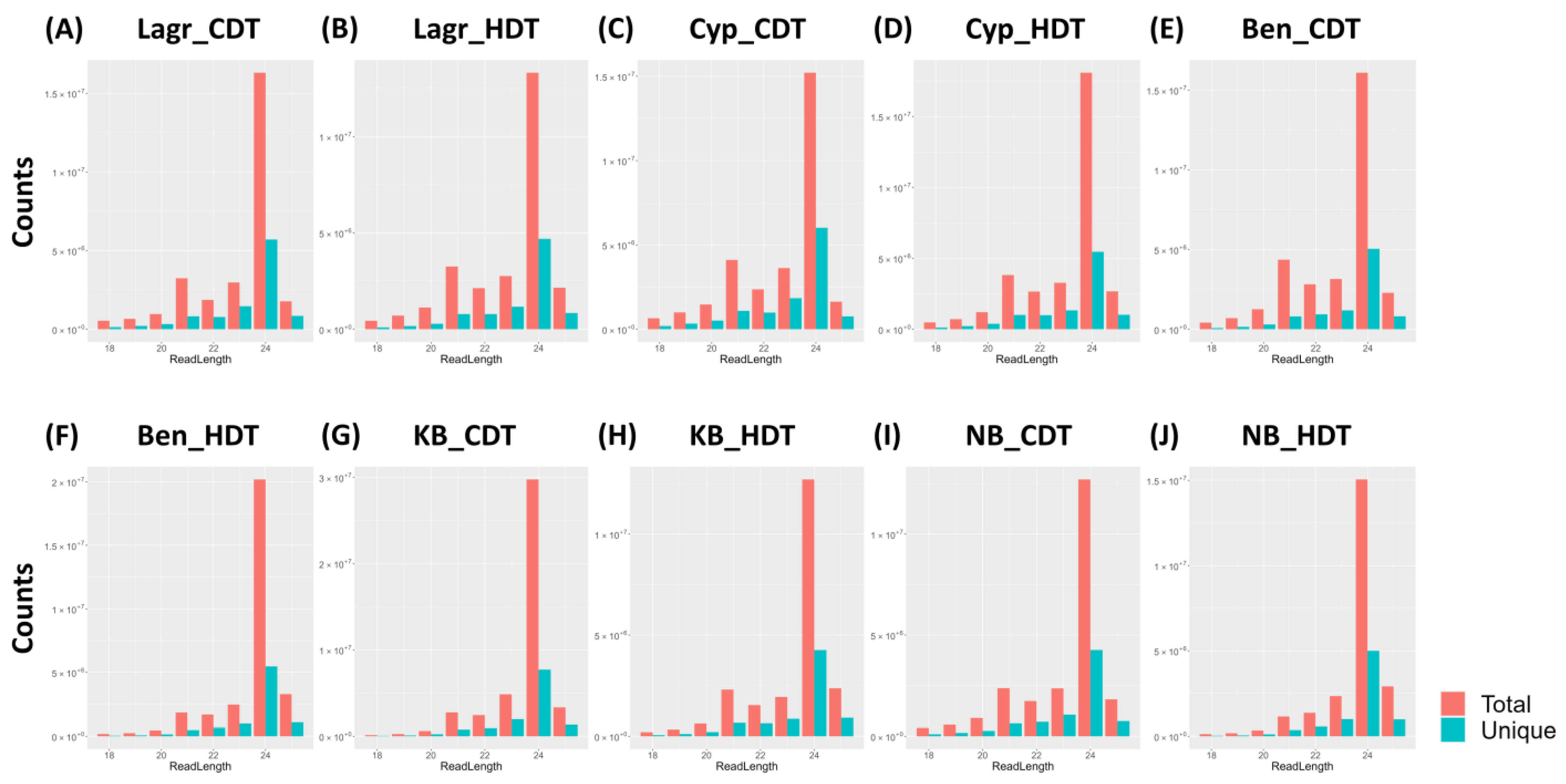
2.2. Summary of sRNA Libraries
2.3. Identification of Novel miRNAs
2.4. Known miRNAs and Their Abundances in R6 Stage Caryopsis Samples
2.5. Differential MicroRNA Expression under HDT in Low Chalky and High Chalky Lines
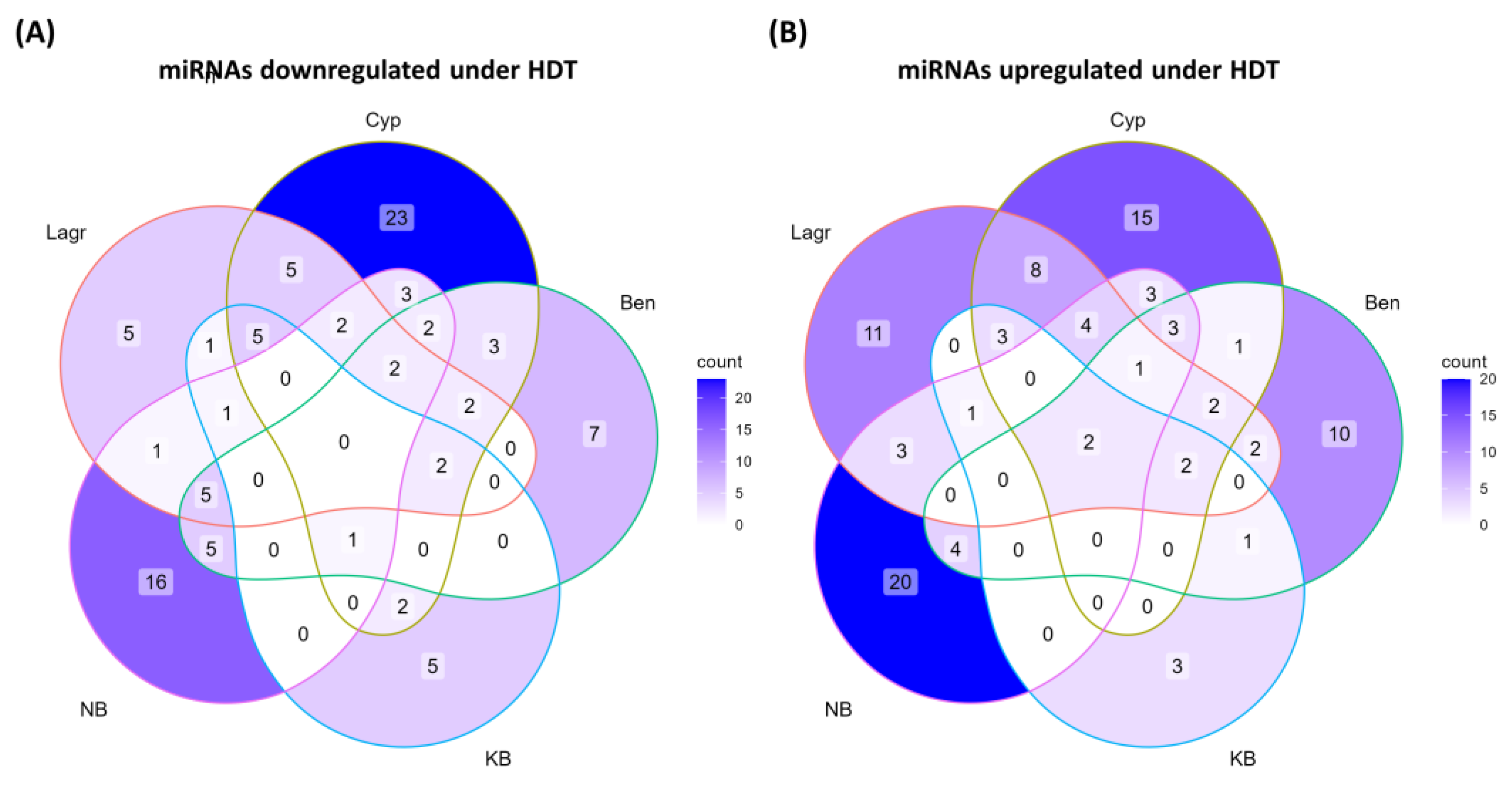
2.5.1. Shared Upregulations among the Low Chalky and High Chalky Genotypes
2.5.2. HDT-Upregulated miRNAs Are Largely Genotype-Specific
2.5.3. Shared Downregulations among the Low Chalky and High Chalky Genotypes
2.5.4. Shared Downregulations among the Low Chalky and High Chalky Genotypes
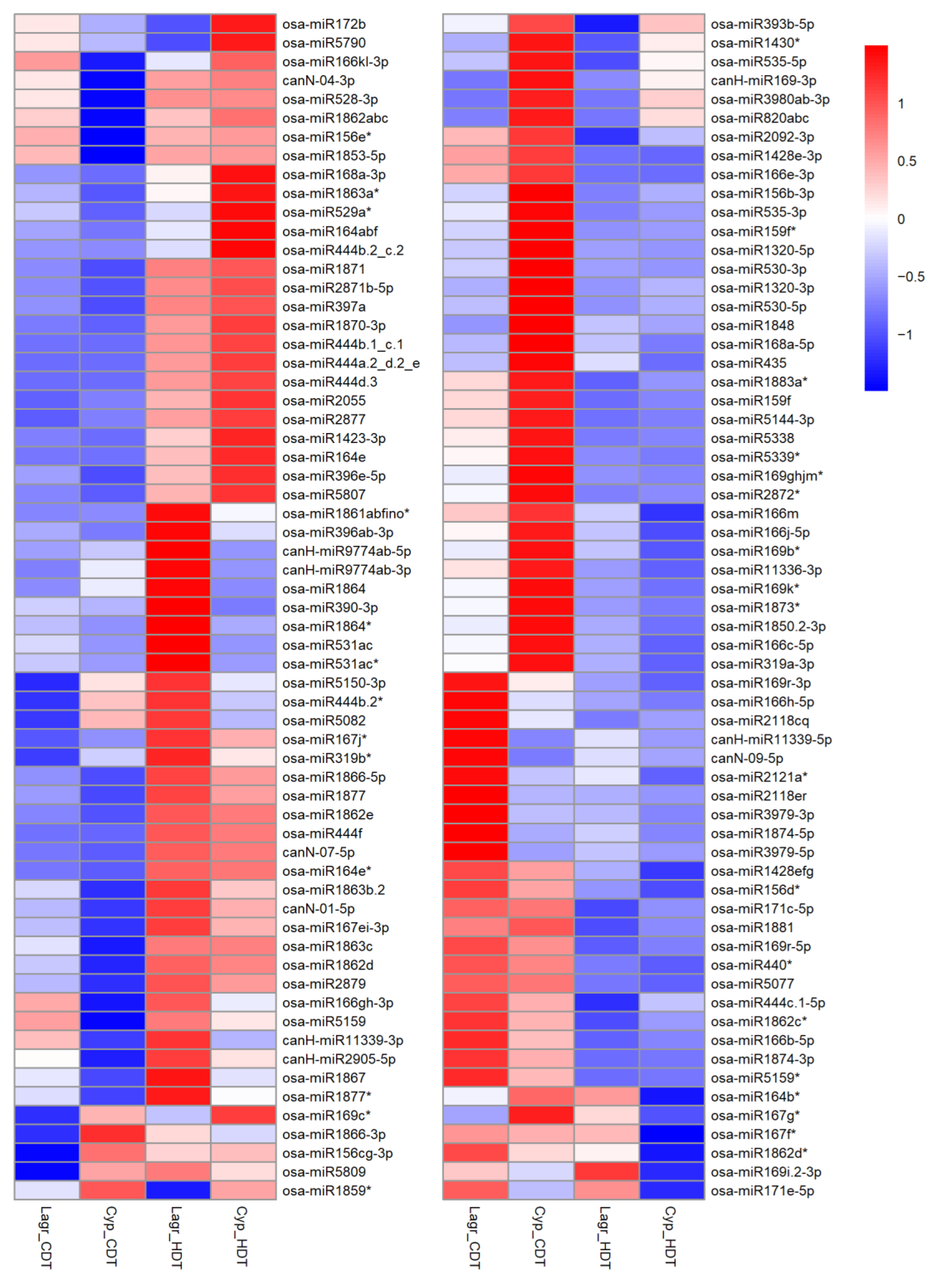
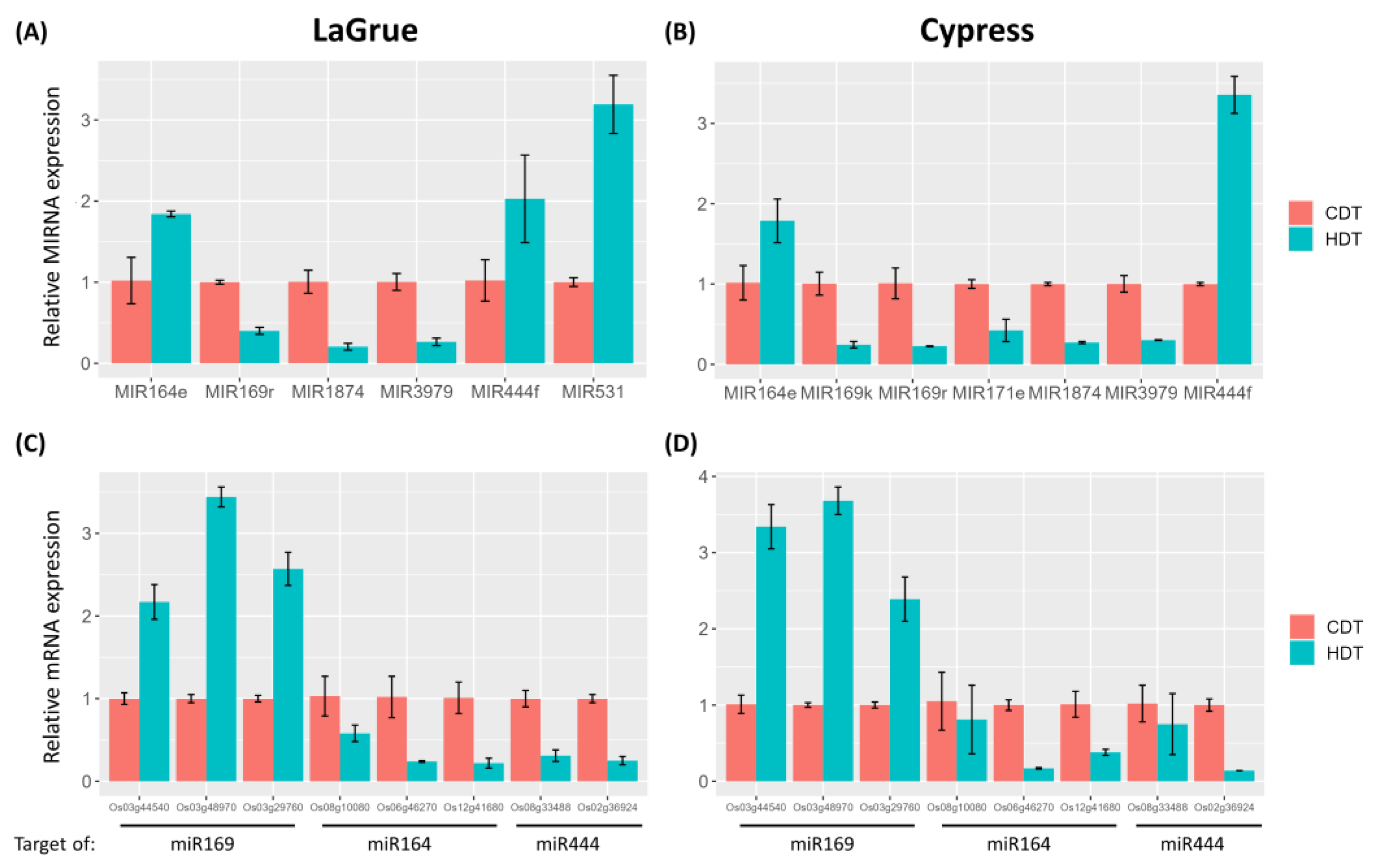
2.6. Comparison of HDT-Responsive miRNAs between Genetically Closely Related Cypress (Low Chalky) and LaGrue (High Chalky) and Their Validations
2.7. Comparison of miRNA Levels between Genetically Closely Related Cypress (Low Chalky) and LaGrue (High Chalky)
2.8. Analysis of the Interaction Term
3. Discussion
3.1. Only Two miRNAs Displayed Shared Regulations among All Five Genotypes under HDT
3.2. HDT-Responsive miRNAs Are Largely Genotype-Specific
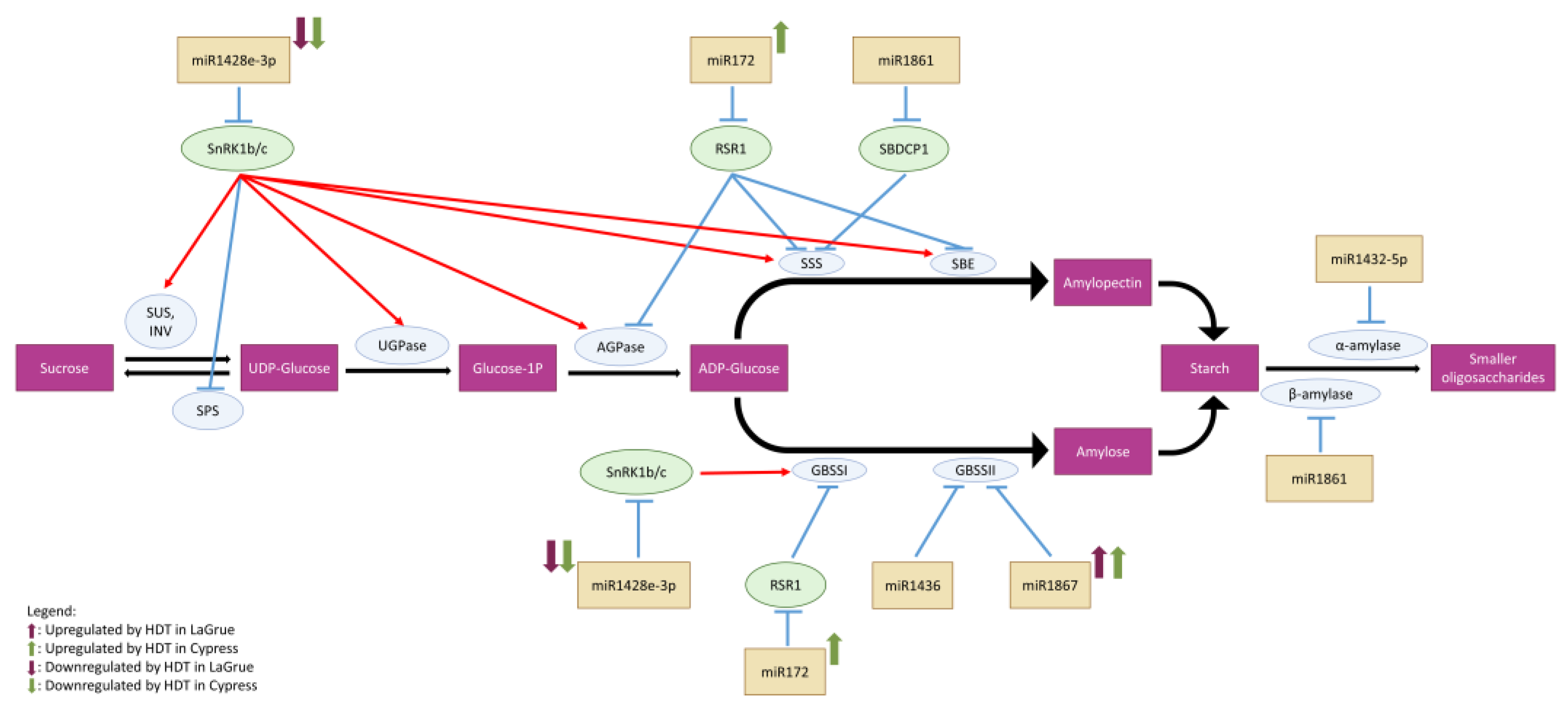
3.3. Differential Regulation of Highly Conserved miRNAs under HDT Suggests an Indirect Role in Grain Filling
3.4. MicroRNA Profiles Differed between Genetically Closely Related Cypress (Low Chalky Line) and LaGrue (High Chalky Line)
3.5. HDT-Responsive miRNA Stars in Rice Caryopsis Tissue
4. Materials and Methods
4.1. Plant Material and Growth Conditions
4.2. Phenotyping, Stress Treatment and Tissue Collection
4.3. Statistical Analysis and Phenotype Evaluation
4.4. Small RNA Libraries and Reads Analysis
4.5. Identification of Novel microRNAs
4.6. MicroRNA Quantification
4.7. Validation of Differentially Expressed miRNAs
4.8. Data Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [Green Version]
- IPCC. Climate Change 2014 Synthesis Report; IPCC: Geneva, Szwitzerland, 2014. [Google Scholar]
- Jagadish, S.K. Heat stress during flowering in cereals--effects and adaptation strategies. New Phytol. 2020, 226, 1567–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Impa, S.; Sunkar, R.; Jagadish, S.K. The neglected other half-role of the pistil in plant heat stress responses. Plant Cell Environ. 2021, 44, 2200–2210. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.; Zhang, H.; Wang, L.; Zhu, Z.; Gao, J.; Li, C.; Zhu, Y. High temperature inhibits the accumulation of storage materials by inducing alternative splicing of OsbZIP58 during filling stage in rice. Plant Cell Environ. 2020, 43, 1879–1896. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Tang, S.; Li, G.; Liu, Z.; Ding, C.; Chen, L.; Wang, S.; Ding, Y. Application of nitrogen fertilizer at heading stage improves rice quality under elevated temperature during grain-filling stage. Crop. Sci. 2017, 57, 2183–2192. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Gupta, C.; Thomas, J.; Pereira, A. Genetic Dissection of Grain Yield Component Traits Under High Nighttime Temperature Stress in a Rice Diversity Panel. Front. Plant Sci. 2021, 12, 712167. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Sasaki, M.; Kuribayashi, N.; Suzuki, H.; Sasuga, Y.; Shiraya, T.; Inomata, T.; Itoh, K.; Baslam, M.; Mitsui, T. Proteomic and glycomic characterization of rice chalky grains produced under moderate and high-temperature conditions in field system. Rice 2016, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Yin, X.; Struik, P.C.; Solis, C.; Xie, F.; Schmidt, R.C.; Huang, M.; Zou, Y.; Ye, C.; Jagadish, S.K. High day-and night-time temperatures affect grain growth dynamics in contrasting rice genotypes. J. Exp. Bot. 2017, 68, 5233–5245. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, T.; Parween, S.; Saito, Y.; Shigemitsu, T.; Yamakawa, H.; Nakazono, M.; Masumura, T.; Nishizawa, N.K.; Kondo, M.; Sreenivasulu, N. Laser microdissection-based tissue-specific transcriptome analysis reveals a novel regulatory network of genes involved in heat-induced grain chalk in rice endosperm. Plant Cell Physiol. 2019, 60, 626–642. [Google Scholar] [CrossRef]
- Chen, L.; Gao, W.; Chen, S.; Wang, L.; Zou, J.; Liu, Y.; Wang, H.; Chen, Z.; Guo, T. High-resolution QTL mapping for grain appearance traits and co-localization of chalkiness-associated differentially expressed candidate genes in rice. Rice 2016, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.T.T.; Ishibashi, Y.; Miyazaki, M.; Tran, H.T.; Okamura, K.; Tanaka, S.; Nakamura, J.; Yuasa, T.; Iwaya-Inoue, M. High Temperature-Induced Repression of the Rice Sucrose Transporter (Os SUT 1) and Starch Synthesis-Related Genes in Sink and Source Organs at Milky Ripening Stage Causes Chalky Grains. J. Agron. Crop. Sci. 2013, 199, 178–188. [Google Scholar] [CrossRef]
- Yamakawa, H.; Hirose, T.; Kuroda, M.Y.; Yamaguchi, T. Comprehensive expression profiling of rice grain filling-related genes under high temperature using DNA microarray. Plant Physiol. 2007, 144, 258–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakata, M.; Kuroda, M.; Miyashita, T.; Yamaguchi, T.; Kojima, M.; Sakakibara, H.; Mitsui, T.; Yamakawa, H. Suppression of α-amylase genes improves quality of rice grain ripened under high temperature. Plant Biotechnol. J. 2012, 10, 1110–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, M.; Araki, M.; Okamura, K.; Ishibashi, Y.; Yuasa, T.; Iwaya-Inoue, M. Assimilate translocation and expression of sucrose transporter, OsSUT1, contribute to high-performance ripening under heat stress in the heat-tolerant rice cultivar Genkitsukushi. J. Plant Physiol. 2013, 170, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.-L.; Zhou, H.-W.; Zhang, H.-Y.; Zhong, P.-A.; Huang, Y.-J. Comparative proteomic analysis of differentially expressed proteins in the early milky stage of rice grains during high temperature stress. J. Exp. Bot. 2014, 65, 655–671. [Google Scholar] [CrossRef] [Green Version]
- Takehara, K.; Murata, K.; Yamaguchi, T.; Yamaguchi, K.; Chaya, G.; Kido, S.; Iwasaki, Y.; Ogiwara, H.; Ebitani, T.; Miura, K. Thermo-responsive allele of sucrose synthase 3 (Sus3) provides high-temperature tolerance during the ripening stage in rice (Oryza sativa L.). Breed. Sci. 2018, 68, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Gann, P.J.; Esguerra, M.; Counce, P.A.; Srivastava, V. Genotype-dependent and heat-induced grain chalkiness in rice correlates with the expression patterns of starch biosynthesis genes. Plant-Environ. Interact. 2021, 2, 165–176. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhu, J.-K. Micro RNAs and short-interfering RNAs in plants. J. Integr. Plant Biol. 2007, 49, 817–826. [Google Scholar] [CrossRef]
- Sunkar, R. MicroRNAs with macro-effects on plant stress responses. Semin. Cell Dev. Biol. 2010, 21, 805–811. [Google Scholar] [CrossRef]
- Sunkar, R.; Chinnusamy, V.; Zhu, J.; Zhu, J.-K. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007, 12, 301–309. [Google Scholar] [CrossRef]
- Sunkar, R.; Li, Y.-F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.-F.; Sunkar, R.; Zhang, W. SeqTar: An effective method for identifying microRNA guided cleavage sites from degradome of polyadenylated transcripts in plants. Nucleic Acids Res. 2012, 40, e28. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zeng, T.; Xia, Q.; Qian, Q.; Yang, C.; Ding, Y.; Chen, L.; Wang, W. Unravelling miRNA regulation in yield of rice (Oryza sativa) based on differential network model. Sci. Rep. 2018, 8, 8498. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.-H.; Spriggs, A.; Matthew, L.; Fan, L.; Kennedy, G.; Gubler, F.; Helliwell, C. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 2008, 18, 1456–1465. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; He, S.; Zhao, N.; Zhai, H.; Liu, Q. A sucrose non-fermenting-1-related protein kinase-1 gene, IbSnRK1, improves starch content, composition, granule size, degree of crystallinity and gelatinization in transgenic sweet potato. Plant Biotechnol. J. 2019, 17, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, F.-F.; Xue, H.-W. Coexpression analysis identifies Rice Starch Regulator1, a rice AP2/EREBP family transcription factor, as a novel rice starch biosynthesis regulator. Plant Physiol. 2010, 154, 927–938. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Z.; Bai, J.; Tao, X.; Wang, L.; Zhang, H.; Zhu, J.-K. Disruption of MIR396e and MIR396f improves rice yield under nitrogen-deficient conditions. Natl. Sci. Rev. 2020, 7, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Teng, Z.; Chen, Y.; Yuan, Y.; Peng, Y.; Yi, Y.; Yu, H.; Yi, Z.; Yang, J.; Peng, Y.; Duan, M.; et al. Identification of microRNAs regulating grain filling of rice inferior spikelets in response to moderate soil drying post-anthesis. Crop. J. 2021, 10, 962–971. [Google Scholar] [CrossRef]
- Peng, T.; Lv, Q.; Zhang, J.; Li, J.; Du, Y.; Zhao, Q. Differential expression of the microRNAs in superior and inferior spikelets in rice (Oryza sativa). J. Exp. Bot. 2011, 62, 4943–4954. [Google Scholar] [CrossRef] [Green Version]
- Chandra, T.; Shaw, B.P. Differential grain filling in apical and basal spikelets of compact panicle rice is associated with difference in expression of miRNAs targeting gene products involved in grain filling. ORYZA-Int. J. Rice 2020, 57, 14–24. [Google Scholar] [CrossRef]
- Misra, G.; Badoni, S.; Parween, S.; Singh, R.K.; Leung, H.; Ladejobi, O.; Mott, R.; Sreenivasulu, N. Genome-wide association coupled gene to gene interaction studies unveil novel epistatic targets among major effect loci impacting rice grain chalkiness. Plant Biotechnol. J. 2021, 19, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Zheng, Y.; Jagadeeswaran, G.; Sunkar, R. Characterization of small RNAs and their target genes in wheat seedlings using sequencing-based approaches. Plant Sci. 2013, 203, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Zheng, Y.; Kudapa, H.; Jagadeeswaran, G.; Hivrale, V.; Varshney, R.K.; Sunkar, R. High throughput sequencing of small RNA component of leaves and inflorescence revealed conserved and novel miRNAs as well as phasiRNA loci in chickpea. Plant Sci. 2015, 235, 46–57. [Google Scholar] [CrossRef]
- Sattar, S.; Song, Y.; Anstead, J.A.; Sunkar, R.; Thompson, G.A. Cucumis melo microRNA expression profile during aphid herbivory in a resistant and susceptible interaction. Mol. Plant-Microbe Interact. 2012, 25, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Hivrale, V.; Zhang, X.; Valliyodan, B.; Lelandais-Brière, C.; Farmer, A.D.; May, G.D.; Crespi, M.; Nguyen, H.T.; Sunkar, R. Small RNA profiles in soybean primary root tips under water deficit. BMC Syst. Biol. 2016, 10, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeve, C.M.; Sunkar, R.; Zheng, Y.; Liu, L.; Liu, Z.; McMullen, M.; Nelson, S.; Sharp, R.E.; Oliver, M.J. Water-deficit responsive microRNAs in the primary root growth zone of maize. BMC Plant Biol. 2019, 19, 126. [Google Scholar] [CrossRef] [Green Version]
- Puli, C.O.R.; Zheng, Y.; Li, Y.-F.; Jagadeeswaran, G.; Suo, A.; Jiang, B.; Sharma, P.; Mann, R.; Ganesan, G.; Gogoi, N.; et al. MicroRNA profiles in Sorghum exposed to individual drought or heat or their combination. J. Plant Biochem. Biotechnol. 2021, 30, 848–861. [Google Scholar] [CrossRef]
- Sunkar, R.; Girke, T.; Jain, P.K.; Zhu, J.-K. Cloning and characterization of microRNAs from rice. Plant Cell 2005, 17, 1397–1411. [Google Scholar] [CrossRef] [Green Version]
- Sunkar, R.; Zhou, X.; Zheng, Y.; Zhang, W.; Zhu, J.-K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.-H.; Park, S.; Zhai, J.; Gurazada, S.G.R.; De Paoli, E.; Meyers, B.C.; Green, P.J. Massive analysis of rice small RNAs: Mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 2011, 23, 4185–4207. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Su, N.; Shen, Y.; Zhang, R.; Wu, F.; Cheng, Z.; Wang, J.; Zhang, X.; Guo, X.; Lei, C.; et al. Identification of novel MiRNAs and MiRNA expression profiling during grain development in indica rice. BMC Genom. 2012, 13, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, T.; Sun, H.; Du, Y.; Zhang, J.; Li, J.; Liu, Y.; Zhao, Y.; Zhao, Q. Characterization and expression patterns of microRNAs involved in rice grain filling. PLoS ONE 2013, 8, e54148. [Google Scholar] [CrossRef] [Green Version]
- Yi, R.; Zhu, Z.; Hu, J.; Qian, Q.; Dai, J.; Ding, Y. Identification and expression analysis of microRNAs at the grain filling stage in rice (Oryza sativa L.) via deep sequencing. PLoS ONE 2013, 8, e57863. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Mishra, S.; Panda, B.B.; Sahu, G.; Dash, S.K.; Shaw, B.P. Study of expressions of miRNAs in the spikelets based on their spatial location on panicle in rice cultivars provided insight into their influence on grain development. Plant Physiol. Biochem. 2021, 159, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, S.; Panigrahy, M.; Kariali, E.; Dash, S.K.; Sahu, B.B.; Sahu, S.K.; Mohapatra, P.K.; Panigrahi, K.C.S. MicroRNAs modulate ethylene induced retrograde signal for rice endosperm starch biosynthesis by default expression of transcriptome. Sci. Rep. 2021, 11, 5573. [Google Scholar] [CrossRef] [PubMed]
- Counce, P.A.; Keisling, T.C.; Mitchell, A.J. A uniform, objective, and adaptive system for expressing rice development. Crop. Sci. 2000, 40, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zheng, Y.; Jagadeeswaran, G.; Li, Y.; Gowdu, K.; Sunkar, R. Identification and temporal expression analysis of conserved and novel microRNAs in Sorghum. Genomics 2011, 98, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Jagadeeswaran, G.; Li, Y.-F.; Sunkar, R. Redox signaling mediates the expression of a sulfate-deprivation-inducible microRNA395 in Arabidopsis. Plant J. 2014, 77, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zheng, Y.; Jagadeeswaran, G.; Ren, R.; Sunkar, R.; Jiang, H. Identification and developmental profiling of conserved and novel microRNAs in Manduca sexta. Insect Biochem. Mol. Biol. 2012, 42, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.M.; Zheng, Y.; Jagadeeswaran, G.; Macmil, S.L.; Graham, W.B.; Roe, B.A.; Desilva, U.; Zhang, W.; Sunkar, R. Cloning, characterization and expression analysis of porcine microRNAs. BMC Genom. 2009, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahuguna, R.N.; Solis, C.A.; Shi, W.; Jagadish, K.S. Post-flowering night respiration and altered sink activity account for high night temperature-induced grain yield and quality loss in rice (Oryza sativa L.). Physiol. Plant. 2017, 159, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-M.; Li, H.-X.; Liu, X.-F.; He, Y.; Zeng, H.-L. Reduction of pyruvate orthophosphate dikinase activity is associated with high temperature-induced chalkiness in rice grains. Plant Physiol. Biochem. 2015, 89, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, K.; Counce, P.; Hardke, J. Rice Growth and Development. In Arkansas Rice Production Handbook; Hardke, J., Goforth, L., Eds.; University of Arkansas, Division of Agriculture, Cooperative Extension Service: Little Rock, AR, USA, 2021. [Google Scholar]
- He, J.; Jiang, Z.; Gao, L.; You, C.; Ma, X.; Wang, X.; Xu, X.; Mo, B.; Chen, X.; Liu, L. Genome-wide transcript and small RNA profiling reveals transcriptomic responses to heat stress. Plant Physiol. 2019, 181, 609–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Li, Y.; Yu, J.; Liu, Y.; Li, C.; Han, X.; Shen, F. Difference in miRNA expression profiles between two cotton cultivars with distinct salt sensitivity. Mol. Biol. Rep. 2012, 39, 4961–4970. [Google Scholar] [CrossRef]
- Mondal, T.K.; Ganie, S.A. Identification and characterization of salt responsive miRNA-SSR markers in rice (Oryza sativa). Gene 2014, 535, 204–209. [Google Scholar] [CrossRef]
- Barrera-Figueroa, B.E.; Gao, L.; Diop, N.N.; Wu, Z.; Ehlers, J.D.; Roberts, P.A.; Close, T.J.; Zhu, J.-K.; Liu, R. Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC Plant Biol. 2011, 11, 127. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, S.; Ragupathy, R.; Edwards, T.; Domaratzki, M.; Cloutier, S. MicroRNA-guided regulation of heat stress response in wheat. BMC Genom. 2019, 20, 488. [Google Scholar] [CrossRef] [Green Version]
- Ragupathy, R.; Ravichandran, S.; Mahdi, M.; Rahman, S.; Huang, D.; Reimer, E.; Domaratzki, M.; Cloutier, S. Deep sequencing of wheat sRNA transcriptome reveals distinct temporal expression pattern of miRNAs in response to heat, light and UV. Sci. Rep. 2016, 6, 39373. [Google Scholar] [CrossRef] [Green Version]
- Mangrauthia, S.K.; Bhogireddy, S.; Agarwal, S.; Prasanth, V.V.; Voleti, S.R.; Neelamraju, S.; Subrahmanyam, D. Genome-wide changes in microRNA expression during short and prolonged heat stress and recovery in contrasting rice cultivars. J. Exp. Bot. 2017, 68, 2399–2412. [Google Scholar] [CrossRef] [Green Version]
- Cakir, B.; Tian, L.; Crofts, N.; Chou, H.-L.; Koper, K.; Ng, C.-Y.; Tuncel, A.; Gargouri, M.; Hwang, S.-K.; Fujita, N.; et al. Re-programming of gene expression in the CS 8 rice line over-expressing ADP glucose pyrophosphorylase induces a suppressor of starch biosynthesis. Plant J. 2019, 97, 1073–1088. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, L.-Q.; Zheng, W.-Y.; Wang, R.-F.; Yang, L.-X. Genome-Wide Identification of Micro RNA s Responsive to High Temperature in Rice (Oryza sativa) by High-Throughput Deep Sequencing. J. Agron. Crop. Sci. 2015, 201, 379–388. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, X.; Liu, Y.; Chen, X. Exploring Heat-Response Mechanisms of MicroRNAs Based on Microarray Data of Rice Post-meiosis Panicle. Int. J. Genom. 2020, 2020, 7582612. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, F.; Cao, H.; Peng, H.; Ni, Z.; Sun, Q.; Yao, Y. TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS ONE 2012, 7, e48445. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.R.; Pathak, H.; Sharma, S.K.; Kala, Y.K.; Nirjal, M.K.; Singh, G.P.; Goswami, S.; Rai, R.D. Novel and conserved heat-responsive microRNAs in wheat (Triticum aestivum L.). Funct. Integr. Genom. 2015, 15, 323–348. [Google Scholar] [CrossRef]
- Zhang, M.; An, P.; Li, H.; Wang, X.; Zhou, J.; Dong, P.; Zhao, Y.; Wang, Q.; Li, C. The miRNA-mediated post-transcriptional regulation of maize in response to high temperature. Int. J. Mol. Sci. 2019, 20, 1754. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Ye, C.; Shen, L.; Cao, Y.; Tu, J.; Yu, J. Unique miRNome in heat tolerant indica rice var. HT54 seedlings. Ecol. Genet. Genom. 2018, 7, 13–22. [Google Scholar] [CrossRef]
- Kruszka, K.; Pacak, A.; Swida-Barteczka, A.; Nuc, P.; Alaba, S.; Wroblewska, Z.; Karlowski, W.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Transcriptionally and post-transcriptionally regulated microRNAs in heat stress response in barley. J. Exp. Bot. 2014, 65, 6123–6135. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Wang, Y.; Yao, Y.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-C.; Xu, H.; Zhu, Y.; Liu, Q.-Q.; Cai, X.-L. OsbZIP58, a basic leucine zipper transcription factor, regulates starch biosynthesis in rice endosperm. J. Exp. Bot. 2013, 64, 3453–3466. [Google Scholar] [CrossRef]
- Pegler, J.L.; Oultram, J.M.; Grof, C.P.; Eamens, A.L. Profiling the abiotic stress responsive microRNA landscape of Arabidopsis thaliana. Plants 2019, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Łyskowski, A.; Jaremko, Ł.; Jaremko, M. Genetic and molecular factors determining grain weight in rice. Front. Plant Sci. 2021, 12, 605799. [Google Scholar] [CrossRef] [PubMed]
- Bello, B.K.; Hou, Y.; Zhao, J.; Jiao, G.; Wu, Y.; Li, Z.; Wang, Y.; Tong, X.; Wang, W.; Yuan, W.; et al. NF-YB 1-YC 12-bHLH 144 complex directly activates Wx to regulate grain quality in rice (Oryza sativa L.). Plant Biotechnol. J. 2019, 17, 1222–1235. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Ren, Y.; Li, W.; Wu, F.; Yang, W.; Huang, X.; Yao, J. NF-YC12 is a key multi-functional regulator of accumulation of seed storage substances in rice. J. Exp. Bot. 2019, 70, 3765–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Parida, S.K.; Agarwal, P.; Tyagi, A.K. Transcription factor OsNF-YB9 regulates reproductive growth and development in rice. Planta 2019, 250, 1849–1865. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, S.; Kazeem, B.B.; Ajadi, A.A.; Luo, J.; Yin, M.; Liu, X.; Chen, L.; Ying, J.; Tong, X.; et al. Five Rice Seed-Specific NF-YC Genes Redundantly Regulate Grain Quality and Seed Germination via Interfering Gibberellin Pathway. Int. J. Mol. Sci. 2022, 23, 8382. [Google Scholar] [CrossRef]
- Abu-Zaitoon, Y.M.; Bennett, K.; Normanly, J.; Nonhebel, H.M. A large increase in IAA during development of rice grains correlates with the expression of tryptophan aminotransferase OsTAR1 and a grain-specific YUCCA. Physiol. Plant. 2012, 146, 487–499. [Google Scholar] [CrossRef]
- Zhao, Y.-F.; Peng, T.; Sun, H.-Z.; Teotia, S.; Wen, H.-L.; Du, Y.-X.; Zhang, J.; Li, J.-Z.; Tang, G.-L.; Xue, H.-W.; et al. miR1432-Os ACOT (Acyl-CoA thioesterase) module determines grain yield via enhancing grain filling rate in rice. Plant Biotechnol. J. 2019, 17, 712–723. [Google Scholar] [CrossRef] [Green Version]
- Panda, S.K.; Sunkar, R. Nutrient-and other stress-responsive microRNAs in plants: Role for thiol-based redox signaling. Plant Signal. Behav. 2015, 10, e1010916. [Google Scholar]
- Zhang, Z.; Wei, L.; Zou, X.; Tao, Y.; Liu, Z.; Zheng, Y. Submergence-responsive microRNAs are potentially involved in the regulation of morphological and metabolic adaptations in maize root cells. Ann. Bot. 2008, 102, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Kanegae, H.; Miyoshi, K.; Hirose, T.; Tsuchimoto, S.; Mori, M.; Nagato, Y.; Takano, M. Expressions of rice sucrose non-fermenting-1 related protein kinase 1 genes are differently regulated during the caryopsis development. Plant Physiol. Biochem. 2005, 43, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Purcell, P.C.; Smith, A.M.; Halford, N.G. Antisense expression of a sucrose non-fermenting-1-related protein kinase sequence in potato results in decreased expression of sucrose synthase in tubers and loss of sucrose-inducibility of sucrose synthase transcripts in leaves. Plant J. 1998, 14, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Ye, Y.; Chen, X.; Wang, J.; Chen, Z.; Zhou, Q. A sucrose non-fermenting-1-related protein kinase 1 gene from potato, StSnRK1, regulates carbohydrate metabolism in transgenic tobacco. Physiol. Mol. Biol. Plants 2017, 23, 933–943. [Google Scholar] [CrossRef]
- McKibbin, R.S.; Muttucumaru, N.; Paul, M.J.; Powers, S.J.; Burrell, M.M.; Coates, S.; Purcell, P.C.; Tiessen, A.; Geigenberger, P.; Halford, N.G. Production of high-starch, low-glucose potatoes through over-expression of the metabolic regulator SnRK1. Plant Biotechnol. J. 2006, 4, 409–418. [Google Scholar] [CrossRef]
- Sugden, C.; Donaghy, P.G.; Halford, N.G.; Hardie, D.G. Two SNF1-related protein kinases from spinach leaf phosphorylate and inactivate 3-hydroxy-3-methylglutaryl-coenzyme A reductase, nitrate reductase, and sucrose phosphate synthase in vitro. Plant Physiol. 1999, 120, 257–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardin, S.C.; Tang, G.-Q.; Scholz, A.; Holtgraewe, D.; Winter, H.; Huber, S.C. Phosphorylation of sucrose synthase at serine 170: Occurrence and possible role as a signal for proteolysis. Plant J. 2003, 35, 588–603. [Google Scholar] [CrossRef] [PubMed]
- Chikano, H.; Ogawa, M.; Ikeda, Y.; Koizumi, N.; Kusano, T.; Sano, H. Two novel genes encoding SNF1-related protein kinases from Arabidopsis thaliana: Differential accumulation of AtSR1 and AtSR2 transcripts in response to cytokinins and sugars, and phosphorylation of sucrose synthase by AtSR2. Mol. Gen. Genet. MGG 2001, 264, 674–681. [Google Scholar] [CrossRef]
- Duncan, K.A.; Hardin, S.C.; Huber, S.C. The three maize sucrose synthase isoforms differ in distribution, localization, and phosphorylation. Plant Cell Physiol. 2006, 47, 959–971. [Google Scholar] [CrossRef] [Green Version]
- Hardin, S.C.; Winter, H.; Huber, S.C. Phosphorylation of the amino terminus of maize sucrose synthase in relation to membrane association and enzyme activity. Plant Physiol. 2004, 134, 1427–1438. [Google Scholar] [CrossRef] [Green Version]
- Ferrero, D.M.; Piattoni, C.V.; Asencion Diez, M.D.; Rojas, B.E.; Hartman, M.D.; Ballicora, M.A.; Iglesias, A.A. Phosphorylation of ADP-glucose pyrophosphorylase during wheat seeds development. Front. Plant Sci. 2020, 11, 1058. [Google Scholar] [CrossRef]
- Tiessen, A.; Prescha, K.; Branscheid, A.; Palacios, N.; McKibbin, R.; Halford, N.G.; Geigenberger, P. Evidence that SNF1-related kinase and hexokinase are involved in separate sugar-signalling pathways modulating post-translational redox activation of ADP-glucose pyrophosphorylase in potato tubers. Plant J. 2003, 35, 490–500. [Google Scholar] [CrossRef]
- Nakata, M.; Fukamatsu, Y.; Miyashita, T.; Hakata, M.; Kimura, R.; Nakata, Y.; Kuroda, M.; Yamaguchi, T.; Yamakawa, H. High temperature-induced expression of rice α-amylases in developing endosperm produces chalky grains. Front. Plant Sci. 2017, 8, 2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, K.; Ou, X.; Tang, H.; Wang, R.; Wu, P.; Jia, Y.; Wei, X.; Xu, X.; Kang, S.-H.; Kim, S.-K.A.O. Rice microRNA osa-miR1848 targets the obtusifoliol 14alpha-demethylase gene Os CYP 51G3 and mediates the biosynthesis of phytosterols and brassinosteroids during development and in response to stress. New Phytol. 2015, 208, 790–802. [Google Scholar] [CrossRef] [Green Version]
- Kushawaha, A.K.; Khan, A.; Sopory, S.K.; Sanan-Mishra, N. Priming by High Temperature Stress Induces MicroRNA Regulated Heat Shock Modules Indicating Their Involvement in Thermopriming Response in Rice. Life 2021, 11, 291. [Google Scholar] [CrossRef]
- Lin, S.-K.; Chang, M.-C.; Tsai, Y.-G.; Lur, H.-S. Proteomic analysis of the expression of proteins related to rice quality during caryopsis development and the effect of high temperature on expression. Proteomics 2005, 5, 2140–2156. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Zeng, X.; Jiao, Z.; Li, M.; Xu, W.; Nong, Q.; Mo, H.; Cheng, T.; Zhang, M. Formation of protein disulfide bonds catalyzed by OsPDIL1; 1 is mediated by microRNA5144-3p in rice. Plant Cell Physiol. 2018, 59, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Wang, Y.; Liu, X.; Jiang, L.; Ren, Y.; Liu, F.; Peng, C.; Li, J.; Jin, X.; Wu, F.; et al. The failure to express a protein disulphide isomerase-like protein results in a floury endosperm and an endoplasmic reticulum stress response in rice. J. Exp. Bot. 2012, 63, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, Y.; Jagadeeswaran, G.; Ren, R.; Sunkar, R.; Jiang, H. Identification of conserved and novel microRNAs in Manduca sexta and their possible roles in the expression regulation of immunity-related genes. Insect Biochem. Mol. Biol. 2014, 47, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Pokoo, R.; Ren, S.; Wang, Q.; Motes, C.M.; Hernandez, T.D.; Ahmadi, S.; Monteros, M.J.; Zheng, Y.; Sunkar, R. Genotype-and tissue-specific miRNA profiles and their targets in three alfalfa (Medicago sativa L.) genotypes. BMC Genom. 2018, 19, 115–131. [Google Scholar] [CrossRef]
- Liu, W.-W.; Meng, J.; Cui, J.; Luan, Y.-S. Characterization and function of microRNA∗s in plants. Front. Plant Sci. 2017, 8, 2200. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, H.; Gao, S.; Wang, W.-C.; Katiyar-Agarwal, S.; Huang, H.-D.; Raikhel, N.; Jin, H. Arabidopsis Argonaute 2 regulates innate immunity via miRNA393∗-mediated silencing of a Golgi-localized SNARE gene, MEMB12. Mol. Cell 2011, 42, 356–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devers, E.A.; Branscheid, A.; May, P.; Krajinski, F. Stars and symbiosis: microRNA-and microRNA*-mediated transcript cleavage involved in arbuscular mycorrhizal symbiosis. Plant Physiol. 2011, 156, 1990–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A. Development and Characterization of Rice Genotypes for Water Use Efficiency and Drought Resistance. Ph.D. Dissertation, University of Arkansas, Fayetteville, AR USA, 2017. [Google Scholar]
- Ghadirnezhad, R.; Fallah, A. Temperature effect on yield and yield components of different rice cultivars in flowering stage. Int. J. Agron. 2014, 2014, 846707. [Google Scholar] [CrossRef]
- Counce, P.A.; Siebenmorgan, T.J.; Ambardekar, A.A. Rice reproductive stage thermal time and calendar day intervals for six US cultivars in the Grand Prairie, Arkansas, over 4 years. Ann. App. Biol 2015, 3, 4746. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 12 October 2022).
- Wickham, H.; Chang, W.; Henry, L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D. Package ‘’ggplot2’’. ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics. Available online: https://github.com/tidyverse/ggplot2 (accessed on 13 October 2022).
- Revelle, W. Package ‘’psych’’. The Comprehensive R Archive Network. Available online: https://CRAN.R-project.org/package=psych (accessed on 13 October 2022).
- Leggett, R.M.; Ramirez-Gonzalez, R.H.; Clavijo, B.; Waite, D.; Davey, R.P. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Front. Genet. 2013, 4, 288. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.; Li, Y.; Song, T.; Wu, Y.; Fang, S.; Bu, D.; Li, H.; Sun, L.; Pei, D.; et al. NONCODEV6: An updated database dedicated to long non-coding RNA annotation in both animals and plants. Nucleic Acids Res. 2021, 49, D165–D171. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Meyers, B.C. Revisiting criteria for plant microRNA annotation in the era of big data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C (T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Allaire, J.J. RStudio: Integrated Development Environment for R; RStudio, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Kolde, R.; Kolde, M.R. pheatmap: Pretty Heatmaps. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 13 October 2022).
- Gao, C.-H.; Yu, G.; Cai, P. GgVennDiagram: An intuitive, easy-to-use, and highly customizable R package to generate venn diagram. Front. Genet. 2021, 12, 706907. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Five Prime Sequence | Three Prime Sequence |
|---|---|---|
| canH 1-MIR11339 | AGCAUUGCCCAUAUUCACAUAGAU | AUAUAUGAAUGUGGCCAAUGCUAA2 |
| canH-MIR169 | AGCCAAGGAUGACUUGCCGGC | CGGCAAGUUUGUCCUUGGCUAC |
| canH-MIR2905 | GUCUUUAUCACUGACAUGUAGGCC | CUCACAUGUCAAUGACAAAGGCAC |
| canH-MIR9774a | AGAAAUACCCAAUAUCUUGCUGA | AGCAAGAUAUUGGGUAUUUCUUU |
| canH-MIR9774b | AGAAAUACCCAAUAUCUUGCUGA | AGCAAGAUAUUGGGUAUUUCUUU |
| canN 1-01 | AUGUGGGUCGCUGACAUAUGGGCC | CCCACAUGUCAGCCACUCAAAGUCA |
| canN-02 | UACUCGAUUAGAUACCACCUAAUA | CUAGGUGGUGUCUACUCGAGUUUU |
| canN-03 | ACUCAGCCUAGGAGGGGAUGCGAC | CACAUCCCCUCAUCGGUUGAGUUU |
| canN-04 | GGACCAUCCACAUGUGAGGAGCUA | AGUCCUCACGUGGGCAUAGUCUCC |
| canN-05 | UACUAGAGUUACUUCCACUUUGAA | UAAAGUGUAGGCACCUCUAGUACA |
| canN-06 | AAUGGCUUGUCUUGUUUUGUGUGC | ACGCAAAACGAGCCAAGUCAUUAU |
| canN-07 | UCGCCGCGGCUGGCAUCAGCA | CUGGCGCGAGCGGCGGCGAGG |
| canN-08 | GAACUGCAUGGGAAAUUUUGUU | CAAAAUUUUCCAUGCACUUCGA |
| canN-09 | GAAGUGCAUGGGGAAUUUUGUU | CGAAAUUUUCCAUGCACUUCGA |
| Name | Mature Sequence |
|---|---|
| Osa-miR164e | UGGAGAAGCAGGGCACGUGAG |
| Osa-miR169k | UAGCCAAGGAUGACUUGCCUG |
| Osa-miR169r | UAGCCAAGGAUGAUUUGCCUG |
| Osa-miR171e | UGAUUGAGCCGUGCCAAUAUC |
| Osa-miR444f | UGCAGUUGUUGCCUCAAGCUU |
| Osa-miR531ac | CUCGCCGGGGCUGCGUGCCGCCAU |
| Osa-miR1874 | UAUGGAUGGAGGUGUAACCCGAUG |
| Osa-miR3979 | CUUCGGGGGAGGAGAGAAGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Payne, D.; Li, Y.; Govindan, G.; Kumar, A.; Thomas, J.; Addo-Quaye, C.A.; Pereira, A.; Sunkar, R. High Daytime Temperature Responsive MicroRNA Profiles in Developing Grains of Rice Varieties with Contrasting Chalkiness. Int. J. Mol. Sci. 2023, 24, 11631. https://doi.org/10.3390/ijms241411631
Payne D, Li Y, Govindan G, Kumar A, Thomas J, Addo-Quaye CA, Pereira A, Sunkar R. High Daytime Temperature Responsive MicroRNA Profiles in Developing Grains of Rice Varieties with Contrasting Chalkiness. International Journal of Molecular Sciences. 2023; 24(14):11631. https://doi.org/10.3390/ijms241411631
Chicago/Turabian StylePayne, David, Yongfang Li, Ganesan Govindan, Anuj Kumar, Julie Thomas, Charles A. Addo-Quaye, Andy Pereira, and Ramanjulu Sunkar. 2023. "High Daytime Temperature Responsive MicroRNA Profiles in Developing Grains of Rice Varieties with Contrasting Chalkiness" International Journal of Molecular Sciences 24, no. 14: 11631. https://doi.org/10.3390/ijms241411631