
Multiomics Reveals the Regulatory Mechanisms of Arabidopsis Tissues under Heat Stress
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
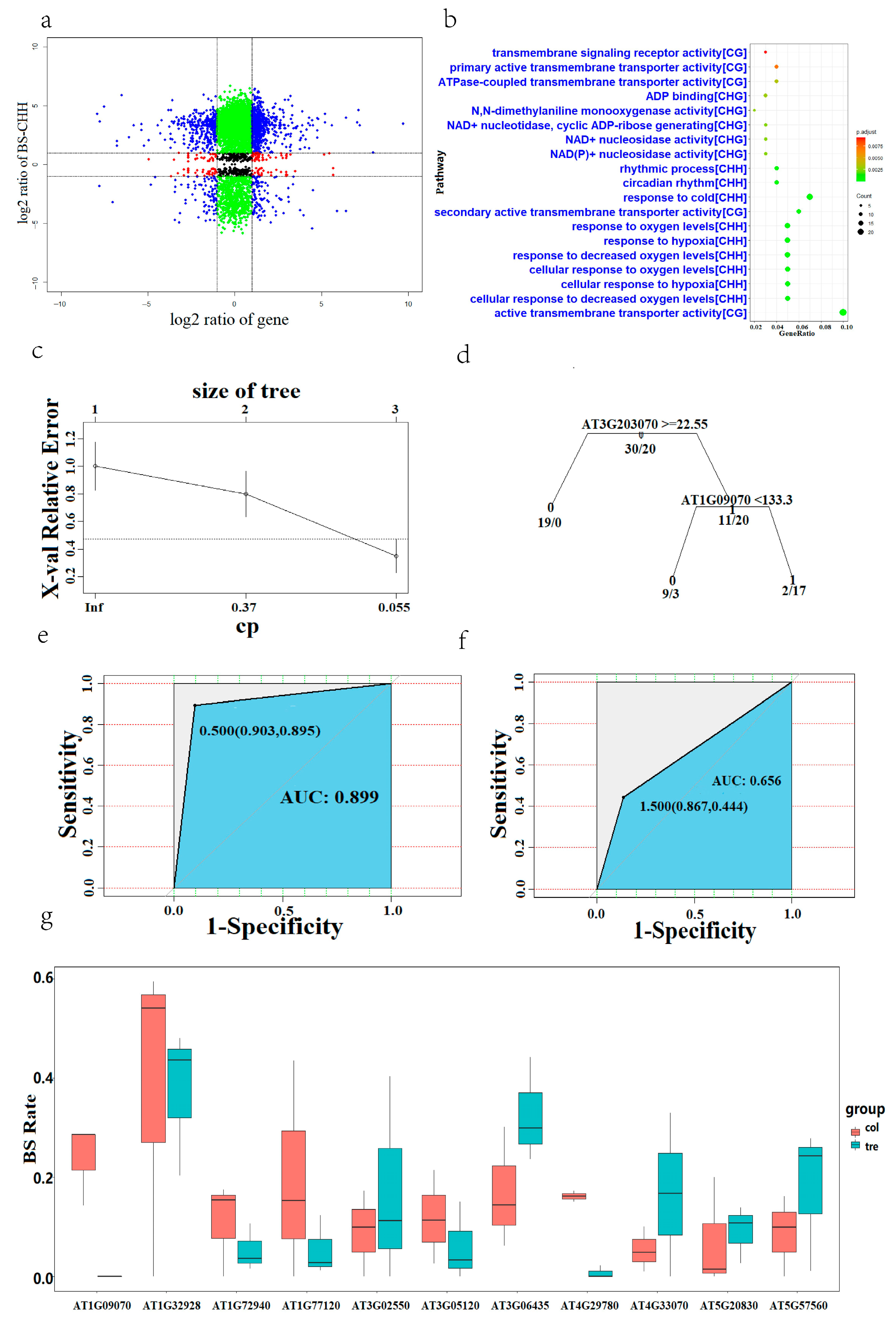{kind=link}
Abstract
:1. Introduction
2. Results
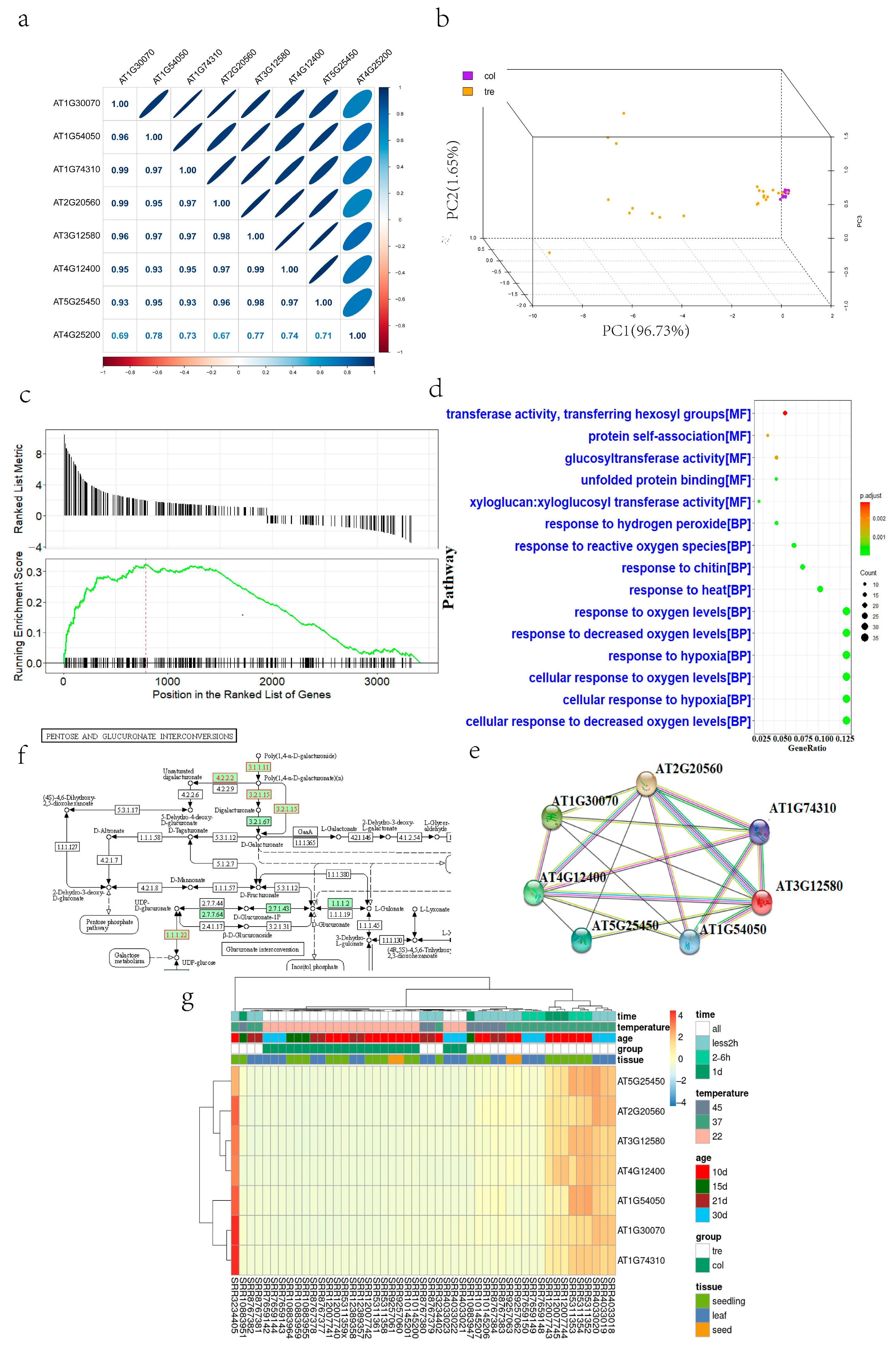
2.1. Transcriptome Advanced Analysis of Heat-Stressed Arabidopsis
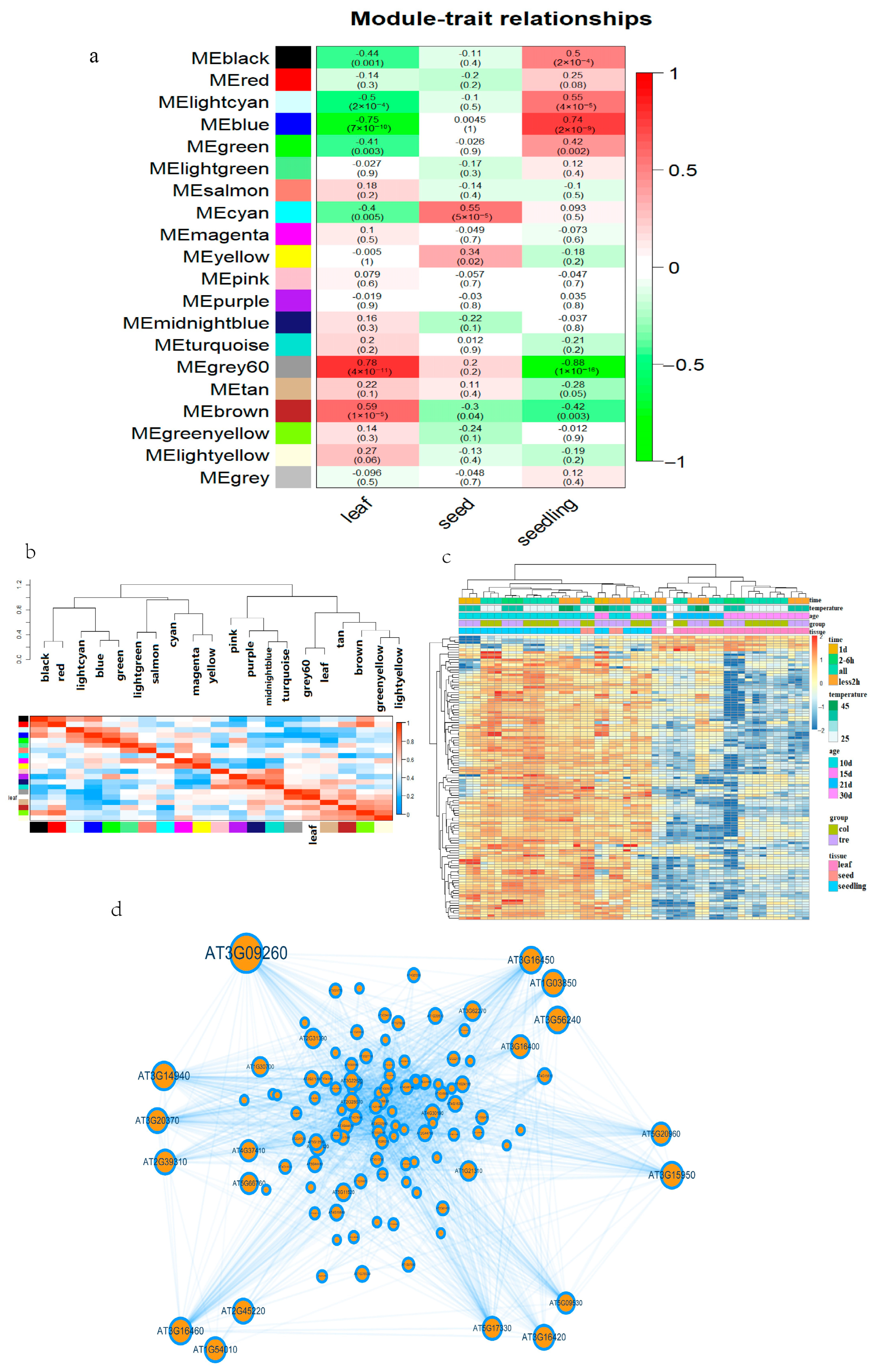
2.2. Weighted Gene Co-Expression Network Analysis (WGCNA) of the Transcriptome
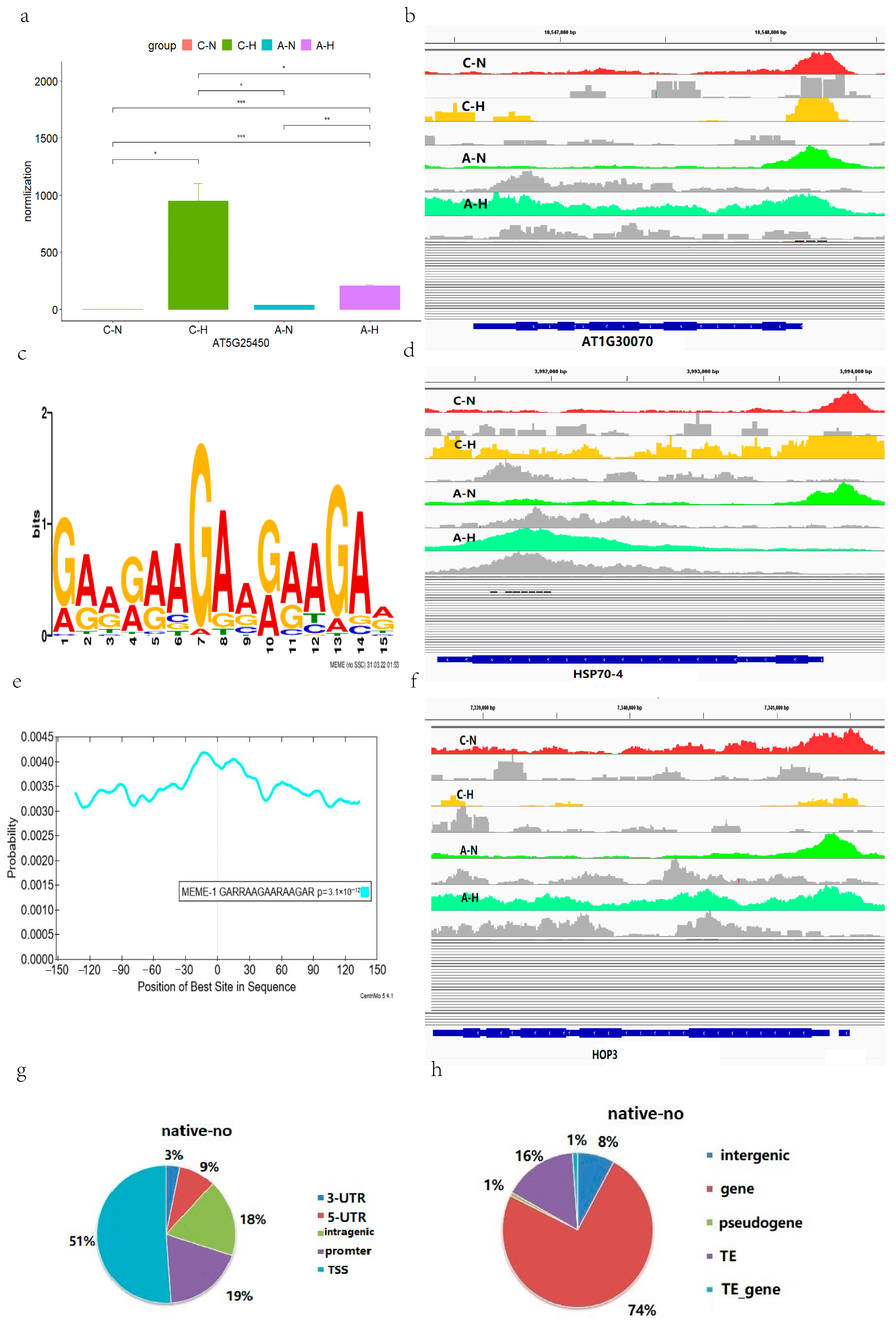
2.3. Regulation Patterns of Transcription Factor A1B in Arabidopsis
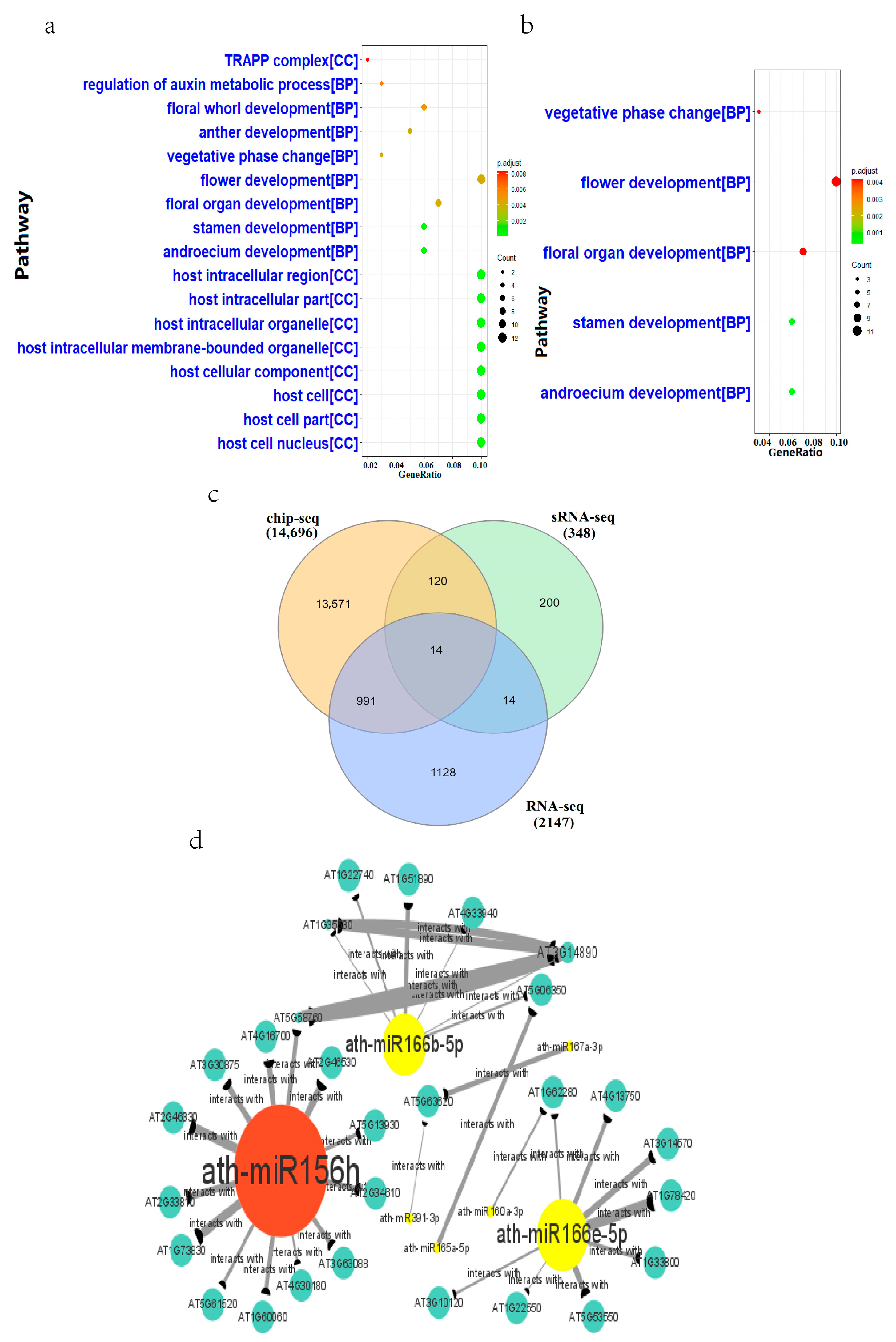
2.4. MicroRNA Regulatory Pathways in Heat-Stressed Arabidopsis
2.5. DNA Methylation Regulation Patterns Exist in Heat-Stressed Arabidopsis
3. Discussion
3.1. Core Genes and Tissue Specificity in Heat-Stressed Arabidopsis
3.2. Altered Binding Profiles of Transcription Factor A1B in Heat-Stressed Arabidopsis
3.3. Core microRNAs in Heat Stressed Arabidopsis
3.4. CHH Methylation in Response to Heat Stress in Arabidopsis
4. Materials and Methods
4.1. Screening of Differential Genes in the Transcriptome of Heat-Stressed Arabidopsis thaliana
4.2. Weighted Gene Co-Expression Network Analysis (WGCNA) of Transcriptome
4.3. Enrichment Analysis of Heat Stress in Arabidopsis
4.4. PCA and String of Core Genes
4.5. Chip-Seq Was Used to Explore Transcription Factor Changes in Response to Heat Stress
4.6. Exploration of Core microRNAs as Well as Their Regulatory Pathways
4.7. Probing Patterns of Methylation Regulation
4.8. Search for Core Regulatory Genes Using Decision Trees
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zioutopoulou, A.; Patitaki, E.; Xu, T.; Kaiserli, E. The Epigenetic Mechanisms Underlying Thermomorphogenesis and Heat Stress Responses in Arabidopsis. Plants 2021, 10, 2439. [Google Scholar] [CrossRef]
- Zuo, Z.-F.; He, W.; Li, J.; Mo, B.; Liu, L. Small RNAs: The Essential Regulators in Plant Thermotolerance. Front. Plant Sci. 2021, 12, 726762. [Google Scholar] [CrossRef]
- Lee, H.; Yoo, S.J.; Lee, J.H.; Kim, W.; Yoo, S.K.; Fitzgerald, H.; Carrington, J.C.; Ahn, J.H. Genetic framework for flower-ing-time regulation by ambient temperature-responsive miRNAs in Arabidopsis. Nucleic Acids Res. 2010, 38, 3081–3093. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Huebner, M.; Weber, A.P. Transcriptional profiling of Arabidopsis heat shock proteins and transcription fac-tors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genom. 2010, 8, 125. [Google Scholar]
- Baniwal, S.K.; Chan, K.Y.; Scharf, K.-D.; Nover, L. Role of Heat Stress Transcription Factor HsfA5 as Specific Repressor of HsfA. J. Biol. Chem. 2007, 282, 3605–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [Green Version]
- Scharf, K.-D.; Berberich, T.; Ebersberger, I.; Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 104–119. [Google Scholar] [CrossRef]
- Perrella, G.; Bäurle, I.; van Zanten, M. Epigenetic regulation of thermomorphogenesis and heat stress tolerance. New Phytol. 2022, 234, 1144–1160. [Google Scholar] [CrossRef]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollunder, J.; Meins, F., Jr.; Kovalchuk, I. Transgenera-tional adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 2010, 5, e9514. [Google Scholar] [CrossRef]
- Chan, S.W.-L.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef]
- Liu, S.; de Jonge, J.; Trejo-Arellano, M.S.; Santos-González, J.; Köhler, C.; Hennig, L. Role of H1 and DNA methylation in selective regulation of transposable elements during heat stress. New Phytol. 2021, 229, 2238–2250. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Wang, Y.; Yao, Y.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Diverse set of microRNAs are responsive to powdery mildew in-fection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barciszewska-Pacak, M.; Milanowska, K.; Knop, K.; Bielewicz, D.; Nuc, P.; Plewka, P.; Pacak, A.M.; Vazquez, F.; Karlowski, W.; Jarmolowski, A.; et al. Arabidopsis microRNA expression regulation in a wide range of abiotic stress responses. Front. Plant Sci. 2015, 6, 410. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhu, J.; Dong, Z. Immediate transcriptional responses of Arabidopsis leaves to heat shock. J. Integr. Plant Biol. 2021, 63, 468–483. [Google Scholar] [CrossRef]
- Sun, L.; Jing, Y.; Liu, X.; Li, Q.; Xue, Z.; Cheng, Z.; Wang, D.; He, H.; Qian, W. Heat stress-induced transposon activation corre-lates with 3D chromatin organization rearrangement in Arabidopsis. Nat. Commun. 2020, 11, 1886. [Google Scholar] [CrossRef] [Green Version]
- Furey, T.S. ChIP-seq and beyond: New and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 2012, 13, 840–852. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.; Muiño, J.M.; Østerås, M.; Farinelli, L.; Krajewski, P.; Angenent, G.C. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat. Protoc. 2010, 5, 457–472. [Google Scholar] [CrossRef]
- Yoo, C.Y.; Miura, K.; Jin, J.B.; Lee, J.; Park, H.C.; Salt, D.E.; Yun, D.-J.; Bressan, R.A.; Hasegawa, P.M. SIZ1 Small Ubiquitin-Like Modifier E3 Ligase Facilitates Basal Thermotolerance in Arabidopsis Independent of Salicylic Acid. Plant Physiol. 2006, 142, 1548–1558. [Google Scholar] [CrossRef] [Green Version]
- Catala, R.; Ouyang, J.; Abreu, I.A.; Hu, Y.; Seo, H.; Zhang, X.; Chua, N.-H. The Arabidopsis E3 SUMO Ligase SIZ1 Regulates Plant Growth and Drought Responses. Plant Cell 2007, 19, 2952–2966. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Chen, C.; Xia, S.; Liu, J.; Shu, J.; Nguyen, V.; Lai, J.; Cui, Y.; Yang, C. Chromatin-associated SUMOylation controls the transcriptional switch between plant development and heat stress responses. Plant Commun. 2020, 2, 100091. [Google Scholar] [CrossRef]
- Cortijo, S.; Charoensawan, V.; Brestovitsky, A.; Buning, R.; Ravarani, C.; Rhodes, D.; van Noort, J.; Jaeger, K.E.; Wigge, P.A. Transcriptional Regulation of the Ambient Temperature Response by H2A.Z Nucleosomes and HSF1 Transcription Factors in Arabidopsis. Mol. Plant 2017, 10, 1258–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, T.-F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-Wide Demethylation of Arabidopsis Endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korotko, U.; Chwiałkowska, K.; Sańko-Sawczenko, I.; Kwasniewski, M. DNA Demethylation in Response to Heat Stress in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 22, 1555. [Google Scholar] [CrossRef]
- Malabarba, J.; Windels, D.; Xu, W.; Verdier, J. Regulation of DNA (de)Methylation Positively Impacts Seed Germination during Seed Development under Heat Stress. Genes 2021, 12, 457. [Google Scholar] [CrossRef]
- Prändl, R.; Hinderhofer, K.; Eggers-Schumacher, G.; Schöffl, F. HSF3, a new heat shock factor from Arabidopsis thaliana, derepresses the heat shock response and confers thermotolerance when overexpressed in transgenic plants. Mol. Genet. Genom. 1998, 258, 269–278. [Google Scholar] [CrossRef]
- Bechtold, U.; Albihlal, W.S.; Lawson, T.; Fryer, M.J.; Sparrow, P.A.; Richard, F.; Persad, R.; Bowden, L.; Hickman, R.; Martin, C.; et al. Arabidopsis HEAT SHOCK TRANSCRIPTION FACTORA1b overexpression enhances water productivity, resistance to drought, and infection. J. Exp. Bot. 2013, 64, 3467–3481. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.S.; Crisp, P.A.; Estavillo, G.M.; Cole, B.; Hong, F.; Mockler, T.C.; Pogson, B.J.; Chory, J. Subset of heat-shock transcrip-tion factors required for the early response of Arabidopsis to excess light. Proc. Natl. Acad. Sci. USA 2013, 110, 14474–14479. [Google Scholar] [CrossRef]
- Albihlal, W.S.; Obomighie, I.; Blein, T.; Persad, R.; Chernukhin, I.; Crespi, M.; Bechtold, U.; Mullineaux, P.M. Arabidopsis HEAT SHOCK TRANSCRIPTION FACTORA1b regulates multiple developmental genes under benign and stress conditions. J. Exp. Bot. 2018, 69, 2847–2862. [Google Scholar] [CrossRef] [Green Version]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Boston, R.S.; Viitanen, P.V.; Vierling, E. Molecular chaperones and protein folding in plants. Plant Mol. Biol. 1996, 32, 191–222. [Google Scholar] [CrossRef]
- Charng, Y.-Y.; Liu, H.-C.; Liu, N.-Y.; Chi, W.-T.; Wang, C.-N.; Chang, S.-H.; Wang, T.-T. A Heat-Inducible Transcription Factor, HsfA2, Is Required for Extension of Acquired Thermotolerance in Arabidopsis. Plant Physiol. 2007, 143, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonsor, S.J.; Scott, C.; Boumaza, I.; Liss, T.R.; Brodsky, J.L.; Vierling, E. Heat shock protein 101 effects in A. thaliana: Genetic variation, fitness and pleiotropy in controlled temperature conditions. Mol. Ecol. 2008, 17, 1614–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.C.; Nakaminami, K.; Matsui, A.; Kobayashi, S.; Kurihara, Y.; Toyooka, K.; Tanaka, M.; Seki, M. Oligouridylate Binding Protein 1b Plays an Integral Role in Plant Heat Stress Tolerance. Front. Plant Sci. 2016, 7, 853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, D.Y.; Vierling, E.; Guy, C.L. Comprehensive Expression Profile Analysis of the Arabidopsis Hsp70 Gene Family. Plant Physiol. 2001, 126, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Bautista, N.; Fernández-Calvino, L.; Muñoz, A.; Castellano, M.M. HOP3 a new regulator of the ER stress response in Arabidopsis with possible implications in plant development and response to biotic and abiotic stresses. Plant Signal. Behav. 2017, 12, e1317421. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.V.; Wigge, P. H2A.Z-Containing Nucleosomes Mediate the Thermosensory Response in Arabidopsis. Cell 2010, 140, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Theologis, A.; Ecker, J.R.; Palm, C.J.; Federspiel, N.A.; Kaul, S.; White, O.; Alonso, J.; Altafi, H.; Araujo, R.; Bowman, C.L.; et al. Sequence and analysis of chromosome 1 of the plant Arabidopsis thaliana. Nature 2000, 408, 816–820. [Google Scholar] [CrossRef]
- Zsigmond, L.; Rigó, G.; Szarka, A.; Székely, G.; Ötvös, K.; Darula, Z.; Medzihradszky, K.F.; Koncz, C.; Koncz, Z.; Szabados, L. Arabidopsis PPR40 Connects Abiotic Stress Responses to Mitochondrial Electron Transport. Plant Physiol. 2008, 146, 1721–1737. [Google Scholar] [CrossRef] [Green Version]
- Stief, A.; Altmann, S.; Hoffmann, K.; Pant, B.D.; Scheible, W.R.; Bäurle, I. Arabidopsis miR156 Regulates Tolerance to Recur-ring Environmental Stress through SPL Transcription Factors. Plant Cell 2014, 26, 1792–1807. [Google Scholar] [CrossRef] [Green Version]
- Lämke, J.; Brzezinka, K.; Altmann, S.; Bäurle, I. A hit-and-run heat shock factor governs sustained histone methylation and transcriptional stress memory. EMBO J. 2016, 35, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D. Shotgun bisulphite sequencing of the Arabidop-sis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salanoubat, M.; Lemcke, K.; Rieger, M.; Ansorge, W.; Unseld, M.; Fartmann, B.; Valle, G.; Blöcker, H.; Perez-Alonso, M.; Obermaier, B.; et al. Sequence and analysis of chromosome 3 of the plant Arabidopsis thaliana. Nature 2000, 408, 820–822. [Google Scholar] [PubMed]
- Oufattole, M.; Park, J.H.; Poxleitner, M.; Jiang, L.; Rogers, J.C. Selective Membrane Protein Internalization Accompanies Movement from the Endoplasmic Reticulum to the Protein Storage Vacuole Pathway in Arabidopsis. Plant Cell 2005, 17, 3066–3080. [Google Scholar] [CrossRef] [Green Version]
- Siren, J.; Valimaki, N.; Makinen, V. Indexing Graphs for Path Queries with Applications in Genome Research. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Shekhawat, K.; Saad, M.M.; Sheikh, A.; Mariappan, K.; Al-Mahmoudi, H.; Abdulhakim, F.; Eida, A.A.; Jalal, R.; Masmoudi, K.; Hirt, H. Root endophyte induced plant thermotolerance by constitutive chro-matin modification at heat stress memory gene loci. EMBO Rep. 2021, 22, e51049. [Google Scholar] [CrossRef]
- Li, N.; Bo, C.; Zhang, Y.; Wang, L. PHYTOCHROME INTERACTING FACTORS PIF4 and PIF5 promote heat stress induced leaf senescence in Arabidopsis. J. Exp. Bot. 2021, 72, 4577–4589. [Google Scholar] [CrossRef]
- Wang, L.; Ma, K.-B.; Lu, Z.-G.; Ren, S.-X.; Jiang, H.-R.; Cui, J.-W.; Chen, G.; Teng, N.-J.; Lam, H.-M.; Jin, B. Differential physiological, transcriptomic and metabolomic responses of Arabidopsis leaves under prolonged warming and heat shock. BMC Plant Biol. 2020, 20, 86. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-Protein Interaction Networks, Integrated Over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Zang, C.; Schones, D.E.; Zeng, C.; Cui, K.; Zhao, K.; Peng, W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009, 25, 1952–1958. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visu-alization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.Y.; Pellegrini, M. BS-Seeker2: A versatile aligning pipe-line for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Kalderstam, J.; Edén, P.; Ohlsson, M. Finding risk groups by optimizing artificial neural networks on the area under the survival curve using genetic algorithms. PLoS ONE 2015, 10, e0137597. [Google Scholar] [CrossRef] [PubMed]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.M.; Gordon-Raagas, H.B.; Mulvey, M.A. Conditioning of Uropathogenic Escherichia coli for Enhanced Colonization of Host. Infect. Immun. 2009, 77, 2104–2112. [Google Scholar] [CrossRef] [Green Version]
- Zandalinas, S.I.; Fichman, Y.; Devireddy, A.R.; Sengupta, S.; Azad, R.K.; Mittler, R. Systemic signaling during abiotic stress combination in plants. Proc. Natl. Acad. Sci. USA 2020, 117, 13810–13820. [Google Scholar] [CrossRef] [PubMed]
- Balfagón, D.; Sengupta, S.; Gómez-Cadenas, A.; Fritschi, F.B.; Azad, R.K.; Mittler, R.; Zandalinas, S.I. Jasmonic Acid Is Required for Plant Acclimation to a Combina-tion of High Light and Heat Stress. Plant Physiol. 2019, 181, 1668–1682. [Google Scholar] [CrossRef] [Green Version]
- Rawat, V.; Abdelsamad, A.; Pietzenuk, B.; Seymour, D.K.; Koenig, D.; Weigel, D.; Pecinka, A.; Schneeberger, K. Improving the Annotation of Arabidopsis lyrata Using RNA-Seq Data. PLoS ONE 2015, 10, e0137391. [Google Scholar] [CrossRef]
- Pietzenuk, B.; Markus, C.; Gaubert, H.; Bagwan, N.; Merotto, A.; Bucher, E. Recurrent evolution of heat-responsiveness in Brassicaceae COPIA elements. Genome Biol. 2016, 17, 209. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Bassil, E.; Hamilton, J.S.; Inupakutika, M.A.; Zandalinas, S.I.; Tripathy, D.; Luo, Y.; Dion, E.; Fukui, G.; Kumazaki, A.; et al. ABA Is Required for Plant Acclimation to a Combination of Salt and Heat Stress. PLoS ONE 2016, 11, e0147625. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Guo, M.; Cui, M.; Yu, Y.; Cui, J.; Liang, C.; Liu, L.; Mo, B.; Gao, L. Multiomics Reveals the Regulatory Mechanisms of Arabidopsis Tissues under Heat Stress. Int. J. Mol. Sci. 2023, 24, 11081. https://doi.org/10.3390/ijms241311081
Chen H, Guo M, Cui M, Yu Y, Cui J, Liang C, Liu L, Mo B, Gao L. Multiomics Reveals the Regulatory Mechanisms of Arabidopsis Tissues under Heat Stress. International Journal of Molecular Sciences. 2023; 24(13):11081. https://doi.org/10.3390/ijms241311081
Chicago/Turabian StyleChen, Haolang, Mingxi Guo, Mingyang Cui, Yu Yu, Jie Cui, Chao Liang, Lin Liu, Beixin Mo, and Lei Gao. 2023. "Multiomics Reveals the Regulatory Mechanisms of Arabidopsis Tissues under Heat Stress" International Journal of Molecular Sciences 24, no. 13: 11081. https://doi.org/10.3390/ijms241311081